Synchrotron radiation for direct analysis of metalloproteins on electrophoresis gels
Richard
Ortega
*
Groupe d’Imagerie Chimique Cellulaire et Spéciation, Laboratoire de Chimie Nucléaire Analytique et Bioenvironnementale (CNAB), CNRS UMR5084, Université de Bordeaux 1, chemin du Solarium, 33175 Gradignan Cedex, France. E-mail: ortega@cenbg.in2p3.fr; Fax: +33 557 120 900; Tel: +33 557 120 907
First published on 24th February 2009
Abstract
Metalloproteomics requires analytical techniques able to assess and quantify the inorganic species in metalloproteins. The most widely used methods are hyphenated techniques, based on the coupling of a high resolution chromatographic method with a high sensitivity method for metal analysis in solution. An alternative approach is the use of methods for solid sample analysis, combining metalloprotein separation by gel electrophoresis and direct analysis of the gels. Direct methods are based on beam analysis, such as lasers, ion beams or synchrotron radiation beams. The aim of this review article is to present the main features of synchrotron radiation based methods and their applications for metalloprotein analysis directly on electrophoresis gels. Synchrotron radiation X-ray fluorescence has been successfully employed for sensitive metal identification, and X-ray absorption spectroscopy for metal local structure speciation in proteins. Synchrotron based methods will be compared to ion beam and mass spectrometry for direct analysis of metalloproteins in electrophoresis gels.
 Richard Ortega Richard Ortega | Richard Ortega is currently a Research Director at CNRS (National Center for Scientific Research) and Bordeaux University, France. His research interests include the development of analytical methods based on ion beams and synchrotron radiation for the imaging and speciation of metals in cells and proteins. He obtained a chemical engineering degree and masters degree in chemistry from the University of Marseille (1991) and PhD in physics at the University of Bordeaux (1994). He worked as a postdoctoral fellow with Dr Bibudhendra Sarkar at the Research Institute of the Hospital for Sick Children, Biochemistry Department, Toronto, Canada (1995). He has also worked at the European Synchrotron Radiation Facility (2001) as an Invited Scientist. Ortega has received an award from the French Society of Chemistry in Analytical Chemistry (2004). |
Introduction
Metalloproteins are proteins that contain a metal cofactor. This singularity implies that the usual protocols of proteomics may not fully apply to the study of metalloproteins and that an inorganic analytical chemistry approach must be added. A variety of methods have been developed recently for the identification and characterization of metalloproteins as reviewed in references 1–9. A metalloproteomic study usually consists of the application of two methods, a chromatographic technique for protein separation, and a metal analysis for the identification of metalloproteins. Depending on the chromatographic method that is used for protein separation, i.e. based on liquid chromatography or on gel electrophoresis, metal analysis is performed either in solution or on a solid sample. Metalloproteomic technology is more often applied in solution by using inductively coupled plasma mass spectrometry (ICP-MS), which provides a very sensitive and quantitative detection for trace elements in solution, coupled to a separation technique such as high performance liquid chromatography or capillary electrophoresis.10 However this method does not apply to 2D polyacrylamide gel electrophoresis (PAGE) although PAGE is widely used as a component of proteomics for the isolation of proteins and further characterisation by mass spectroscopy.The direct analysis of metalloproteins on electrophoresis gels requires specific analytical methods able to detect metals on solid samples. Autoradiography using radiolabeled elements, laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS), particle induced X-ray emission (PIXE), and synchrotron radiation based methods such as synchrotron radiation X-ray fluorescence spectroscopy (SR-XRF) and X-ray absorption spectroscopy (XAS) have been successfully applied to metalloproteomic studies on electrophoresis gels. SR-XRF is a very sensitive method for metal analysis, more sensitive than PIXE and comparable to LA-ICP-MS, which can be quantitative. On the other hand, XAS is a quasi-unique technique for investigating the local structural environment of metal ions. The aim of this review article is to provide information on the specificities and limitations of SR-XRF and XAS in metalloproteomics, and to compare them to other existing methods such as LA-ICP-MS and PIXE.
Synchrotron radiation X-ray fluorescence
Synchrotron radiation induced X-ray fluorescence enables the quantitative analysis of chemical elements directly on solid samples. The use of a synchrotron X-ray source offers several orders of magnitude higher brilliance than conventional laboratory sources, i.e. rotating anodes. The principle of XRF is based upon the detection of X-rays emitted from sample atoms irradiated with X-rays of higher energy. The energy of the emitted X-rays is characteristic of the excited element enabling the identification of the composition of the atoms in the sample. XRF analysis is multielemental and quantitative; the surface of the fluorescence peaks is directly proportional to the concentration of the elements within the sample. If it is combined with a protein quantification technique it can be used to determine the metal/protein stoichiometry.The feasibility of XRF element detection in proteins separated on polyacrylamide gels was demonstrated by Stone et al.11 using a bench-top microbeam X-ray system. They evaluated the detection of selenium contained in the selenoprotein glutathione peroxidase previously separated using PAGE. Current detection limits of selenium in the gel matrix were as low as 2.1 ng. Two years later, the first XRF measurements of metals in proteins using synchrotron radiation were performed by Homma-Takeda et al.12 for the detection of mercury bound to metallothionein as a model study. The direct measurement of Hg in IEF-PAGE (isoelectrofocusing PAGE) prepared under denaturing conditions was used to prove the covalent binding of Hg to metallothionein. Very importantly, the use of IEF-PAGE showed that the isoelectric point of the protein was sensitive to the metallation state.12 Later on SR-XRF was used on more complex protein extracts to determine the metalloprotein content as, for example, in human liver cytosol.13,14 These studies indicated the importance of the protocols for protein separation in the preservation of the protein metallation state. A larger number of metalloproteins could be detected by SXRF analysis of proteins separated with gel filtration chromatography and thin layer isoelectric focusing, compared to SXRF analysis of the same protein fraction separated by SDS-PAGE. Denaturing conditions using SDS alter the binding of weakly bound hetero-atoms in proteins. Therefore, the comparative analysis of metals from proteins separated either under denaturing or under non-denaturing conditions can be used to study the affinity of the metal binding to the proteins.
An example of study under denaturing conditions is the SDS-PAGE combined with SR-XRF analysis of the selenoproteins in rat testis homogenate.15,16 Selenium peaks determined by SR-XRF, corresponding to a selenium amount in the nanogram range, were identified. The major peak was attributed to the seleno-enzyme phospholipid hydroperoxide glutathione peroxidase, which accounts for about 80% of the selenium present in the testis homogenate. Because selenium binds covalently to the protein its detection is still possible even after SDS treatment. However the method was less sensitive than a radiometric method using 75Se and gel autoradiography. Of the nine selenium containing proteins found in the autoradiogram, only four could be detected by SR-XRF. This study indicates that denaturing conditions, i.e. use of SDS, are suitable for the study of firmly bound trace elements in proteins such as selenoproteins. It also indicates that when radiometric methods can be used they might offer a better sensitivity than SR-XRF.
However, the use of radiolabeled elements is not always possible, especially for human studies. Moreover, in most cases SDS treatment will disrupt the metal binding to the protein. This is why non-denaturing conditions should be advocated for the general study of metalloprotein identification. IEFprotein separation under non-denaturing conditions followed by SR-XRF has been successfully applied for the differentiation of metalloprotein content between tumoural and normal surrounding tissues from hepatocellular carcinoma patients.17 A decrease of Fe and Zn metalloprotein content in tumoural tissue was suggested, although the analysis was not fully quantitative due to the lack of appropriate calibration standards. The decrease of Zn protein content in tumour tissue was mostly attributed to a decrease in the Zn metallothioneins, identified by the position of high Zn and S content at isoelectric point 5–6.
SR-XRF analysis of proteins separated by gel electrophoresis has also been used for environmental studies. An original contribution to the field is the application of the Kohonen neural network to exploratory analyses of SR-XRF measurements of metalloproteins in plants exposed to various metals.18 A recent study investigated the mercury, arsenic, and selenium-containing proteins in fish liver from a mercury polluted area of Guizhou Province in China.19 The results indicated that Se and As were found in some of the Hg-containing protein bands. The bands may correspond to the antagonistic effect of Se against the toxicity of Hg and As. In addition, Hg and As often coexist in the same band, suggesting that the two elements may be involved in the same metabolic processes.
X-Ray absorption spectroscopy
XAS is one of the most widely used techniques for investigating the local structural environment of metal ions. XAS is usually applied at synchrotron radiation facilities providing intense and tuneable X-ray beams. X-Ray absorption spectra are obtained by tuning the photon energy around the absorption edge of a specific element and by measuring the photons transmitted through the sample. Each core electron has a well-defined binding energy, and when the energy of the incident X-ray is scanned across one of these energies, there is an abrupt increase in the absorption coefficient. Local structural information on the absorber element can be deduced by analyzing oscillations in X-ray absorption versus photon energy that are caused by the scattering of the X-ray excited photoelectron. XAS can be divided into X-ray absorption near edge structure (XANES), which provides information primarily about geometry and oxidation state, and extended X-ray absorption fine structure (EXAFS), which provides information about metal site ligation.The application of XAS in metalloproteomics has so far been closely associated with structural genomics .4,20–23 Several experimental setups have been implemented, or are being implemented, at synchrotron radiation facilities to perform XAS at the same time as X-ray protein crystallography.4,24–26 Such an integrated structural approach confers important additional information in proteomics. The combination of proteincrystallography and XAS methods on the same metalloprotein single crystal yields a structural model of the protein with exceptional active-site resolution.4,20,27EXAFS data are obtained with higher resolution than proteincrystallography data and therefore can help to determine the exact structure of the metal binding site such as bond lengths and coordination number. XANES can also be used for the identification of metal redox states in protein crystals, as the energy of the X-ray absorption edge depends on the oxidation state of the absorbing atom. This is unique information that is generally impossible to obtain crystallographically.
Recently, our group has proposed the use of XAS for the analysis of non-crystalline metalloproteins directly on electrophoresis gels.28 This method allows the direct analysis of the metal structural environment in its native state as the gel electrophoresis is performed under non-denaturing conditions. Using XANES on IEF gels we were able to determine the oxidation state of copper and zinc in bovine and human copper zinc superoxide dismutase (CuZnSOD) (Fig. 1). The results obtained are in good agreement with XAS analysis of bovine CuZnSOD crystals,29 and indicate the existence of mixed Cu(I)/Cu(II) oxidation states. In addition, the use of IEF enables the separation of isoelectric point isoforms of CuZnSOD, which could not be resolved in protein crystals, and indicates that in a minor isoform copper was present as Cu(II), while in the major isoform as a mixed redox state Cu(I)/Cu(II). Overall, the combination of IEF and XANES offers unique capabilities for the characterization of metalloproteins because metal oxidation state can be obtained in proteins using non-denaturing electrophoresis conditions, thus preserving their native state. In addition, IEF separation enables the speciation of protein isoforms with distinct isoelectric points. Further developments are in progress in order to apply EXAFS to electrophoresis gels.
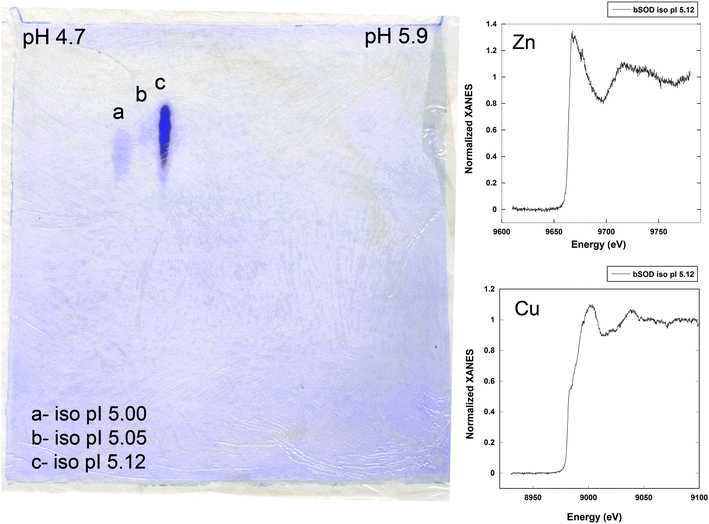 | ||
| Fig. 1 Left: 2D gel electrophoresis of commercial bovine CuZnSOD run under non-denaturing conditions showing three pI isoforms at 5.00, 5.05 and 5.12. XANES spectra were recorded on an unstained gel prepared simultaneously to the stained gel presented here. Right: zinc and copper absorption K-edge spectra of bovine CuZnSOD isoform at pI 5.12.28 | ||
Other methods for direct analysis of proteins in gels
At least five analytical methods have been applied to identify metalloproteins directly on electrophoresis gels: autoradiography using a radiolabeled element, PIXE, SR-XRF, XAS and LA-ICP-MS; their main characteristics are summarized in Table 1. Autoradiography is a very sensitive method, but is limited to the study of few radiolabeled elements such as 75Se. Therefore it has been used mainly for selenoprotein identification on 2D electrophoresis gels.7,15PIXE allows a sensitive, multi-elemental analysis, appropriate to study the content of trace elements in diluted samples. The principle of PIXE is similar to XRF except that the incoming particles are accelerated ions, such as protons, instead of photons. PIXE analysis has been proposed as a tool to study metalloproteins separated on electrophoresis gels.30 Qualitative and/or quantitative information can be provided, according to the concentration of the inorganic element of interest. PIXE has been performed to identify metals bound to proteins or protein subunits after separation by SDS-PAGE.31–36LA-ICP-MS in combination with gel electrophoresis has also been used for the speciation of metal binding to proteins.37–41 The laser ablation is carried out on the gel and the ablated sample is directly blown into the ICP collision cell by a stream of argon gas. LA-ICP-MS offers a greater sensitivity and isotopic resolution compared to PIXE analysis. The use of sulfur analysis from protein cystein residues has been proposed in order to use sulfur quantitative analysis as an internal standard when the protein structure is known.41 However quantification of metal content using LA-ICP-MS can sometimes be difficult due to matrix effects.| Analytical technique | Detection limit/μg g−1 | Selectivity | Quantification | Analytical depth/μm |
|---|---|---|---|---|
| Autoradiography | <0.01 | Monoelemental (radiolabeled isotope) | Quantitative | — |
| Particle induced X-ray emission (PIXE) | 1 | Multielemental | Quantitative | 10–100 |
| Synchrotron radiation X-ray fluorescence (SR-XRF) | 0.1 | Multielemental | Quantitative | >100 |
| X-Ray absorption spectroscopy (XAS) | 100 | Chemical species | Quantitative | >100 |
| Laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) | 0.01 | Multielemental and isotopic | Quantitative (matrix effects) | 200 |
Specificities and limitations of synchrotron radiation based methods
XAS has been widely applied to study metalloproteins in the last few years because it is a quasi-unique method used to provide chemical and structural information about ‘spectroscopically quiet’ metals.20EXAFS is commonly used to determine the coordination number and bond lengths of the absorbing elements. EXAFS also enables the determination of the nature of the ligands binding to the metal centre. However there are some limitations, EXAFS enables us to distinguish only between ligands that differ by one row in the periodic table. In metalloprotein studies, EXAFS is able to tell whether the metals in a protein are bound together in a multinuclear cluster or are bound at separate sites. It also enables us to identify metal to sulfur bounds but cannot distinguish between oxygen and nitrogen metal binding. On the other hand, XANES can be used to determine the oxidation state of a metal in a protein crystal, or a protein band from electrophoresis gels. In this case, a special procedure should be applied to preserve oxidation states during storage and analysis by using cryogenic methods.28 Such structural and chemical information cannot be obtained by the other techniques for direct metal analysis on electrophoresis gels (Table 1).A second advantage of synchrotron radiation based methods for metalloprotein studies is the possibility to obtain the identification, quantification, and speciation of inorganic elements in protein bands simultaneously by combining SR-XRF and XAS on the same sample. The detection limit offered by SR-XRF, about 0.1 μg g−1, is better than PIXE analysis, but worse than autoradiography and LA-ICP-MS, meaning that only highly expressed proteins can be identified, especially if XAS must be applied. A general limitation of XAS analysis is that only the average structure can be determined. If the metal of interest is present in multiple environments, such as different oxidation states, the information determined by XAS will represent the mixture of these states.28 Another obvious limitation of synchrotron radiation based methods is the access to the synchrotron beam, although this point is improving with the construction of new facilities worldwide. Nevertheless, XAS analysis remains time consuming and although high-throughput analyses of metalloproteins are under development for protein crystal analysis4 such a systematic approach has not been tested for proteins on electrophoresis gels.
Conclusions and challenges
Two dimensional gel electrophoresis is a powerful and widely used technique for proteome analysis. Using 2D gel electrophoresis it is, for example, possible to identify isoforms of metalloproteins of different isoelectric points.28 However, such a protein separation protocol is not very often used to study metalloproteins. The aim of this review article was to underline the potential of direct analytical methods for metal identification, detection, and speciation on 2D electrophoresis gels, and to estimate the specific role of synchrotron radiation based methods. The main analytical methods for direct metal analysis of metalloproteins on electrophoresis gels are autoradiography, PIXE, SR-XRF, XAS and LA-ICP-MS. These methods are suited to the identification of metal cofactors in proteins. One important common feature of these five analytical methods is that they can be applied directly after PAGE, avoiding the decomposition of the matrix to liberate the metal. Autoradiography, LA-ICP-MS and SR-XRF are the most sensitive methods and should be used preferentially for metal identification after PAGE. XAS is the unique technique that can achieve structural data on the chemical structure of the metal active-site. XAS can be used to identify the oxidation state, coordination, the nature of the ligands and inter-atomic distances of the metal cofactor. Although XAS has been mainly applied so far to the analysis of protein crystals in combination with X-ray protein crystallography, it has also great potential for the study of non-crystalline metalloproteins after PAGE.At least two important challenges must be overcome in the future to enlarge the application of SR-XRF and XAS to the direct analysis of proteins on 2D gels. The first one regards the detection limits, especially of XAS. Some technical improvements to the efficiency of X-ray detectors and application of XAS with a micro-beam to small protein bands should enable the investigation of weakly expressed metalloproteins. The second challenge is more general and concerns the sample preparation. Treatment with SDS is denaturing and results in a loss of non-covalent bound metals. This is why metal analysis must be performed following non-denaturing IEF and 2D PAGE in order to identify metalloproteins with non-covalent bound metals. However non-denaturing conditions are known to alter the resolution power of 2D-PAGE. One solution to this problem could be to separate protein fractions under native conditions before 2D PAGE by using, for example, steric exclusion chromatography, or other related chromatographic methods that would lower the number of proteins in the electrophoresisgel .
References
- J. Szpunar, Analyst, 2000, 125, 963–988 RSC.
- R. Ma, C. W. McLeod, K. Tomlinson and R. K. Poole, Electrophoresis, 2004, 25, 2469–2477 CrossRef CAS.
- J. Szpunar, Anal. Bioanal. Chem., 2004, 378, 54–56 CAS.
- I. Ascone, J. Synchrotron Radiat., 2005, 12, 1–3 CAS.
- R. A. Scott, J. E. Shokes, N. J. Cosper, F. E. Jenney and M. W. W. Adams, J. Synchrotron Radiat., 2005, 12, 19–22 CAS.
- P. P. Kulkarni, Y. M. She, S. D. Smith, E. A. Roberts and B. Sarkar, Chem.–Eur. J., 2006, 12, 2410–2422 CrossRef CAS.
- J. S. Garcia, C. Schmidt de Magalhaes and M. A. Zezzi Arruda, Talanta, 2006, 69, 1–15 CrossRef CAS.
- Y. Gao, C. Chen and Z. Chai, J. Anal. At. Spectrom., 2007, 22, 856–866 RSC.
- W. Shi and M. R. Chance, Cell. Mol. Life Sci., 2008, 1–9.
- R. Lobinski, C. Moulin and R. Ortega, Biochimie, 2006, 88, 1591–1604 CrossRef CAS.
- S. F. Stone, G. Bernasconi, N. Haselberger, M. Makarewicz, R. Ogris, P. Wobrauschek and R. Zeisler, Biol. Trace. Elem. Res., 1994, 43–45, 299–307 CrossRef CAS.
- S. Homma-Takeda, M. Shinyashiki, I. Nakai, C. Tohyama, Y. Kumagai and N. Shimojo, Anal. Lett., 1996, 29, 601–611 CAS.
- Y. Gao, C. Chen, Z. Chai, J. Zhao, J. Liu, P. Zhang, W. He and Y. Huang, Analyst, 2002, 127, 1700–1704 RSC.
- Y. Gao, C. Chen, P. Zhang, Z. Chai, W. He and Y. Huang, Anal. Chim. Acta, 2003, 485, 131–137 CrossRef CAS.
- G. Weseloh, M. Kühbacher, H. Bertelsmann, M. Özaslan, A. Kyriakopoulos, A. Knöchel and D. Behne, J. Radioanal. Nucl. Chem., 2004, 259, 473–477 CrossRef CAS.
- M. Kuhbacher, G. Weseloh, A. Thomzig, H. Bertelsmann, G. Falkenberg, M. Radtke, H. Riesemeier, A. Kyriakopoulos, M. Beekes and D. Behne, X-Ray Spectrom., 2005, 34, 112–117 CrossRef CAS.
- Y. Gao, Y. Liu, C. Chen, B. Li, W. Wei, Y. Huang and Z. Chai, J. Anal. At. Spectrom., 2005, 20, 473–475 RSC.
- J. S. Garcia, G. A. da Silva, M. A. Z. Arruda and R. J. Poppi, X-Ray Spectrom., 2007, 36, 122–129 CrossRef CAS.
- L. Li, G. Wu, J. Sun, B. Li, Y. Li, C. Chen, Z. Chai, A. Iida and Y. Gao, J. Toxicol. Environ. Health, 2008, 18, 1266–1269 CrossRef.
- J. E. Penner-Hahn, Coord. Chem. Rev., 2005, 249, 161–177 CrossRef CAS.
- S. S. Hasnain and K. O. Hogdson, J. Synchrotron Radiat., 1999, 6, 852–864 CrossRef CAS.
- S. S. Hasnain and R. W. Strange, J. Synchrotron Radiat., 2003, 10, 9–15 CAS.
- R. W. Strange and M. C. Feiters, Curr. Opin. Struct. Biol., 2008, 18, 1–8.
- M. Cianci, S. Antonyuk, N. Bliss, M. W. Bailey, S. G. Buffey, K. C. Cheung, J. A. Clarke, G. E. Derbyshire, M. J. Ellis, M. J. Enderby, A. F. Grant, M. P. Holbourn, D. Laundy, C. Nave, R. Ryder, P. Stephenson, J. R. Helliwel and S. S. Hasnain, J. Synchrotron Radiat., 2005, 12, 455–466 CrossRef CAS.
- M. J. Latimer, K. Ito, S. E. McPhillips and B. Hedman, J. Synchrotron Radiat., 2005, 12, 23–27 CAS.
- M. J. Ellis, S. G. Buffey, M. A. Hough and S. S. Hasnain, J. Synchrotron Radiat., 2008, 15, 433–439 CrossRef CAS.
- A. Arcovito, M. Benfatto, M. Cianci, S. S. Hasnain, K. Nienhaus, G. U. Nienhaus, C. Savino, R. W. Strange, B. Vallone and S. Della Longa, Proc. Natl. Acad. Sci., U. S. A., 2007, 15, 6211–6216 CrossRef.
- S. Chevreux, S. Roudeau, A. Fraysse, A. Carmona, G. Devès, P. L. Solari, T. C. Weng and R. Ortega, J. Anal. At. Spectrom., 2008, 23, 1117–1120 RSC.
- M. A. Hough and S. S. Hasnain, J. Mol. Biol., 1999, 287, 579–592 CrossRef.
- M. Bertrand, G. Weber and B. Schoefs, Trends Anal. Chem., 2003, 22, 254–262 CrossRef CAS.
- Z. Szökefalvi-Nagy, I. Demeter, Cs. Bagyinka and K. L. Kovács, Nucl. Instrum. Methods Phys. Res., Sect. B, 1987, 22, 156–158 CrossRef.
- Z. Szökefalvy-Nagy, C. Bagyinka, I. Demeter, K. L. Kovács and L. H. Quynh, Biol. Trace Elem. Res., 1990, 26, 93–101 CrossRef.
- Z. Szökefalvy-Nagy, C. Bagyinka, I. Demeter, K. Hollós-Nagyand and I. Kovács, Fresenius J. Anal. Chem., 1999, 363, 469–473 CrossRef CAS.
- C. Solis, A. Oliver and E. Andrade, Nucl. Instrum. Methods Phys. Res., Sect. B, 1998, 136–138, 928–931 CrossRef CAS.
- C. Solis, A. Oliver, E. Andrade, J. L. Ruvalcaba-Sil, I. Romero and H. Celis, Nucl. Instrum. Methods Phys. Res., Sect. B, 1999, 150, 222–225 CrossRef CAS.
- D. Strivay, B. Schoefs and G. Weber, Nucl. Instrum. Methods Phys. Res., Sect. B, 1998, 136–138, 932–935 CrossRef CAS.
- J. L. Neilsen, A. Abildtrup, J. Christensen, P. Watson, A. Cox and C. W. McLeod, Spectrochim. Acta B, 1998, 53, 339–345 CrossRef.
- T. W. M. Fan, E. Pruszkowski and S. Shuttleworth, J. Anal. At. Spectrom., 2002, 17, 1621–1623 RSC.
- M. R. Binet, R. Ma, C. W. McLeod and R. K. Poole, Anal. Biochem., 2003, 318, 30–38 CrossRef CAS.
- G. Ballihaut, L. Tastet, C. Pecheyran, B. Bouyssiere, O. F. X. Donard, R. Grimaud and R. Lobinski, J. Anal. At. Spectrom., 2005, 20, 493–499 RSC.
- J. Sa. Becker, M. Zoriy, J. Su. Becker, C. Pickhardt, E. Damoc, G. Juhacz, M. Palkovits and M. Przybylski, Anal. Chem., 2005, 77, 5851–5860 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |