Metallomic EPR spectroscopy
Wilfred R.
Hagen
*
Department of Biotechnology, Delft University of Technology, Julianalaan 67, 2628BC Delft, The Netherlands. E-mail: w.r.hagen@tudelft.nl; Tel: +31-15-278-5051
First published on 28th July 2009
Abstract
Based on explicit definitions of biomolecular EPR spectroscopy and of the metallome, this tutorial review positions EPR in the field of metallomics as a unique method to study native, integrated systems of metallobiomolecular coordination complexes subject to external stimuli. The specific techniques of whole-system bioEPR spectroscopy are described and their historic, recent, and anticipated applications are discussed.
 Wilfred R. Hagen Wilfred R. Hagen | Fred Hagen is professor of Enzymology in the Department of Biotechnology at Delft University of Technology. He received his PhD (1982) from the University of Amsterdam working with E. C. Slater and S. P. J. Albracht. After a post-doc at The University of Michigan with W. R. Dunham and R. H. Sands, he returned to the Netherlands to join C. Veeger at Wageningen University, where he later became professor of Bioinorganic Chemistry. In 1995 he was also appointed professor of Physical Chemistry at Nijmegen University. In 2000 he moved to Delft for his present position. His research interest is in the biochemistry of metal ions at large. |
Metallomics and spectroscopy
As a first step in positioning biomolecular EPR spectroscopy in the methodological toolbox of metallomics, the concept of the metallome is pinned down, below, in two alternative definitions, with emphasis, respectively, on analytical versus structural aspects. The latter one is the preferred framework for metallomic EPR spectroscopy.In analogy with well-established notions of the genome and the proteome, the term ‘metallome’ was coined by R. J. P. Williams to mean ‘an element distribution in a cell resulting from the elements’ paths’.1 These ‘paths’ are the combined and integrated action on environmentally presented metal ions, of cellular sequestering, passive and active transport, storage and release, regulation of expression, and insertion into biomacromolecules, altogether resulting in a particular distribution of metals over compartments in terms of concentration and speciation (coordination chemistry, oxidation state). Williams and Fraústo da Silva also proposed that this distribution set can be alternatively represented by ‘the free element content of the cell in every one of its compartments’ when combined with the set of binding constants to all bio(macro)molecules (and their intrinsic biological activities) in the cell.1–6 Several authors have interpreted this statement as equating the free element content per se to the metallome, and they have subsequently formulated their extensions of this restricted definition.7–9 Williams and Fraústo da Silva explicitly noted that the metallome is a function not only of the cellular genome and proteome, but also of the surrounding environment including its energy supplying capacity (ref. 1,2,5,6; see also ref. 10,11). One could add that this function is of a recurrent nature because the proteome (and on a significantly longer time scale also the genome ) is itself a function of the environment.
Thus, metallomics, or the study of the metallome in terms of external trigger induced responses in cellular metal concentrations, localizations, and speciations, can potentially provide a rich, integrative body of cell physiological and bioinorganic chemical knowledge. However, its Achilles’ heel is in the ‘speciation’ part which implies structural and thermodynamic knowledge at the (sub)molecular level, a commodity of limited availability and uneven distribution, because acquiring this knowledge is a long-term work in progress. For example, for the metalloproteomic subset of the metallome this is reflected in the present-day’s very asymmetric annotation of genomes : a usually modest part of the encoded proteins has been biochemically scrutinized, a larger part has been annotated (presumably with numerous errors) by in silicogene comparison, and an often even larger part is made up of ‘possible genes’ also known as putative proteins or conserved hypothetical proteins.
Frequently, the complexity of ‘speciation’ is reduced by simplifying it to mean associations between metal ions and, e.g., specific proteins (or even proteins in general), and thus the metallomic experiment becomes an analytical chemical determination of concentrations of elements in (possibly compartmentalized) ‘pools’ of solvent, small molecules, and macromolecules. One could call this approach the study of the ‘elemental’ metallome to discriminate it from the more ambitious coordination chemical study of the ‘molecular’ metallome. Zerkle et al. have further reduced the elemental metallome to exclude all non-protein metal complexes, and have used annotated microbial genomes to construct ‘the model metallome’ of putative transition-ion metalloproteins.12 Dupont et al. have reduced the elemental metallome even further to also exclude all (putative) metalloproteins that cannot be assigned with confidence to abundant and well-characterized fold superfamilies and/or fold families, and they have named this restricted subset ‘the reconstructed metallome’.13
In preparing to position EPR spectroscopy in metallomics, it is useful to note that proteomics has gradually evolved from the study of the proteome per se into attempts to develop integrated views of all pair-wise interactions of all cellular proteins and other biomacromolecules. The latter is usually named ‘functional proteomics’. Likewise, one can study the metallome (be it elemental or molecular) initially in a reductionistic approach, and then subsequently delve into the broader field of functional metallomics, which encompasses interactions between the members of the metallome. Thus, the metallome can be scrutinized at multiple levels of complexity (Fig. 1). A key question in the present context is whether or not biomolecular metal complexes are destroyed in the act of study (as, e.g., in 2D gel electrophoresis or mass spectrometry of tryptic digests), because spectroscopy only makes sense when applied to intact systems.
