Unusual flexibility of distal and proximal histidine residues in the haem pocket of Drosophila melanogasterhaemoglobin†
Anda Iulia
Ioanitescu
a,
Sabine
Van Doorslaer
*a,
Sylvia
Dewilde
b and
Luc
Moens
b
aDepartment of Physics, University of Antwerp, Universiteitsplein 1, Wilrijk-Antwerp, Belgium. E-mail: sabine.vandoorslaer@ua.ac.be; Fax: +32 3 820 2470; Tel: +32 3 820 2461
bDepartment of Biomedical Sciences, University of Antwerp, Universiteitsplein 1, Wilrijk-Antwerp, Belgium. E-mail: luc.moens@ua.ac.be; Fax: +32 3 820 2248; Tel: +32 3 820 2323
First published on 16th April 2009
Abstract
Several pH-dependent low-spin ferric haem forms are identified in a frozen solution of the ferric 121Cys→Ser mutant of Drosophila melanogasterhaemoglobin (DmHb1*) using electron paramagnetic resonance (EPR) techniques. Different forms with EPR parameters typical of bis-histidine coordinated haem iron centers were observed. Strong pH-dependent changes in the EPR signatures were observed related to changes in the haem pocket. The pulsed EPR data indicate that both the distal and proximal histidine exhibit a large libration around the Fe–NHis axis. The resonance Raman spectra of the CO-ligated ferrous form of Drosophila melanogasterhaemoglobin are typical of an open conformation, with little stabilization of the CO ligand by the surrounding amino-acid residues. The EPR data of the cyanide-ligated ferric DmHb1* indicates a close similarity with cyanide-ligated ferric myoglobin. The structural characteristics of DmHb1* are found to clearly differ from those of other bis-histidine-coordinated globins.
Introduction
It was long assumed that most insects do not have respiratory proteins because the tracheal system should be sufficient for oxygen supply.1 Recently, intracellularhaemoglobins (Hbs) have been identified in different insects,2 including Drosophila melanogaster.3,4Drosophila species harbor three distinct globingenes,4 of which the globin 1 of Drosophila melanogaster (DmHb1) has the highest level of expression.2DmHb1 is a monomeric intracellularhaemoglobin of 153 amino acids (17 kDa) expressed at low concentration in the tracheal system and in the fat body of embryonic, larval and adult flies.5 Previous UV/Vis-absorption experiments showed that the haem-iron atom of DmHb1 is hexacoordinated in its ferric and ferrous forms.5 An X-ray study of the ferric form of the 121Cys→Ser mutant of DmHb1 confirmed the 3-on-3 α-helical fold characteristic of globins and a F8His–Fe(III)–E7His haem-binding mode6 The same study also revealed that the Fe–Nε coordination bond is of comparable length for both proximal and distal histidines (F8/E7His, 1.97 Å/2.02 Å). Moreover, the proximal/distal histidine planes show a staggering relative to the haempyrrole-nitrogen atoms with a dihedral angle of 88° between the two histidine planes.The X-ray structure of the cyanide-ligated ferric form of DmHb1 (DmHb1CN) is also available.7 The distance between the haem-iron atom and the proximal-histidinenitrogen (the carbon atom of the distal ligated cyanide) is 2.05 Å (2.03 Å). Moreover, the cyanide ligand is almost orthogonal to the haem plane. In DmHb1CN, the E7His is moved 2.7 Å away from the haem in comparison to the bis-histidine coordinated form, but has not swung out of the haem pocket. This is due to the strong hydrogen bonding between the E7His NE2 atom and the N atom of the cyanide ligand.7
Ligand-binding kinetics studies showed that the observed oxygen affinity of ferrous DmHb1 is much lower than the intrinsic oxygen affinity of the pentacoordinated form because of competition with the internal E7His residue.5 The P50 value of ferrous DmHb1 (0.12 torr)5 is comparable to that found for Gasterophilus Hb (0.15 torr).8
The available data on DmHb1 suggest that its primary role is that of a respiratory protein involved in oxygen storage and transport.5 However, other functions, such as oxygen sensing or involvement in nitric-oxide synthesis cannot a priori be excluded.
In this work, we present a resonance Raman (RR) and electron paramagnetic resonance (EPR) study of different forms of the 121Cys→Ser mutant of DmHb1, which we will indicate in this work as DmHb1*. We chose this mutant in order to be able to compare unambiguously with the previous X-ray data,6,7 but control experiments were done on wild-type DmHb1 (ESI† ). We will show that, despite the fact that a lot of structural data is already available on this protein, novel information can be obtained from these experiments that puts some of the earlier results in a new light.
Materials and methods
Materials
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 10 min. DmHb1* was refolded as follows. Hemin was dissolved in 0.1 M NaOH and 1/10 diluted with 50 mM Tris–HCl pH 7.5. A 1.4× excess of hemin solution was added to the solubilized protein. This solution was then dialyzed against 5 mM Tris–HCl pH 8.5. Precipitate formed during dialyses was removed by centrifugation at 10000g for 10 min. The refolded DmHb1* solution was concentrated by Amicon filtration (PM10) and passed through a Sephacryl S200 column. The pure DmHb1* fractions were pooled, concentrated and stored at 253 K.
000g for 10 min. DmHb1* was refolded as follows. Hemin was dissolved in 0.1 M NaOH and 1/10 diluted with 50 mM Tris–HCl pH 7.5. A 1.4× excess of hemin solution was added to the solubilized protein. This solution was then dialyzed against 5 mM Tris–HCl pH 8.5. Precipitate formed during dialyses was removed by centrifugation at 10000g for 10 min. The refolded DmHb1* solution was concentrated by Amicon filtration (PM10) and passed through a Sephacryl S200 column. The pure DmHb1* fractions were pooled, concentrated and stored at 253 K.
Since it was noticed from the EPR data that DTT can bind to the haem iron of DmHb1*, an additional acetoneprecipitation of the protein was performed. For this, cold acetone (5 ml) was added to the pure fractions and incubated for 60 min at −20 °C. The insoluble material was removed by centrifugation (10000g for 10 min). The pellets were then left at room temperature to allow the acetone to evaporate. The material was solubilised in 6 M guanidinium chloride and 50 mM Tris–HCl pH 7.5. The refolding procedure was repeated as described above.
For the pH dependence, separate buffer solutions for pH 5.8 (0.5 M citratebuffer), pH 8.5 (0.5 M Tris–HCl buffer) and pH 10 (0.5 M glycine–NaOHbuffer) were prepared.
The protein (haem) concentration used for optical and RR measurements was typically ∼30 μM. For all EPR measurements, 25% glycerol was added to all samples as a cryoprotectant. Protein (haem) concentrations of ∼0.5–2 mM were used for these experiments.
Methods
Electron-spin-echo (ESE)-detected EPR. 10 The spectra were recorded using the pulse sequence π/2–τ–π–τ–echo, with pulse lengths tπ/2 = 16 ns and tπ = 32 ns. The ESE was monitored as a function of the magnetic field for different τ values.
HYSCORE (hyperfine sublevel correlation) spectroscopy experiments 11 were carried out with the pulse-sequence π/2–τ–π/2–t1–π–t2–π/2–τ–echo. An eight-step phase cycle was performed to eliminate unwanted echo contributions. The following parameters were used: pulse lengths of tπ/2 = 16 or 24 ns and tπ = 16 ns, starting times of 96 ns for t1 and t2 and time increments of 16 ns (data matrix 250 × 250). Different τ values were taken to reduce the blind spots. In some cases, the second and third π/2 pulses were replaced by matching pulses.12 Furthermore, SMART HYSCORE experiments13 (HTA-ti–π–t2–HTA–τ–π–τ–echo) were performed. Specifications about the matching conditions are given in the figure captions.
Results
The UV/Vis-absorption spectrum of ferric DmHb1* indicates that the protein is dominantly in a ferric low-spin (LS) form5 (ESI, Fig. S1† ). Fig. 1 depicts the resonance Raman spectra of the ferric DmHb1* at pH 5.8 and 8.5. The change in pH has no drastic influence on the RR spectra. The occurrence of the markers at ν4 = 1374 cm−1 and ν3 = 1506 cm−1 confirms the presence of a hexa-coordinated ferric LS haem form. A small peak at 1471 cm−1 (ν3) suggests the presence of a small fraction of a ferric high-spin (HS) form. Two vinyl modes are present at 415 and 431 cm−1, with a slightly increased intensity of the latter for DmHb1* at pH 5.8. The presence of two distinct vinyl bands together with the occurrence of weak out-of-plane modes γ7 (307 cm−1) and γ12 (508 cm−1) indicate a small out-of-plane displacement of the haem iron, although this displacement is smaller than observed for aquometmyoglobin.15 The band at 380 cm−1 is assigned to the haem-propionate bending mode δ(CβCcCd) and is higher than found for aquometmyoglobin (376 cm−1),15 which indicates a stronger interaction between the haempropionate and nearby amino-acid residues for ferric DmHb1*.16 In the X-ray diffraction (XRD) structure of ferric DmHb1* a solvent-accessable region was found to remain partly open between the side chain of the E3Arg and the heme D propionate, where two ordered water molecules are located.6 The presence of these water molecules may explain the observed shift of the haem-propionate bending modes. | ||
| Fig. 1 (A) Low-frequency and (B) high-frequency regions of the resonance Raman spectra of different forms of DmHb1*. The RR spectra of the as-isolated (ferric) form at pH 8.5 (upper trace) and pH 5.8 (middle trace), and of the deoxy ferrous form at pH 8.5 (bottom trace) are presented. The laser-excitation wavelength was 413.1 nm at 17 mW power. | ||
The RR spectrum of the deoxy ferrous form of DmHb1* at pH 8.5 (Fig. 1, lower trace) is characteristic of a hexa-coordinated ferrous LS haem form: ν4 = 1360 cm−1, ν3 = 1490 cm−1, ν2 = 1578 cm−1. This agrees with the absorption data (ESI† ). The small peaks at 1471 cm−1 (ν3) and 1545 cm−1 (ν2) suggest the presence of a small fraction of a ferrous HS form. The propionate bending mode δ(CβCcCd) is found at a slightly higher frequency (385 cm−1) than compared to the ferric form, indicating strong hydrogen bonding between the haempropionate and the surrounding residues.16
Fig. 2 shows extracts from the RR spectra of the CO-ligated ferrous form of DmHb1* as a function of laser power. As the laser power augments, the ν4-peak at 1374 cm−1 decreases in intensity and a ν4-signal appears at 1362 cm−1 (Fig. 2B). This is typical for an increased photo-dissociation of the Fe–CO bond. This photo-dissociation process can be used to identify the Fe–CO stretching modes in the RR spectra. Inspection of the low-frequency region of the RR spectra reveals that the peak at 493 cm−1 decreases dramatically in intensity with increasing laser-power (Fig. 2A). This signal can thus be ascribed to the νFe–CO stretching mode. This mode can be linked directly to the haem-pocket structure of the CO-ligated ferrous form (see discussion section).
 | ||
| Fig. 2 Low-frequency (A) and high-frequency (B) parts from the resonance Raman spectra of the CO-ligated ferrous form of DmHb1* as a function of the laser power: (a) 0.5 mW, (b) 1 mW and (c) 1.5 mW. | ||
In order to reveal more information about the ferric forms of DmHb1*, different EPR experiments were set up. In the CW-EPR spectrum of a frozen solution of cyanide-ligated ferric DmHb1* only the low-field component at gmax = 3.43 is resolved (ESI, Fig. S2† ). This spectrum is typical of cyanide-ligated haem proteins and the value of gmax indicates that the proximal histidine (F8His) has a neutral and not an imidazolate character.17
Fig. 3 shows the CW-EPR spectra of frozen solutions of ferric DmHb1* at pH values 5.8, 8.5 and 10. In the ESI (Fig. S3† ), the corresponding CW-EPR spectrum of wild-type DmHb1 at pH 8.5 is given for comparison. The peak indicated with an asterisk in Fig. 3 is due to extra-haem iron and is of no biological interest.18 All EPR spectra depict a mixture of signals stemming from HS and LS ferric haem complexes. The measurement conditions were carefully chosen so as not to saturate the different EPR contributions. The difference in noise between the three spectra is due to the lower protein concentrations used at pH 5.8 and 10.
 | ||
| Fig. 3 Experimental (top) and simulation (bottom) of the X-band CW-EPR spectra of ferric DmHb1* at pH 5.8 (a), pH 8.5 (b) and pH 10 (c). The high-spin (HS) and the four low-spin signals (LS1, LS2, LS3 and LS4) are marked. The asterisk indicates the typical signal due to extra-haem iron. | ||
The EPR contribution of HS ferric DmHb1* can be described by a rhombic effective g matrix: g1,eff = 5.92, g2,eff = 5.88 and g3,eff = 1.993. Several other bis-histidine-coordinated ferric haem proteins show contributions of HS ferric haem forms in their CW-EPR spectra.18–22 In the case of ferric human cytoglobin (CYGB), the appearance of the HS form was found to be batch dependent.22 Ageing of the ferric DmHb1* sample invariably results in a strong increase of the HS signal, indicating that the HS form is most probably linked to a first denaturation step of the DmHb1* protein, as was also supported by other denaturation indications.
In the CW-EPR spectrum of ferric DmHb1* at pH 8.5 (Fig. 3b), three LS ferric forms, LS1, LS2 and LS3, can be recognized besides the HS component. LS1 and LS2 form the most dominant contributions (Fig. 3b and 4b). The CW-EPR contributions of the different LS forms were found to disappear from the spectrum at different temperatures (not shown), which allowed for a determination of some of the principal g values (Table 1). The X-ray structure of ferric DmHb1* showed the presence of only one bis-histidine-coordinated haem form. Ferric haem proteins exhibiting bis-histidine coordination of the haem iron are in an LS state and principal g values similar to the ones found for LS1, LS2 and LS3 have been reported for these cases.19–23 The observation of three LS forms in the EPR spectra contrasts the XRD findings.
 | ||
| Fig. 4 Low-field region from the experimental X-band CW-EPR spectra of DmHb1* shown in Fig 3. Spectra taken at (a) pH 5.8, (b,c) pH 8.5 and (d) pH 10. (a,b,d) DmHb1* whereby 25% glycerol was added as a cryoprotectant, (c) DmHb1* without addition of glycerol. All the spectra were recorded at a temperature of 10 K. The low-field components of LS1, LS2 and LS3 are indicated. | ||
| g z | g y | g x | Ref. | |
|---|---|---|---|---|
| a Abbreviations: NGB = human neuroglobin, CYGB = human cytoglobin, n.d. = cannot be determined accurately. | ||||
| DmHb1* LS1 | 3.178 (±0.005) | 2.07 (±0.04) | n.d. | This work |
| DmHb1* LS2 | 2.988 (±0.005) | 2.19 (±0.02) | n.d. | This work |
| DmHb1* LS3 | 3.50 (±0.04) | N.d. | n.d. | This work |
| DmHb1* LS4 | 2.630 (±0.005) | 2.155 (±0.005) | 1.845 (±0.005) | This work |
| NGB LSA | 3.26 | 2.06 | n.d. | 20 |
| NGB LSB | 3.10 | 2.17 | 1.30 | 20 |
| CYGB | 3.20 | 2.08 | n.d. | 20 |
| Tomato Hb | 2.98 | 2.23 | 1.44 | 21 |
In order to rule out that some of the observed LS forms could be induced by the addition of the cryoprotectant glycerol, the CW-EPR spectrum of ferric DmHb1* without glycerol was also recorded (Fig. 4c). In the absence of glycerol, the features of all LS components are still present, although the EPR lines are now broader. The latter is expected, since the addition of glycerol reduces the inhomogeneous signal broadening due to ice-crystal formation. A similar effect was also observed for the CW-EPR spectra of ferric cytochromec6 with and without glycerol.24 Furthermore, the three LS forms were observed for different batches of ferric DmHb1* and for wild-type DmHb1 (Fig. S3† ), excluding a batch-specific effect.
Since the single crystals used in the X-ray study of de Sanctis et al.6 were grown from a solution at pH 10–11.5, the pH dependence of the CW-EPR spectrum of ferric DmHb1* was investigated (Fig. 3 and 4). At pH 5.8 and 10, the three LS forms were again found, albeit with a different relative intensity (Fig. 3a, c, 4a and d). Furthermore, a fourth LS feature appears in the spectrum at high pH (LS4, Table 1). The principal g values of LS4 are typical of a F8His–Fe(III)–OH− form induced by the high pH.25 The simulations of the CW-EPR spectra taken at different pH values using the parameters given in Table 1 and Table S1 (ESI† ), are included in Fig. 3. In the ESI† (Fig. S4), these simulations are amplified and overlaid with the experimental spectra allowing the reader to evaluate the quality of the simulations.
The largest principal g value of bis-imidazole-coordinated ferric porphyrin complexes is found to depend on many factors. In a recent work, F. A. Walker and co-workers showed that the dihedral angle between the axial ligand planes plays an important factor23 and that progressively larger gmax values are usually found for larger dihedral angles. Since gmax also strongly depends on other parameters, such as H-bonding of the axial histidines, it is impossible to get a direct quantification of the dihedral angle from the maximal principal g value. However, a dihedral angle of 88° as found in the X-ray structure of ferric DmHb1*,6 has been known to lead to so-called HALS (highly anisotropic LS)-type or ‘type-I’ CW-EPR spectra,23 characterized by a maximal g value larger than or equal to 3.20 and a CW-EPR spectrum that is not detectable above 40 K. In many cases, the gx and gy features of the HALS-type ferric forms are not detectable in the CW-EPR spectra. LS3 and possibly LS1 agree with this, but the EPR characteristics of LS2 are more in agreement with a so-called ‘type-II’ LS form that is generally found for bis-histidine-coordinated haem proteins with smaller dihedral angles between the histidine planes.
Since the high-field signals of LS1 to LS3 are not resolved, it is impossible to determine the spin concentrations of the individual contributions accurately. However, using the spectral simulations performed in the previous section, a first rough estimate of the relative contributions could be made based on the spin quantitation method of Svistunenko et al.26 (ESI). LS1 is found to be the dominant fraction (42–47%) and its contribution depends only marginally on pH (small decrease with pH). LS3 contributes 9–16% to the spectra. The HS component constitutes about 21–28% of the sample at pH 5.8–8.5, but decreases strongly at pH 10 (∼9%). The contribution of LS2 shows a rather curious pH dependence. It makes up 9% of the total ferric forms at pH 5.8, 26% at pH 8.5 and only 8% at pH 10. LS4 is only visible at pH 10 (26%).
The observed pH dependence seems to indicate that LS1 and LS3 constitute quite stable ferric forms of the protein that are largely pH-independent. LS2 is more favoured at a higher pH (increase in relative contribution from pH 5.8 to 8.5). The strong decrease of the contribution of LS2 at pH 10 probably has to be linked to the appearance of the LS4 signal. This indicates that the distal histidine in LS2 can easily be replaced by a hydroxide ion at high pH. In addition, LS4 is also formed from the HS form, as has been reported earlier for other globins.26,27 It should be noted that other pH-dependence scenarios cannot be ruled out on the basis of the EPR quantification alone.
In an attempt to gain more information about the LS1 to LS3 forms, pulsed EPR experiments were set up. In our previous work, we derived a pulsed EPR strategy to study different bis-histidine ferric haem complexes.21,22,28,29 In principle, the situation in the present study is more complicated because of the overlap of the EPR spectra of the different LS forms, which means that, for most magnetic-field settings, the ESEEM (electron spin echo envelope modulation) spectra will contain features stemming from various LS species. In order to exploit the pH-dependent ratio, pulsed-EPR experiments were performed on frozen solutions of ferric DmHb1* at different pH values.
In a first experiment, two-pulse ESEEM spectra were recorded as a function of magnetic field for DmHb1* at the different pH values. Fig. 5 shows, for the case of DmHb1* at pH 5.8, the field-dependent spectrum obtained by Fourier-transformation of the different two-pulse ESEEM spectra and summing over the frequency range 0–7.8 MHz. This is the region in which the 14N nuclear frequencies lie. The proton region was omitted in this sum for two reasons. First, the Cu(II) background signal typical of the Bruker dielectric cavity is known to exhibit a small proton modulation and, secondly, proton ESEEM signals are hard to detect for magnetic fields above 350 mT (shallow modulation depth). Including the proton region will therefore inevitably lead to a misinterpretation of the relative contributions of the different LS forms. Although the CW-EPR spectra showed that LS1 is the dominant contribution at this pH, the signals of LS2 dominate the field-dependent ESEEM spectra (Fig. 5; LS1:LS2:LS3 = 30:65:5). This is due to the longer relaxation times of LS2 compared to LS1 and LS3, which is also reflected by the different saturation behavior and temperature-dependent behavior of their CW-EPR spectra. The ESEEM spectra of ferric DmHb1* at higher pH values were also found to be dominated by the LS2 form (see also ESI, Fig. S5† ).
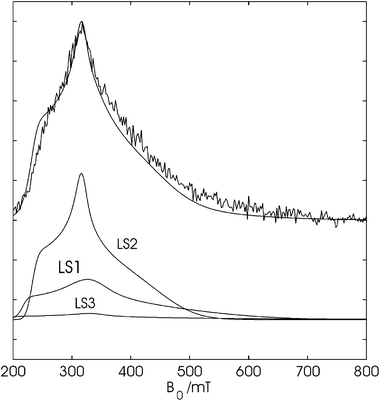 | ||
| Fig. 5 Top: ESE-detected EPR spectrum of DmHb1* (pH 5.8) taken at 301 τ values, Fourier-transformed in the ESEEM dimension and summed over the frequency range 0–7.8 MHz overlaid by the simulation with LS1:LS2:LS3 = 30:65:5. Bottom: individual components used in the simulation. | ||
Fig. 6a and b show the experimental 14N HYSCORE spectraof a frozen solution of ferric DmHb1* (pH 8.5) taken at two magnetic-field positions. Additionally, different 14N HYSCORE spectra recorded at other magnetic-field settings are shown in the ESI.† No HYSCORE spectracould be recorded at magnetic-field settings larger than 470 mT due to the low spin-echo signal at these fields. In this way, the gx regions of LS1 and LS3 could not be probed. 14N HYSCORE spectrawere taken at a low-field observer position where LS1 and LS3 dominantly contribute to the spectra (216.7 mT) and compared to the 14N HYSCORE spectrataken at the observer position corresponding to g = gz for LS2 (230.7 mT) (ESI,† Fig. S6 and S7). No clear differences were found between these spectra other than those resulting from the change in magnetic field. Furthermore, HYSCORE spectraof ferric DmHb1* at pH 5.8 and 10 were also recorded at selected positions (not shown) and compared to the corresponding spectra of ferric DmHb1* at pH 8.5. No major differences were found. This confirms the dominance of LS2 in the ESEEM spectra, but indicates also that LS1 and LS3 exhibit very similar 14N interactions, which limits the amount of information that can be obtained from the HYSCORE spectra.
As shown in our earlier work, the orientation of the nuclear-quadrupole tensor of the pyrrole nitrogens in the principal g-axes frame can be used to determine the relative orientation of the in-plane principal g-axes versus the molecular frame.21,22,28,29 Indeed, the largest nuclear quadrupole value of the haemnitrogens is found to lie invariably in the haem plane, perpendicular to the Fe–Nhaem bond. HYSCORE simulations were performed using the principal g values of LS2 (Fig. S6, ESI† ). These simulations indicate that the in-plane gx axis deviates maximally 10° from the Nhaem–Fe–Nhaem direction (Table S2, ESI† ).
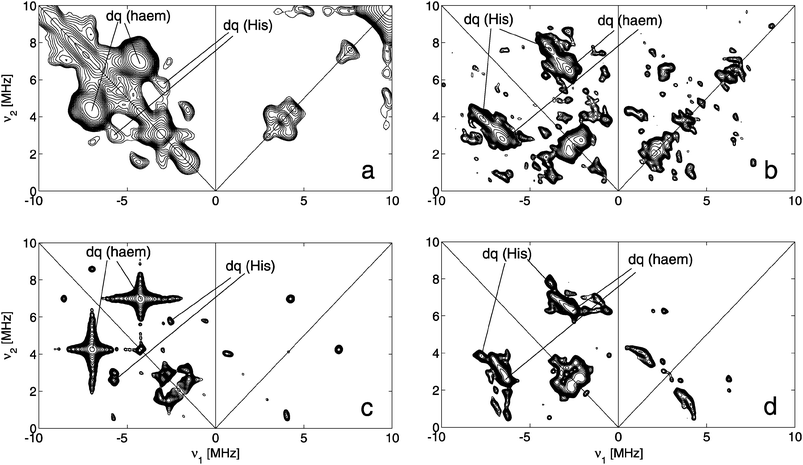 | ||
| Fig. 6 (a) Experimental SMART HYSCOREspectrum of a frozen solution of ferric DmHb1* at pH 8.5 taken at an observer position corresponding to g ≈ gz of LS2 (B0 = 230.7 mT). tπ = 16 ns, tπ/2 = 16 ns, pulse length of high-turning angle (HTA) pulse is 24 ns with ν1 = 15.625 MHz. τ = 96 ns. (b) Experimental standard HYSCOREspectrum of a frozen solution of ferric DmHb1* at pH 8.5 taken at an observer position corresponding to g ≈ gy of LS2 (B0 = 324.1 mT). tπ/2 = 16 ns, tπ = 16 ns, τ = 176 ns. (c) Simulation of (a). (d) Simulation of (b). The simulations were performed assuming a four-spin system (S = 1/2, two haem nitrogen nuclei and one Histidinenitrogen). The simulation parameters are given in Table 2. | ||
| A1/MHz | A2/MHz | A3/MHz | P1/MHz | P2/MHz | P3/MHz | Ref. | |
|---|---|---|---|---|---|---|---|
| a Abbreviations: mNgb = mouse neuroglobin, CYGB = human cytoglobin. b β(gz, A3) = 0° ± 5°, α(gx, A1) = (0 + n90)° ± 10°, β(gz, P3) = 0° ± 5°, α (gx, P1) = (0 + n90)° ± 10° (n = 0,1,2,3). c β (gz, A3) = 0° ± 5°, α (gx, A1) = 20° ± 20°, β (gz, P3) = 0° ± 5°, α (gx, P1) = 20° ± 20°. d The 14N hyperfine and nuclear quadrupole parameters of the distal and proximal histidine were found to be different for this protein.29 | |||||||
| Pyrrole nitrogens | |||||||
| Ferric DmHb1* | −4.1b (±0.2) | −4.2b (±0.2) | −5.5b (±0.2) | −0.49b (±0.05) | 0.90b (±0.05) | −0.41b (±0.05) | This work |
| Ferric wt mNgb | −4.8 | −4.8 | −5.7 | −0.52 | 0.95 | −0.43 | 29 |
| Ferric tomato Hb | −4.5 | −3.6 | −5.4 | −0.46 | 0.88 | −0.42 | 21 |
| Ferric wt CYGB | −4.0 | −4.1 | −5.4 | −0.50 | 0.92 | −0.42 | 22 |
| His imidazole nitrogens (NE2) | |||||||
| Ferric DmHb1* | −5.0b (±0.5) | −5.8b (±0.4) | −4.2b (±0.3) | 0.14c (±0.05) | 0.74c (±0.2) | −0.90c (±0.1) | This work |
| Ferric wt mNgbd | −5.65 | −5.25 | −4.9 | 0.16 | 0.64 | −0.80 | 29 |
| −5.65 | −5.25 | −4.9 | 0.30 | 0.55 | −0.85 | 29 | |
| Ferric tomato Hb | −5.1 | −6.4 | −4.9 | 0.04 | 076 | −0.80 | 21 |
| Ferric wt CYGB | −5.9 | −4.7 | −5.0 | 0.34 | 0.56 | −0.85 | 22 |
Furthermore, in all HYSCORE spectra, only one set of double-quantum (dq) cross peaks ascribed to the coordinating Hisnitrogens (NE2) can be found, indicating that E7His and F8His bind equally strongly to the haem iron (Fig. S6, ESI† ). It has been shown earlier that in such a case, the following counter-rotation principle will hold: if the mean of the two imidazole-plane orientations rotates about the NE2His–Fe axis over a given angle away from the Nhaem–Fe–Nhaem direction, the gx axis will counter-rotate in the haem plane. For LS2, the imidazole mean will thus lie close to the Nhaem–Fe–Nhaem axis, since gx quasi eclipses this axis. In the X-ray structure, the mean of the two histidineimidazole planes also lies along the Nhaem–Fe–Nhaem direction.6
In principle, the hyperfine and nuclear-quadrupole tensors of the coordinating histidinenitrogens can be used to determine the dihedral angle between the two histidine planes. However, this analysis is, in practice, hampered because the single-quantum cross peaks of the NE2His contributions are masked by the strong cross peaks of the pyrrolenitrogens.22,29 Furthermore, where the pyrrolenitrogen hyperfine values are found to vary only slightly for different bis-histidine-coordinated globins, significant differences in the histidinenitrogen hyperfine values have been found depending on the relative orientation of the histidine planes (Table 2). The difference in the principal g values of LS1, LS2 and LS3 indicates a significant difference of the dihedral angle between the two His planes for the three forms, which will introduce a broadening of the cross peaks of the NE2His contributions in the HYSCORE spectraat hand, although the dominant contribution will stem from LS2. Finally, since good-quality proton HYSCORE spectracould only be obtained for the low-field observer positions (ESI† ), an accurate determination of the orientation of the histidine planes is not possible from the HYSCORE data. Satisfactory agreements between the available experimental and simulated 14N and 1H HYSCORE spectraare found assuming a dihedral angle between the two imidazole planes of 20–40° (Fig. S6, Table S2, ESI† ), but this is insufficient to prove this angle due to the above-mentioned problems.
Discussion
The νFe–CO stretching mode at 493 cm−1 found in the RR spectrum of CO-ligated ferrous DmHb1* (Fig. 2) has been observed earlier in different Hbs and Mbs at acidic pH values.30–32 It is characteristic of an open haem pocket with little interaction between the CO ligand and the surrounding amino-acid residues. This contrasts the cyanide-ligated ferric form of DmHb1*, for which a hydrogen bond is found to link the E7His NE2 atom with the nitrogen of the cyanide ligand.7 It also disagrees with the CO-ligation modes found up till now for other globins that display endogeneous bis-histidine coordination of the haem iron in their wild-type ferrous and ferric forms. The RR spectrum of the CO-ligated form of ferrous wt human neuroglobin (NGB) revealed three configurations for the Fe–CO unit: besides the ‘open’ conformation of the haem pocket (νFe–CO = 494 cm−1), an ‘intermediate’ conformation (νFe–CO = 505 cm−1), and a ‘closed’ conformation of the haem pocket, where the CO is stabilized by one or more surrounding amino-acids (νFe–CO = 521 cm−1), were observed.33 The νFe–CO modes detected for CO-ligated ferrous human cytoglobin (CYGB) agreed with two ‘closed’ conformations of the protein, whereby CO interacts with the E7His residue (νFe–CO = 510/518 cm−1), and one ‘open’ conformation (νFe–CO = 494 cm−1).34 Barley hemoglobin has a dominant Fe–CO stretching mode at 534 cm−1 at pH 7.4 and only a weak signal at 493 cm−1 agreeing with a dominant stabilization of the CO ligand by the E7His.35 Finally, two CO-coordination modes can be distinguished in the CO-ligated form of the nerve hemoglobin of the mollusc Spisula solidissima: an open conformation (75%, νFe–CO = 494 cm−1) and a closed form (25%, νFe–CO = 510 cm−1).36 In this respect, the haem pocket of DmHb1*CO clearly has the most open configuration.Comparison of the X-ray data of ferric DmHb1* and cyanide-ligated ferric DmHb1* already showed that the bis-histidine-coordinated state is a strained state (E7His is closing on the haem), whereby DmHb1*CN relaxes to a backbone conformation that is more closely related to that of the cyanide derivative of ferric sperm whale Mb.6,7 In this respect, the EPR characteristics of ferric DmHb1*CN are similar with those of MbCN (gmax = 3.4518). Our RR experiments prove that there is some doming of the haem in ferric DmHb1* at room temperature in solution (see vinyl and out-of-plane modes in the RR spectrum in Fig. 1), although this doming is far less than in aquometmyoglobin.
The observation that ferric DmHb1* very easily degrades to a HS ferric form with subsequent protein denaturation (Fig. 3) shows that the bond between the E7His nitrogen and the haem iron is strained and quite unstable, reflecting the protein’s tendency to an open penta-coordinated state. In agreement with this, the E7His binding affinity in ferrous DmHb1* is found to be much lower than that of neuroglobin.5
Undoubtedly the most interesting observation in this work is the fact that different LS forms are found in the frozen solutions of ferric DmHb1* (LS1–LS4, Fig. 3). The LS1 to LS3 forms have principal g values that agree with a bis-histidine ligation of the haem iron. The large differences in the gmax point to a change in the dihedral angle between the distal and proximal His planes, whereby the higher gmax values will correspond to the larger dihedral angle. The earlier published crystal structure reported only one form whereby the two axial ligands are quasi perpendicular to each other and the E7His imidazole ring is almost perfectly staggered relative to the haempyrrole nitrogen atoms, being oriented towards the haem’s meso-carbons.6 The EPR charateristics of LS1 and LS3 are typical for large dihedral angles between the His planes (type-I or HALS-type ferric forms37). The XRD analysis also revealed that the ND1 and CE1 atoms of the E7His and F8His imidazole rings displayed anisotropic displacements, whereby the histidine nitrogen atoms directly coordinated to the haem iron (NE2) display nearly spherical isotropy. This led the authors to suggest that there is a preferential libration of both imidazole rings around the NE7His–Fe–NF8His direction. Our present findings corroborate such a suggestion.
The strong pH-dependence of LS2 and the ease with which the distal E7His ligand can be replaced by a hydroxide ion at high pH, seems to indicate that in LS2, the haem pocket is more accessible by the solvent than the other two forms. De Sanctis et al. found two protein cavities on the distal side of the haem pocket. They suggested that the two cavities may merge during a dynamic process, yielding a single elongated apolar cavity forming an access path for small ligands. This would involve modest conformational shifts in some of the protein residues. Possibly, this may have happened in the LS2 form, explaining its difference to the LS1 and LS3 forms. Note, that a pH dependence quite similar to that observed for LS2 has been reported for an LS form of methaemoglobin.26 In methaemoglobin, the dominant ferric haem form is the aquometform. However, small fractions of ferric low-spin haem complexes have been observed involving a bis-histidine ligation. Also in this case, the decrease of the LS form at high pH was paralleling the formation of an F8His–Fe(III)–OH− form.
The ESEEM spectra were found to have features predominantly stemming from LS2. This form has the EPR characteristics of a type-II ferric haem complex, which is characterized by smaller dihedral angles between the imidazole planes of the ligating histidines. The HYSCORE analysis reveals that the gx axis is approximately along the Nhaem–Fe–Nhaem axis. The orientation of the gx axis shows that both E7His and F8His have rotated in an opposite direction leaving the mean of the Hisimidazole planes along the Nhaem–Fe–Nhaem axis. Since no additional cross peaks were observed in the HYSCORE spectrathat could be ascribed to the pyrrolenitrogens of LS1 and LS3, this seems to be the case for all three LS forms. This implies that the difference between the three LS forms involves rotation of both the E7His and F8His, as is also suggested from the X-ray data. This unusual flexibility of the F8His may be related to the occurrence of a Pro at position F10 that strongly perturbs the local hydrogen-bonding pattern.6
When comparing X-ray data with EPR data of ferric haem proteins, we need to consider the possibility of photoreduction occurring in the former case. In this case, the two sets of data cannot be compared, since the X-ray data would concern the ferrous instead of the ferric system. However, in the case at hand, the X-ray data of cyanide-ligated DmHb1* are typical for the ferric case.7 Indeed, if photoreduction occurs, the Fe–CN atoms are no longer in a straight line and the Fe–C distance changes.38 In this way, photoreduction in cyanide-ligated DmHb1* can be ruled out, and therefore, photoreduction of the bis-histidine coordinated DmHb1* is also less likely to happen.
The dihedral angle between the imidazole planes found in the different static structures must be thus governed by external factors, such as crystal packing (XRD data), temperature (EPR data), pH, etc. This proves that the haem region of DmHb1* is very flexible and easy to perturb. It is clear that in the case of DmHb1*, for which a respiratory function is proposed, a high flexibility of the haem pocket region is needed in order to allow efficient O2 binding and controlled O2 release. Surprisingly, the Hb of the botfly G.intestinalis, with which DmHb1 shows striking similarities (the two Hb genes are diverged by speciation), exhibits a pentacoordinated haem iron.39
It is clear that the local haem-pocket structure of ‘bis-histidine-coordinated’ globins is very diverse and that no general structural or functional signature for these globins can be given. Interestingly, the EPR spectrum of a frozen solution of ferric wild-type NGB also revealed the presence of two LS forms, albeit with different principal g values than ferric DmHb1* (ref. 20, Table 1, LSA and LSB). In the NGB case, the occurrence of LSA could be linked to the formation of a disulfide bridge between the cysteines at positions CD7 and D5.20 It is clear that this mechanism cannot be the one governing the observation of different LS forms in ferric DmHb1* (no Cys present). Nevertheless, in both cases the considerable flexibility in the haem region seems to be related to the protein function and may well be one of the key points of functional differentiation in bis-histidine-coordinated globins.
Conclusions
The EPR analysis of the frozen solutions of ferric 121Cys→Ser mutant of Drosophila melanogasterhaemoglobin (DmHb1*) revealed the coexistence of three low-spin ferric haem forms. The EPR parameters of these ferric forms are typical for a bis-histidine coordination of the haem iron with a difference in the dihedral angle between the histidineimidazole planes. This points to large liberation of the distal and proximal histidine around the Fe–NHis axis, which may be related to the need to control O2 uptake and release. The resonance Raman spectra of CO-ligated ferrous DmHb1* indicate an open structure of the haem-pocket with little stabilization of the distal ligand by the surrounding amino-acid residues.Acknowledgements
This work was supported by the Fund for Scientific Research-Flanders (FWO) and the University of Antwerp (TOP-BOF grant). S.D. is a postdoctoral fellow of the FWO.References
- P. Willmer, G. Stone and I. Johnston, Environmental Physiology of Animals, Blackwell, Oxford, 2000 Search PubMed.
- T. Burmester and T. Hankeln, J. Insect Physiol., 2007, 53, 285–294 CrossRef CAS.
- T. Burmester and T. Hankeln, Mol. Biol. Evol., 1999, 16, 1809–1811 CAS.
- T. Burmester, J. Storf, A. Hasenjäger, S. Klawitter and T. Hankeln, FEBS J., 2006, 273, 468–480 CrossRef CAS.
- T. Hankeln, V. Jaenicke, L. Kiger, S. Dewilde, G. Ungerechts, M. Schmidt, J. Urban, M. C. Marden, L. Moens and T. Burmester, J. Biol. Chem., 2002, 277, 29012–29017 CrossRef CAS.
- D. de Sanctis, S. Dewilde, C. Vonrhein, A. Pesce, L. Moens, P. Ascenzi, T. Hankeln, T. Burmester, M. Ponassi, M. Nardini and M. Bolognesi, J. Biol. Chem., 2005, 280, 27222–27229 CrossRef CAS.
- D. de Sanctis, P. Ascenzi, A. Bocedi, S. Dewilde, T. Burmester, T. Hankeln, L. Moens and M. Bolognesi, Biochemistry, 2006, 45, 10054–10061 CrossRef CAS.
- S. Dewilde, M. Blaxter, M. L. Van Hauwaert, K. Van Houte, A. Pesce, N. Griffon, L. Kiger, M. C. Marden, S. Vermeire, J. Vanfleteren, E. Esmans and L. Moens, J. Biol. Chem., 1998, 273, 32467–32474 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2005, 177, 390–403.
- A. Schweiger and G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance, University Press Oxford, Oxford, 2001 Search PubMed.
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132, 279–282 CrossRef.
- G. Jeschke, R. Rakhmatullin and A. Schweiger, J. Magn. Reson., 1998, 131, 261–271 CrossRef CAS.
- L. Liesum and A. Schweiger, J. Chem. Phys., 2001, 114, 9478–9488 CrossRef CAS.
- Z. L. Madi, S. Van Doorslaer and A. Schweiger, J. Magn. Reson., 2002, 154, 181–191 CrossRef CAS.
- S. Z. Hu, K. M. Smith and T. G. Spiro, J. Am. Chem. Soc., 1996, 118, 12638–12646 CrossRef CAS.
- T. Uchida, E. Sato, A. Sato, I. Sagami, T. Shimizu and T. Kitagawa, J. Biol. Chem., 2005, 280, 21358–21368 CrossRef CAS.
- W. E. Blumberg, J. Peisach, B. A. Wittenberg and J. B. Wittenberg, J. Biol. Chem., 1968, 243, 1854–1862 CAS.
- J. Peisach, W. E. Blumberg, S. Ogawa and E. A. Rachmilewitz, J. Biol. Chem., 1971, 246, 3342–3355 CAS.
- M. Fahnenschmidt, H. K. Rau, R. Bittl, W. Haehnel and W. Lubitz, Chem.–Eur. J., 1999, 5, 2327–2333 CrossRef CAS.
- E. Vinck, S. Van Doorslaer, S. Dewilde and L. Moens, J. Am. Chem. Soc., 2004, 126, 4516 CrossRef CAS.
- A. I. Ioanitescu, S. Dewilde, L. Kiger, M. C. Marden, L. Moens and S. Van Doorslaer, Biophys. J., 2005, 89, 2628–2639 CrossRef CAS.
- A. I. Ioanitescu, S. Van Doorslaer, S. Dewilde, B. Endeward and L. Moens, Mol. Phys., 2007, 105, 2073–2086 CrossRef CAS.
- L. A. Yatsunyk, A. Dawson, M. D. Carducci, G. S. Nichol and F. A. Walker, Inorg. Chem., 2006, 45, 5417–5428 CrossRef CAS.
- I. García-Rubio, M. Medina, R. Cammack, P. J. Alonso and J. I. Martinez, Biophys. J., 2006, 91, 2250–2263 CrossRef CAS.
- F. A. Walker, D. Reis and V. L. Balke, J. Am. Chem. Soc., 1984, 106, 6888–6898 CrossRef CAS.
- D. A. Svistunenko, M. A. Sharpe, P. Nicholls, M. T. Wilson and C. E. Cooper, J. Magn. Reson., 2000, 142, 266–275 CrossRef CAS.
- M. Ikeda-Saito, H. Hori, L. A. Andersson, R. C. Prince, I. J. Pickering, G. N. George, C. R. Sanders, R. S. Lutz, E. J. McKelvey and R. Mattera, J. Biol. Chem., 1992, 267, 22843–22852.
- E. Vinck and S. Van Doorslaer, Phys. Chem. Chem. Phys., 2004, 6, 5324–5330 RSC.
- E. Vinck, S. Van Doorslaer, S. Dewilde, G. Mitrikas, A. Schweiger and L. Moens, J. Biol. Inorg. Chem., 2006, 11, 467–475 CrossRef CAS.
- R. Mourant, D. P. Braunstein, K. Chu, H. Frauenfelder, G. U. Nienhaus, P. Ormos and R. D. Young, Biophys. J., 1993, 65, 1496–1507 CrossRef.
- J. T. Sage, D. Morikis and P. M. Champion, Biochemistry, 1991, 30, 1227–1237 CrossRef CAS.
- J. Ramsden and T. G. Spiro, Biochemistry, 1989, 28, 3125–3128 CrossRef CAS.
- H. Sawai, M. Makino, Y. Mizutani, T. Ohta, H. Sugimoto, T. Uno, N. Kawada, K. Yoshizato, T. Kitagawa and Y. Shiro, Biochemistry, 2005, 44, 13257–13265 CrossRef CAS.
- H. Sawai, N. Kawada, K. Yoshizato, H. Nakajima, S. Aona and Y. Shiro, Biochemistry, 2003, 42, 5133–5142 CrossRef CAS.
- T. K. Das, H. C. Lee, S. M. G. Duff, R. D. Hill, J. Peisach, D. L. Rousseau, B. A. Wittenberg and J. B. Wittenberg, J. Biol. Chem., 1999, 274, 4207–4212 CrossRef CAS.
- S. Dewilde, B. Ebner, E. Vinck, K. Gilany, T. Hankeln, T. Burmester, J. Kreiling, C. Reinisch, J. R. Vanfleteren, L. Kiger, M. C. Marden, C. Hundahl, A. Fago, S. Van Doorslaer and L. Moens, J. Biol. Chem., 2006, 281, 5364–5372 CAS.
- F. A. Walker, Coord. Chem. Rev., 1999, 185–186, 471–534 CrossRef CAS.
- M. Bolognesi, C. Rosano, R. Losso, A. Borassi, M. Rizzi, J. B. Wittenberg, A. Boffi and P. Ascenzi, Biophys. J., 1999, 77, 1093–1099 CrossRef CAS.
- T. Burmester and T. Hankeln, News Physiol. Sci., 2004, 19, 1010–1113 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV/Vis spectra, CW-EPR spectrum of cyanide-ligated ferric DmHb1*, CW-EPR spectrum of ferric wild-type DmHb1, simulations of CW-EPR spectra of DmHb1* and simulation parameters, relative contributions of ferric haem forms, pH-dependence of ESE-detected EPR spectra, full HYSCORE analysis. See DOI: 10.1039/b902059b |
| This journal is © The Royal Society of Chemistry 2009 |