 | ||
| Fig. 1 The metallome can be studied at different levels of complexity. In this scheme dotted lines are compartmentalizing membranes, the outermost line delineating a cell or a protoplast (this schematic cell contains two organelles). Open solid circles/ellipses are outlines of biomacromolecules. ‘M’ is a metal, ‘L’ is a ligand, and three types of coordination complexes, or pools, are indicated: prosthetic groups in biomacromolecules (ML4 in a circle), small-molecule complexes (free ML4), and aqua cations (free M). The red numbers 1–5 are labels for increasing levels of resolution: (1) the presence of a metal in whole cells; (2) distribution over different organelles; (3) association with particular pools (macromolecules, small molecules, free ions); (4) bound to a specific class of bio(macro)molecules; and (5) as the prosthetic group of a particular protein with detailed specific coordination chemical structure. | ||
EPR spectroscopy
Electron Paramagnetic Resonance (EPR) spectroscopy is a method to determine the electronic structure of molecules by measuring the magnetism of electrons in an external magnetic field plus the modifications of this magnetism by virtue of the binding of these electrons to molecular structures. The conceptual reference point of the spectroscopy is a free electron not subjected to any interaction except (i) with a static magnetic field of flux density B, (ii) with a thermal ‘surrounding’ that gives the electron a temperature T, and (iii) with a monochromatic radiation with frequency ν in the microwave range. These three interactions are called, respectively, the electronic Zeeman interaction , the spin–lattice relaxation, and the EPR. The quantum-mechanical nature of the electron is manifest in the fact that its Zeeman interaction defines exactly two stable energy states, which, when the electron is viewed as a minuscule bar magnet, correspond to parallel and antiparallel orientation in the dipolar magnetic field. In zero field the two states are degenerate; in finite field the state of antiparallel orientation has the lowest energy, i.e. the ground state. Transition from the ground state to the excited state is possible by absorption of microwave radiation of energy hν when exactly equal to the Zeeman energy difference between the states of geβB. This resonance condition: hν = geβB, in which h and β are fundamental constants and the dimensionless proportionality constant ge = 2.00232, can be written as ge = (h/β)(ν/B), or: 2.00232 = 0.714484ν[MHz]/B[gauss]. Thus, for a common microwave frequency of 9.5000 GHz the resonance field B of a free electron is 3389.9 gauss (or 0.33899 tesla), which is easily attainable with an electromagnet.When bound to a molecule the electron will orbit around one or more positively charged nuclei, and it will therefore feel an internal magnetic field in addition to the external field B. This is usually expressed as a ‘shift’ in the g-value proportionality constant: hν = (ge + Δg)βB, or simply hν = gβB, in which the deviation from ge contains chemical information on the molecular structure. For the same fixed microwave frequency of 9.5 GHz the resonance absorption of microwaves for a molecule-bound electron occurs at a field value different from that of the free electron. The plot of microwave absorption versus field (i.e. the EPR spectrum) is further complicated, in other words: has more chemical information, because: (1) the Zeeman energy gβB is a function of the orientation of the non-spherical molecule in the external magnetic field, which results in three g-values along the molecular axes: gx, gy, gz, (2) interaction between the observed electron and other electrons in the molecule (and possibly in nearby molecules), and (3) interaction between the observed electron and magnetic nuclei in the molecule. These complications are called, respectively, g-anisotropy, zero-field interaction, and hyperfine interaction. Together they determine the molecular EPR spectrum as illustrated in Fig. 2. A few monographs are available on the subject of molecular14 or biomolecular15EPR spectroscopy.
 | ||
| Fig. 2 Schematic analysis of a molecular EPR spectrum. Typically, the EPR spectrum from a randomly oriented (bio)molecule is analyzed in terms of three main magnetic interactions: (1) the line positions expressed as g-values from the Zeeman interaction between unpaired electrons and an external field; (2) the line splittings from the hyperfine interaction between electrons and magnetic nuclei; (3) the line shifts from the zero-field interaction between different unpaired electrons (including dipolar interaction between different paramagnets). | ||
Metallomic EPR spectroscopy
Paramagnetic biomolecules are either radicals or transition-ion complexes. The observation that these systems in biomacromolecules are usually located ‘where the action is’, i.e. in active centers, confers an added relevance to biomolecular EPR spectroscopy. Paramagnetic biological transition ion coordination complexes can be S = 1/2 single unpaired electron low-spin systems (e.g., cupric proteins) or S = n/2 high-spin systems with an odd number of unpaired electrons (e.g., high-spin ferric proteins), or S = n integer spin with an even number of unpaired electrons (e.g., high-spin ferrous proteins). Radicals are predominantly S = 1/2 or single unpaired electron systems, e.g., amino acid based (tyrosyl, tryptophanyl, cysteinyl, glycyl), semiquinone organic (co)factors (flavin, quinone), or small inorganic molecules (nitric oxide, superoxide). This division into four classes (i.e.S = 1/2 radicals and S = 1/2, S = n/2, S = nmetal ions) is practical because it grosso modo corresponds to four types of EPR spectra and four different sets of experimental conditions.16 Note that complex biomolecules can have several, or all, of these systems in a single molecule, that a single center can be converted from one class to the other by redox chemistry, and that some single systems are substoichiometric mixtures of classes.Because EPR occurs between molecular levels that differ only by a small amount of energy (0.3 cm−1 for 9.5 GHz) the concentration sensitivity of the spectroscopy is disappointing from an analytical chemical viewpoint. Detection limit ballpark numbers range from circa 10−7 M for radicals, via 10−6–10−5 M for S = n/2 metalloproteins, to 10−3 M or worse for the most demanding systems (e.g., Ni2+ with S = 1). On the other hand, maximum concentration is only limited by molecular solubility and not by the spectrometer detection system. Biomolecular EPR spectroscopy is widely applied because its limited concentration sensitivity is counterbalanced by high spectral resolution of structures in active sites. Application of EPR spectroscopy to the characterization of metal centers in proteins and model compounds has been recently reviewed.16EPR spectroscopy of biomolecules concerns for a large part studies on isolated, purified, and concentrated protein samples, and this suggests a possible source of friction for its application in metallomics.
The term functional metallomics implies that the study of a metallome leads to an integrative overview of a complex network of metal homeostasis. How then does the method of EPR spectroscopy, with its established suitability for the characterization of single, well-defined systems of high concentration and purity, fit into a metallomics toolbox? Of course, there is nothing wrong with singling out a particular system as part of a metallomics study, and scrutinizing that isolated system with EPR spectroscopy, but this would not be different from a conventional experiment outside a metallomics framework. Here, on the contrary, we consider a specific branch of the spectroscopy with particular relevance to metallomics, namely its application to integral systems, such as whole cells or cellular organelles, carrying multiple paramagnetic entities that are typically not isolated, not purified, and not highly concentrated.
The application to integral biosystems is not new and in fact dates back to the very early history of biomolecular EPR spectroscopy. In recent times, however, the field has not only experienced a renaissance, but also a very significant extension in the spectroscopy of recombinant overexpression systems. Finally, the development of metallomics also provides an exciting new frame of reference for the spectroscopy, suggesting a broader interpretation of results as well as the design of new types of experiments.
Overview of methodologies
The very early history of biomolecular EPR spectroscopy attests to its potential as a tool in functional metalloproteomics. While the elements molybdenum and copper were long known to be present in biological preparations, their catalytic functionality was only clearly indicated for the first time in the fifties when MoV and CuIIEPR signals were detected in redox enzymes xanthine oxidase, cytochrome c oxidase, and laccase with anisotropic lineshapes (implying specific coordination) and with amplitudes responding to addition of substrate (implying functional changes in oxidation state).17–19 Even more striking are the historical discoveries of important new subclasses of the metallome, e.g., iron-sulfur proteins and nickel proteins on the basis of unusual EPR signals detected from membrane preparations and attributed to iron20–22 and nickel.23,24The development of functional metalloproteomic EPR spectroscopy is strongly linked to studies of integral systems of respiratory-chain complexes. Respiratory chains are metallomics objects par excellence because they are high-density submetallomes whose metal centers are functionally related. The typical aerobic , eukaryotic respiratory chain (Fig. 325) consists of the four complexes NADH:coenzyme Q oxidoreductase, succinate:coenzyme Q oxidoreductase, reduced coenzyme Q: ferricytochrome c oxidoreductase, and ferrocytochrome c: O2oxidoreductase, possibly with the addition of complexes that connect fatty acid breakdown to the coenzyme Q pool. The sub-metallome of such a system encompasses a dozen or more different iron-sulfur clusters, half a dozen or more heme groups, and two copper centers, and these all work together in an integrated supramolecular system for the transduction of Gibbs free energy from redox chemistry into the equivalent energy associated with a proton-motive force. EPR spectroscopy quantitatively ‘sees’ all these centers simultaneously in (frozen) action. Since each individual metal prosthetic group is a one-electron donor/acceptor, it has an EPR spectrum either in its oxidized state or in its reduced state, and the two states are interconvertible via an externally adjustable bulk potential (see below). With reference to a canonical respiratory chain, we will now illustrate that a metallomic EPR analysis can be carried out at different levels of increasing sophistication.
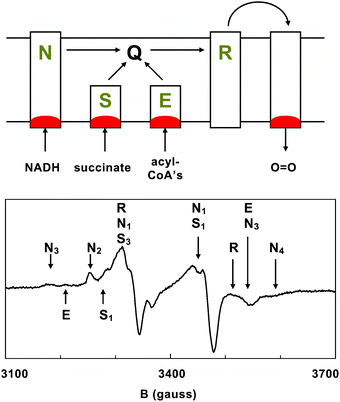 | ||
| Fig. 3 Metallomic EPR spectroscopy of a rat heart. When the respiring cells of a rat heart are frozen in action with a liquid-nitrogen cooled Wollenberger tong, the resulting EPR spectrum is dominated by the sub-metallome of the partially reduced aerobic respiratory chain showing a variety of iron-sulfur centers from (N) NADH dehydrogenase, (S) succinate dehydrogenase, (E) electron-transferring flavoprotein dehydrogenase, and (R) the Rieske protein in complex-III or ubiquinone: cytochrome c oxidoreductase (data modified from ref. 25). | ||
In an elemental metallomic EPR study, metal speciation can be determined at the level of classes of prosthetic groups plus enzyme association: each EPR signal can be associated with a group (iron-sulfur, heme, etc.) and with an enzyme complex (NADH dehydrogenase, etc.). Individual assignment is facilitated by the use of specific oxidants and/or reductants (NADH, succinate, cytochrome c, molecular oxygen) possibly in combination with site-specific inhibitors of electron transfer. The concentration of each individual metal group can, in principle, also be determined and these values together define intramolecular stoichiometries as well as enzyme complex stoichiometries. The ‘in principle’ is to indicate that the complexity of these determinations goes well beyond that of analytical element measurements. The methodology and its pitfalls are treated, below, in a separate paragraph on quantitative EPR analysis of complex systems.
At the next level of sophistication, the static structures of the prosthetic groups as coordination complexes can be included in the metallome analysis. The ‘powder pattern’ in the EPR spectrum from a frozen solution is a reflection of these structures and can be used to determine the symmetry of a complex (axial, orthorhombic, or lower), and, by comparison with known structures and their spectra, to determine ligation (deviations from all-Cys for Fe/S clusters; axial ligands for hemes). Furthermore, the working mechanism of inhibitors, when operative through direct interaction with metal centers, can be determined (e.g., 2,3-dimercaptopropanol specifically destroys the Rieske [2Fe-2S] cluster in complex III26). Finally, mutual electronic interaction between metal centers can be mapped from EPR spectra by analysis of through-space dipolar interactions and through-bond exchange interactions. This last step, however, requires considerable theoretical insight and practical experience (e.g. Ch. 11 in ref. 15).
Rather more straightforwardly, mutual thermodynamic relations between metal centers can be established by the determination of their individual reduction potentials by means of monitoring individual EPR signal amplitudes as a function of applied bulk potential in mediator-equilibrated redox titrations. When carried out at different pH values the analysis can also provide information on protonation of metal centers and on the dependence of reduction potentials on pKa values. Knowledge of all individual reduction potentials allows a sorting of metal centers according to increasing potential, and this typically provides a clear indication of the functional order in which reducing equivalents are transferred through the system.
Sorting according to reduction potential is not flawless because complex biological metal redox systems typically employ a small number of ‘uphill jumps’ in which the linear order of Em’s is broken, e.g., for specific regulation such as the diode-mimicking electron gating in succinate dehydrogenase.27 In these cases, the actual order of electron transfer can only be determined unequivocally in pre-steady-state kinetic studies. Such endeavors can furthermore identify short-lived reaction intermediates and thus contribute to a yet higher level of understanding of the metallome (see below).
Finally, all or part of the above methods are applicable in a wider frame of functional metallomics when used in comparative studies of (parts of) cells grown under different conditions (e.g., aerobic versusanaerobic 28), or poised under different conditions (energized versus resting mitochondria29,30), or from different lineages or species (the respiratory chain of mitochondriaversus that of the ‘mitochondrion-like’ bacterium Paracoccus denitrificans31) or, more recently, with differentially induced expression levels of metalloproteins.
Choosing and preparing the system
We have already noted that a key limiting factor in applications of EPR spectroscopy to biological systems is its relatively limited concentration sensitivity. For example, a typical detection limit for a garden variety dinuclear [2Fe-2S] or cubane [4Fe-4S] S = 1/2 iron-sulfur cluster is, say, 5 μM, which is obviously easily attainable, and surpassable, with preparations of purified metalloproteins: it corresponds to a protein concentration of 0.5 mg ml−1 for a 100 kDa protein, and aqueous solutions of reasonably soluble proteins can usually be concentrated without serious difficulty up to 100 mg ml−1. However, it poses a significant challenge with integral (sub)metallomes such as cell organelles. Aside from a few exceptional cases, such as the millimolar concentration of hemoglobin iron in red blood cells or the 50–100 μM pool concentration of some simple electron-transfer proteins (ferredoxin, c-type cytochrome) in microorganisms, the concentration of specific metal complexes (including members of the free metallome) in cells will be less by one, or more, or many more, orders of magnitude, hence the general need to concentrate metallome samples as much as possible before EPR scrutiny.To concentrate protein, a variety of methods is available including filtering in combination with overpressure of an inert gas, or in combination with centrifugation, freeze drying plus redissolution in minimal volume, or simply ‘wet drying’ by application of a ‘mild vacuum’ (i.e. pumping to circa 0.1 atm under pressure). None of these methods are particularly suitable for integral systems: filters will easily foul, a freeze/thaw cycle compromises membrane integrity, and concentration by pumping on high ionic strength solutions is inefficient. A useful approach is the method of centrifugation: low-g force for whole cells, intermediate-g for organelles, and ultracentrifugation for ‘fragments’ e.g., sub-mitochondrial particles. The resulting pellets can be re-suspended by careful swirling with a rounded glass rod in minimal buffer volume (i.e. approximately equal to one or a few times the volume of the pellet). With whole-cell preparations a significant (i.e. order of magnitude) extra concentration can be achieved in a standard EPR tube (typically 14 cm in length and 5.0 and 4.7 mm outer and inner diameter, respectively) as follows: fill the tube to the rim with a thick cell suspension (circa 1.5 ml), place it in a table centrifuge for reaction tubes with sufficient support to avoid breaking the tube, spin at low g, and remove the upper liquid (circa 1.3 ml) by suction with flexible tubing.
Whole organs (Fig. 3) can be ‘caught in the act’ in a specific metabolic state by freeze clamping, for example, by means of a device called a Wollenberger tong cooled in liquid nitrogen, followed by grinding the sample in a mortar filled with some liquid nitrogen.25 Note that, although in the literature this grinding procedure has frequently been associated with the creation of radicals, there is no compelling evidence to conclude that this notion is more than folkloristic superstition.
Freezing and thawing of aqueous solutions does not usually affect protein structure in an irreversible manner, but it does lead to massive disruption of membranes, as evidenced, e.g., by complete uncoupling of oxidative phosphorylation. Addition of circa 10% by volume of a cryoprotectant such as glycerol largely prevents the disruption: with pigeon-heart mitochondria over 90% of ADP phosphorylation with NADH reduction is thus retained after a freeze/thaw cycle (my unpublished observations). On the other hand, one may also deliberately make cells or organelles ‘leaky’ by means of one, or a few freeze/thaw cycles, in order to make the metallome available for conditioning such as global poising of the redox potential with added reductant/oxidant.
Quantitative EPR analysis of complex systems
A major goal of metallomic EPR analysis of complex systems is the quantitative deconvolution of spectra in identifiable components (spectra from individual metal species such as a particular prosthetic group, Fig. 3), and this includes determination of the relative stoichiometries of all components. One usually tries to disentangle complex spectra by recording differential responses of components to externally applied perturbations in particular to changes in microwave power, in sample temperature, or in poised redox potential.Changing the microwave power and/or the sample temperature is experimentally simple, but the results can be very difficult to interpret. The underlying concept is that of a difference in relaxation rate between paramagnetic centers: at sufficiently low temperature, T, increasing the microwave power, P, eventually leads to saturation of the absorption, and this happens at different T, P values for different metal centers.16 Thus spectral amplitudes of individual centers can be subsequently reduced by partial saturation, while amplitudes of other centers with fast relaxation rates remain relatively unaffected, and deconvolution is straightforward by making difference spectra as illustrated in Fig. 4A&B. A fundamental weakness of this approach is in the tacit assumption that each single-component spectrum saturates homogeneously, i.e. each part is equally affected. In practice this is almost never true because spectra of biological metal complexes are inhomogeneously broadened by virtue of conformational distributions, in the EPR literature also known as g-strain: each molecule in a real sample has a slightly different 3D structural conformation, therefore a slightly different EPR spectrum, therefore a slightly different relaxation rate, therefore a slightly different saturation behavior.32 By consequence, for different parts of the spectrum the extent of saturation will differ, and, therefore, the overall shape of the EPR spectrum will change (deform) under partial saturation, with each value of the applied microwave power giving rise to its own modified spectrum (Fig. 4C&D). An analysis based on taking difference spectra for different power settings can result in a large number of artifactual components. Remarkably, although complex saturation behavior associated with g-strain was identified 27 years ago,32 up until this date its notion is broadly ignored in metallomic EPR studies.
 | ||
| Fig. 4 The saturation problem in quantitative EPR of complex systems. The problem is illustrated by the simple case of the overlapping spectra, blue and green in trace A, from two hypothetical iron-sulfur centers, with the blue spectrum more readily saturable than the green one. In trace B, the grey lines give their sum-spectra without saturation (solid line) and with the blue component’s intensity half reduced by saturation (broken line). The red trace recovers the spectrum of the green component by taking a proper difference between the grey spectra. Trace C shows the effect of g-strain related change in line shape (broken line) due to inhomogeneous saturation for the blue component (solid line). In trace D the method of taking the difference between spectra taken at different saturation levels can now be seen to not only recover the green component, but also an artifactual component created by ignoring the saturation-induced change in the line shape of the blue component. | ||
The pitfalls of inhomogeneous saturation can be avoided by analyzing non-saturated spectra by means of multi-component simulation. Unfortunately, this approach is a tedious one due to theoretical and numerical difficulties that originate in the complex nature of the g-strain phenomenon.33,34 In summary, a simplifying (i.e.g-strain ignoring) approach to multicomponent EPR spectral deconvolution is at this time still widely divulged (e.g., ref. 35,36), and this practice can result in sub-optimal resolution power. An illustrative example of what this can lead to, may be found in the recent vigorous debate on sub-metallome EPR analysis of the multiple iron-sulfur centers in the respiratory complex-I of Escherichia coli.37,38 The problems associated with inhomogeneous saturation and with involved lineshape analysis can be alleviated by using a rather different, experimentally more demanding approach: deconvolution based on differences in reduction potential.
Clamping metallomes
Typically, in a functional-proteomics experiment proteomes are compared from cells subject to different environmental constraints such as in aerobic versusanaerobic microbial growth. In the spectroscopic application of metalloproteomic EPR, a conceptually similar differentiation is possible by clamping the system before freezing by means of a physico-chemical parameter at two or more values, e.g., temperature, pressure, pH, ionic strength. At this time, the literature on the subject is still limited. We consider two parameters for which results have been reported, namely: (i) bulk redox potential and (ii) time of incubation with a ‘global’ substrate.The bulk potential of a biological sample can be set by anaerobic stepwise substoichiometric additions of a relatively low-potential reductant (e.g., dithionite anion) or high-potential oxidant (e.g., ferricyanide) provided all components in the system (including a measuring and a reference electrode) equilibrate with the reductant/oxidant within a practical time, say minutes. This condition is approached by the addition of a ‘redox cocktail’, i.e. a mixture of several redox dyes whose standard reduction potentials form a ladder of values with an average step of circa 50 mV.15,39 Equilibration time can be reduced by an increase in the concentration of the dyes and/or the number of different dyes. A complicating factor with whole cells and with cell organelles may be the fact that each dye has its own partition coefficient over the water versuslipid phase. Remarkably, promising results of early studies by Ohnishi on mammalian mitochondria29,40 do not appear to have found follow-up studies on (sub)metallomes in more recent times, in spite of our dramatically increased knowledge on the 3D structure of, e.g., respiratory chain complexes, and our significantly increased understanding of the shape and saturation behavior of the EPR spectral components. A wide area of potentially rewarding metallomic EPR research appears to be waiting here for exploration.
Addition of a global substrate (i.e. effecting a range of interacting cellular components), such as NADH or O2, and subsequent clamping at defined points in time, affords a collection of intermediates in pre-steady-state kinetics. The methodology of rapid mixing and rapid freezing was developed and applied to purified enzymes by R. C. Bray et al.,41,42 and was subsequently applied to complex subcellular systems notably respiratory chain complexes.43,44 Recent developments have drastically reduced a long-standing dead time of 5–10 milliseconds into the microsecond time domain,45,46 and the first ‘ultrarapid’ studies of complex systems have appeared.47–50 Applications to cell organelles or whole cells have not been reported yet, although no additional technical difficulties are anticipated.
Metallomic de-focusing and re-focusing
Over the years, the study of a particular class of prosthetic groups may swing back and forth between the bird’s-eye view of functional metallomics and the detailed scrutiny of particular coordination chemical structures. An illustrative case in point is the class of [3Fe-4S] clusters whose history traces back four decades to early studies in metallomic EPR analysis of mitochondrial systems of oxidative phosphorylation. Attempts at comprehensive assignment of the many observed individual EPR signals to prosthetic groups in the respiratory chain complexes, would always leave one component unassigned: the g = 2.01 signal.51 In mammalian cells the signal was found to be associated with three sub-metallomes, namely with the mitochondrial soluble fraction, with the respiratory complex II, and, later, with the soluble cytoplasmic fraction. In the following decade, biochemists then turned away from the metallomic viewpoint to focus on identifying individual sources of the unassigned signal. Briefly, g = 2.01 in the soluble mitochondrial fraction was found to be from an inactivated form of the citric acid cycle enzyme aconitase, and was identified with the oxidized form of a ‘cuboidal’ [3Fe-4S] cluster, i.e. a [4Fe-4S] cube with one Fe missing, and liganded by three Cys residues. Addition of Fe2+ under reducing conditions would restore aconitase activity by formation of a cube with the fourth iron, not coordinated by the protein, as a Lewis-acid catalyst for dehydration/hydration of citrate to isocitrate. Subsequently, the cytoplasmic g = 2.01 signal was identified with ‘cytoplasmic aconitase’, which turned out to be the incompletely iron loaded form of the iron responsive element (IRE1) or iron regulatory protein (IRP1), a translation regulatory factor that interacts with mRNAs encoding proteins functional in iron homeostasis, e.g., the transferrrin receptor or the ferritin storage protein (cf. the reviews52,53).Once this detailed knowledge was established, the time had come to step back again for a wider view of the integrated system, and this swing was initiated by a whole-cell study in which the g = 2.01 signal associated with respiratory chain complex II was shown to be a physiologically functional (in electron transfer) [3Fe-4S] prosthetic group in E. coli cells54 (in contrast to the [3Fe-4S] isolation artifact in mitochondrial aconitase). This study is also an early exploration of a technique that is by now frequently used in whole cell or cell organelleEPR studies, namely, the amplification of a particular metal-containing component over the metallome background by plasmid-borne overexpression of relevant proteins in order to compensate for the intrinsically low concentration sensitivity of the EPR method. In follow-up studies, the reduction potential of the cluster in comparison with other clusters in the submetallome of membrane particles was also determined by the method outlined above.55,56
More recently, homologous overexpression has also been used in a study of fumarase repair stimulated by the cysteine desulfurase IscS in whole E. coli cells. Bacterial fumarase is another citric acid cycle enzyme with a Lewis acid catalytic center subject to [4Fe-4S] ↔ [3Fe-4S] conversion. The in vivo breakdown of the cluster due to H2O2 is readily EPR-monitored in the overexpressing cells as the g = 2.01 signal stands out over the rest of the EPR detectable metallome.57
In an approach that could perhaps be seen as the mirror image of the previous procedure of overexpression, a yeast (Saccharomyces cerevisiae) strain that was deficient in mitochondrial aconitase, was used in a whole cell EPR study on the functioning of the cytosolic aconitase (IRP1), which was detected against a metallome background whose EPR was apparently dominated by Mn2+ with additional interference from a tyrosine radical from ribonucleotide reductase, but without mitochondrial aconitase signals.58 The [3Fe-4S] form was found to be a relevant intermediate in the in vivo conversion of IRP1 from apo to [4Fe-4S] protein. This work is instructive as an illustration of typical detection limits (here circa 1 μM aconitase) in whole cell EPR studies under carefully chosen conditions.
Outlook
EPR spectroscopy exclusively detects paramagnetic compounds and, therefore, can only contribute partial data sets to metallome analysis. Furthermore, its relatively modest spectrometric concentration sensitivity of typically 10−7 to 10−3 M excludes the detection of, e.g., metalloproteins of low expression levels unless homologous overexpression approaches are invoked. On the other hand, the afforded spectroscopic information on biological transition ions can be rich and detailed in terms of the coordination chemistry of individual complexes and their dependence on thermodynamic and kinetic conditioning. Moreover, the EPR measurement of a paramagnetic metal complex in a whole cell, or in a fractionated cell, is spectroscopically not in any way more involved than its detection in purified biomolecules except for the obvious limits in concentratability. In the field of bioenergetics, biological EPR spectroscopy has an impressive track record for the scrutiny of sub-metallomes formed by respiratory chains and their individual multi-center complexes. This history holds considerable promise for extended paramagnetic metallome research, in which specific modifiers (notably: redox potential poising, ultra-rapid kinetics, homologous overexpression) will be employed to their full capacity, and in which analysis will be based on our increasing knowledge of biometal coordination chemistry and of the intricacies of EPR spectra from biomolecules. The focus thus far on mitochondrial systems has left many variants of anaerobic microbial life to be explored. From a medical perspective it is noticeable that metallomic type approaches, employing whole-cell EPR spectroscopy for research on standards versus pathological variants of human or animal-model cell types, have recently shown a minor boom in the literature35,59–61 (Fig. 5). Also, combinations with complementary spectroscopies, notably low-temperature magnetic Mössbauer spectroscopy, are beginning to be explored in whole-cell studies.36,62 And several recent attempts have been reported to coordination-chemically define elusive subjects like the “free” metallome of Fe(III), also known as the ‘chelatable iron pool’.63–68 In this probing of biological variation and by searching for fruitful combinations with analytical methodologies, the EPR spectroscopist has irrevocably begun to find his specific niche in the vast field of metallomics. | ||
| Fig. 5 An example of metallomic EPR in medically oriented research. This spectrum of rabbit venous blood is the control in a study on transfusion-induced oxidative damage, and it shows signals identifiable with (1) high spin heme Fe(III) in catalase; (2) high-spin heme Fe(III) in methemoglobin; (3) high-spin non-heme Fe(III) in transferrin; (4) Cu(II) in ceruloplasmin; (5) a globin-associated radical (reproduced with permission, from Dunne et al., 2006, Biochemical Journal 399: 513–524. © The Biochemical Society59). | ||
References
- R. J. P. Williams, Coord. Chem. Rev., 2001, 216–217, 538–595.
- J. J. R. Fraústo da Silva and R. J. P. Williams, The Biological Chemistry of the Elements; The Inorganic Chemistry of Life, Oxford University Press, Oxford, Second edn, 2001 Search PubMed.
- R. J. P. Williams and J. J. R. Fraústo da Silva, J. Theor. Biol., 2003, 220, 323–343 CrossRef CAS.
- R. J. P. Williams, Acta Biochim. Pol., 2004, 51, 281–298 CAS.
- R. J. P. Williams and J. J. Fraústo da Silva, J. Chem. Educ., 2004, 81, 738–749 CrossRef CAS.
- R. J. P. Williams and J. J. R. Fraústo da Silva, The Chemistry of Evolution: The Development of our Ecosystem, Elsevier, Amsterdam, 2006 Search PubMed.
- J. Szpunar, Anal. Bioanal. Chem., 2004, 378, 54–56 CAS.
- H. Haraguchi, J. Anal. At. Spectrom., 2004, 19, 5–14 RSC.
- J. Szpunar, Analyst, 2005, 130, 443–465 Search PubMed.
- L. P. Wackett, A. G. Dodge and L. B. M. Ellis, Appl. Environ. Microbiol., 2004, 70, 647–655 CrossRef CAS.
- D. J. Thiele and J. D. Gitlin, Nat. Chem. Biol., 2008, 4, 145–147 CrossRef CAS.
- A. L. Zerkle, C. H. House and S. L. Brantley, Am. J. Sci., 2005, 905, 467–502 Search PubMed.
- C. L. Dupont, S. Yang, B. Palenik and P. E. Bourne, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17822–17827 CrossRef CAS.
- J. A. Weil and J. R. Bolton, Electron Paramagnetic Resonance: Elementary Theory and Practical Applications, Wiley, Hoboken, NJ, Second edn, 2007 Search PubMed.
- W. R. Hagen, Biomolecular EPR Spectroscopy, CRC Press, Boca Raton, Fl, 2009 Search PubMed.
- W. R. Hagen, Dalton Trans., 2006, 4415–4434 RSC.
- R. C. Bray, B. G. Malmström and T. Vänngård, Biochem. J., 1955, 73, 193–197.
- R. H. Sands and H. Beinert, Biochem. Biophys. Res. Commun., 1959, 1, 175–178 CrossRef CAS.
- B. G. Malmström, R. Mosbach and T. Vänngård, Nature, 1959, 183, 321–322 CrossRef CAS.
- H. Beinert and R. H. Sands, Biochem. Biophys. Res. Commun., 1959, 1, 171–174 CrossRef CAS.
- H. Beinert and R. H. Sands, Biochem. Biophys. Res. Commun., 1960, 3, 41–46 CrossRef CAS.
- R. H. Sands and H. Beienert, Biochem. Biophys. Res. Commun., 1960, 3, 47–52 CrossRef CAS.
- J. R. Lancaster Jr, FEBS Lett., 115, 285–288 Search PubMed.
- S. P. J. Albracht, E.-G. Graf and R. K. Thauer, FEBS Lett., 1982, 140, 311–313 CrossRef CAS.
- A. M. M. van der Kraaij, J. F. Koster and W. R. Hagen, Biochem. J., 1989, 264, 687–694 CAS.
- E. C. Slater and S. de Vries, Nature, 1980, 288, 717–718 CrossRef CAS.
- F. A. Armstrong, H. A. Heering and J. Hirst, J. Am. Chem. Soc., 1997, 26, 169–179 CAS.
- R. Godjieva, F. Mamedov, G. Renger and S. Styring, Biochemistry, 1999, 38, 10578–10584 CrossRef.
- T. Ohnishi, Eur. J. Biochem., 1976, 64, 91–103 CAS.
- A. D. Vonogradov, V. D. Sled, D. S. Burbaev, V. G. Grivennikova, I. A. Moroz and T. Ohnishi, FEBS Lett., 1995, 370, 83–87 CrossRef.
- S. P. J. Albracht, H. W. van Verseveld, W. R. Hagen and M. L. Kalkman, Biochim. Biophys. Acta, 1980, 593, 173–186 CAS.
- W. R. Hagen and S. P. J. Albracht, Biochim. Biophys. Acta, 1982, 702, 61–71 CrossRef CAS.
- W. R. Hagen, D. O. Hearshen, R. H. Sands and W. R. Dunham, J. Magn. Reson., 1985, 61, 220–232 CAS.
- W. R. Hagen, D. O. Hearshen, L. J. Harding and W. R. Dunham, J. Magn. Reson., 1985, 61, 233–244 CAS.
- D. A. Svistunenko, N. Davies, D. Brealey, M. Singer and C. E. Cooper, Biochim. Biophys. Acta, 2006, 1757, 262–272 CAS.
- B. N. Hudder, J. G. Morales, A. Stubna, E. Münck, M. P. Hendrich and P. A. Lindahl, JBIC, J. Biol. Inorg. Chem., 2007, 12, 1029–1053 CrossRef CAS.
- G. Yakovlev, T. Reda and J. Hirst, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 12720–12725 CrossRef.
- T. Ohnishi and E. Nakamaru-Ogiso, Biochim. Biophys. Acta, 2008, 1777, 703–710 CAS.
- P. L. Dutton, Methods Enzymol., 1978, 54, 411–435 Search PubMed.
- W. J. Ingledew and T. Ohnishi, Biochem. J., 1980, 186, 111–117 CAS.
- G. Palmer, R. C. Bray and H. Beinert, J. Biol. Chem., 1964, 239, 2657–2666 CAS.
- R. C. Bray, G. Palmer and H. Beinert, J. Biol. Chem., 1964, 239, 2667–2676 CAS.
- S. de Vries, S. P. J. Albracht, J. A. Berden and E. C. Slater, Biochim. Biophys. Acta, 1982, 681, 41–53 CAS.
- R. van Belzen and S. P. J. Albracht, Biochim. Biophys. Acta, 1989, 974, 311–320 CAS.
- M. Tanaka, K. Matsuura, S. Yoshioka, S. Takahashi, K. Isimori, H. Hori and I. Morishima, Biophys. J., 2003, 84, 1998–2004 CrossRef CAS.
- A. V. Cherepanov and S. de Vries, Biochim. Biophys. Acta, 2004, 1656, 1–31 CAS.
- F. G. M. Wiertz, O.-M. H. Richter, A. V. Cherepanov, F. MacMillan, B. Ludwig and S. de Vries, FEBS Lett., 2004, 575, 127–130 CrossRef CAS.
- F. G. M. Wiertz, O.-M. H. Richter, B. Ludwig and S. de Vries, J. Biol. Chem., 2007, 282, 31580–31591 CrossRef CAS.
- K. A. Sam, M. J. F. Strampraad, S. de Vries and S. J. Ferguson, J. Biol. Chem., 2008, 283, 27403–27409 CrossRef CAS.
- M. L. Verkhovskaya, N. Belevich, L. Euro, M. Wikström and M. I. Verkhovsky, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3763–3767 CrossRef CAS.
- N. R. Orme-Johnson, W. H. Orme-Johnson, R. E. Hansen, H. Beinert and Y. Hatefi, Biochem. Biophys. Res. Commun., 1971, 44, 446–452 CrossRef CAS.
- H. Beinert and M. C. Kennedy, FASEB J., 1993, 7, 1442–1449 CAS.
- H. Beinert, M. C. Kennedy and C. D. Stout, Chem. Rev., 1996, 96, 2335–2373 CrossRef CAS.
- M. K. Johnson, J. E. Morningstar, G. Cecchini and B. A. C. Ackrell, Biochem. Biophys. Res. Commun., 1985, 131, 653–658 CrossRef CAS.
- M. T. Werth, G. Cecchini, A. Manodori, B. A. C. Ackrell, I. Schröder, R. P. Gunsalus and M. K. Johnson, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 8965–8969 CAS.
- A. Manodori, G. Cecchini, I. Schröder, R. P. Gunsalus, M. T. Werth and M. K. Johnson, Biochemistry, 1992, 31, 2703–2712 CrossRef CAS.
- O. Djaman, F. W. Outten and J. A. Imlay, J. Biol. Chem., 2004, 279, 44590–44599 CrossRef CAS.
- N. M. Brown, M. C. Kennedy, W. E. Antholine, R. S. Eisenstrein and W. E. Walden, J. Biol. Chem., 2002, 277, 7246–7254 CrossRef CAS.
- J. Dunne, A. Caron, P. Menu, A. I. Alayash, P. W. Bueler, M. T. Wilson, R. Silaghi-Dumitrescu, B. Faive and C. E. Cooper, Biochem. J., 2006, 399, 513–524 CrossRef CAS.
- J. M. Kolesar, W. R. Schelman, P. G. Geiger, K. D. Holen, A. M. Traynor, D. B. Alberti, J. P. Thomas, C. R. Chitambar, G. Wilding and W. E. Antholine, J. Inorg. Biochem., 2008, 102, 693–698 CrossRef CAS.
- M. Elas, J. Bielanska, K. Pustelny, P. M. Plonka, L. Drelicharz, T. Skorka, U. Tyrankiewicz, M. Wozniak, S. Heinze-Paluchowska, M. Walski, L. Wojnar, D. Fortin, R. Ventura-Clapier and S. Chlopicki, Free Radical Biol. Med., 2008, 45, 321–328 CrossRef CAS.
- M. M. Cosper, G. N. L. Jameson, M. K. Eidsness, B. H. Huynh and M. K. Johnson, FEBS Lett., 2002, 529, 332–336 CrossRef CAS.
- K. Keyer and J. A. Imlay, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 13635–13640 CrossRef CAS.
- C. Srinivasan, A. Liba, J. A. Imlay, J. Selverstone Valentine and E. Butler Gralla, J. Biol. Chem., 2000, 275, 29187–29192 CrossRef CAS.
- G. Wang, R. C. Conover, A. A. Olczak, P. Alamuri, M. K. Johnson and R. J. Maier, Free Radical Res., 2005, 39, 1183–1191 CrossRef CAS.
- J.-F. Jacques, S. Jang, K. Prévost, G. Desnoyers, M. Desmarais, J. Imlay and E. Massé, Mol. Microbiol., 2006 DOI:10.1111/j.1365-2958.2006.05439.x (10 pp).
- W. Gao, Y. Liu, C. S. Giometti, S. I. Tollaksen, T. Khare, L. Wu, D. M. Klingeman, M. W. Fields and J. Zhou, BMC Genomics, 2006 DOI:10.1186/1471-2164-7-76 (13 pp).
- J. C. Toledo Jr, C. A. Bosworth, S. W. Hennon, H. A. Mahtani, H. A. Bergonia and Jack R. Lancaster Jr, J. Biol. Chem., 2008, 283, 28926–28933 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |