Evaluation of metal-ion stress in sunflower (Helianthus annuus L.) leaves through proteomic changes
Jerusa Simone
Garcia
a,
Gustavo Henrique Martins Ferreira
Souza
bc,
Marcos Nogueira
Eberlin
b and
Marco Aurélio Zezzi
Arruda
*a
aUniversidade Estadual de Campinas—Unicamp, Institute of Chemistry, Spectrometry, Sample Preparation and Mechanization Group (GEPAM), P.O. Box 6154, Campinas, São Paulo, 13084-971, Brazil. E-mail: zezzi@iqm.unicamp.br; Fax: +55 (019)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35213023; Tel: +55 (019)35213089
35213023; Tel: +55 (019)35213089
bUniversidade Estadual de Campinas—Unicamp, Institute of Chemistry, ThoMSon Mass Spectrometry Laboratory, P.O. Box 6154, Campinas, São Paulo, 13084-971, Brazil
cUniversidade Estadual de Campinas—Unicamp, Department of Pharmacology, Faculty of Medical Sciences, Campinas, São Paulo, 13083-970, Brazil
First published on 27th November 2008
Abstract
In this work, sunflowers (Helianthus annuus L.) were cultivated using soil and vermicompost as substrate, and plant irrigation was carried out using either a Zn solution or a mixed ions solution (Cd, Cu, Pb and Zn). After plant harvesting, the effects of metal-ion contamination on proteins expression (either up- or down-regulation) in sunflower leaves were evaluated using two-dimensional electrophoresis (2-DE), gel images and mass spectrometry (MALDI-QTOF MS). When Zn or mixed ions solution was added to the substrate, nine proteins showed different expressions. Another twenty-three protein spots also showed considerable variation when both treatments (Zn or mixed ions) were applied. Twelve of these proteins were successfully characterized, six of them being reported for the first time in Helianthus annuus L. Two other proteins showed new sequences that have been downloaded to the protein databank.
Introduction
Metal-ion contamination is a serious type of pollution in the environment. For plants, it can induce development problems such as growth decreases, reduced biomass production and other morphological and biochemical alterations.1,2 Metal sensitivities and toxicities in plants are influenced by the concentration, the type of metal and the stage of plant development. Information concerning metal homeostasis and plant tolerance are therefore important to elucidate the mechanisms affecting plant development. Generally, the mechanisms of metal-ion tolerance involve the exclusion of the metal-ion from uptake or translocation to the shoots, and immobilization of the metal in the cell wall to protect sensitive structures in the cytoplasm. This protection against metals occurs via a complex formation with organic acids and amino acids as well as by their binding to specific proteins. Metal toxicity can, however, inhibit protein activity or disrupt their structures.3,4Another metal detoxification mechanism in plants involves the activation of antioxidative enzymes systems such as catalase, peroxidase and superoxide dismutase. These enzymes are responsible for protecting the plant cells from the toxic effects of the reactive oxygen species that have their production increased during metal contamination and other types of stress.5,6
Plant proteomics use many high-throughput biotechnological approaches to elucidate biological functions of plant proteins in different environments including those resulting from exposure to metal-ion contamination, salinity, drought, air pollutants and extreme temperatures.7 Each condition produces a unique set of proteins in the organisms or a given tissue since protein activity, location and concentration are greatly dependent on environmental, physiological and pathological conditions.8 Plant stresses result in an increase of defense protein expression.7 Proteins related to antioxidative defensive mechanisms may, however, be either down- or up-regulated.7,9
Among the proteins affected due to metal-ion contamination are the metalloproteins, which are responsible for various essential metabolic processes.2Metalloprotein evaluations also can contribute effectively to proteomic studies since they should give a new point of view for the understanding of the mechanisms of these metal-dependent proteins.10
As sunflower (Helianthus annuus L.) is considered the 5th most important culture in the world because of its oil production, has been employed in phytoremediation processes due to its capability to accumulate metals in its tissues,11 only few studies in terms of oxidative stress have been carried out and few entries in the protein data bank related to this specie can be found, then the aim of this work was to evaluate systematically (via 2-DE, images and mass spectrometric techniques) the alterations in sunflower leaf proteomes under metal-ion (Cd, Cu, Pb and Zn) stress and to characterize some of the proteins affected by metal-ion stress. Additionally, investigations using the strategy adopted in this work on sunflower proteome alterations, and caused by metal-ion toxicity are almost unexplored.
Experimental
Plant material and growth
Sunflower seeds (Helianthus annuus L.) were germinated and grown under ambient conditions at temperatures ranging from 18 to 24 °C (night and day, respectively). The seeds were cultivated in plastic pots (1 litre) with a mix of 320 g of soil from Piracaia, Brazil, and 80 g of vermicompost (humic material) from Campinas, Brazil. The plants (one plant per pot) were grown for 40 days using three different treatments. In the first one (control), sunflowers were irrigated with water only. In the second one, the sunflowers were irrigated with a solution containing 760 μmol L−1Zn(NO3)2. Finally, for the last treatment, a mixed-ion solution containing 440 μmol L−1Cd(NO3)2, 790 μmol L−1Cu(NO3)2, 240 μmol L−1Pb(NO3)2 and 760 μmol L−1Zn(NO3)2 was used. Irrigation, on alternate days, was done by adding 30 mL of the appropriate solution to the substrate contained in each pot, totalling 300 mg of each metal-ion after 40 days. The metal quantities added to the substrate were above the phytotoxicity level. At the end of the experiment, ten replicates were obtained for each treatment.Protein extraction and two-dimensional gel electrophoresis
The protein extraction procedure was based on that reported by Garcia et al.11 with minor modifications. For that, sunflower leaves were harvested, immediately frozen in liquid nitrogen and stored at −80 °C until extraction. Ground leaves (1 g) were suspended into 3 mL of a solution containing 125 mmol L−1Tris-HCl (pH 6.8), 1% (m/v) SDS, 1% (v/v) glycerol and 0.5% (v/v) β-mercaptoethanol. The homogenate was then centrifuged at 8500 g for 5 min at 4 °C and the pellet discarded. To precipitate proteins for sample cleaning, 5 mL of a precipitating agent (a solution comprising 20% (m/v) trichloroacetic acid and 0.2% m/v dithiothreitol–DTT in acetone) were added to 1 mL of protein extract. The pellets obtained were washed three times using cold acetone containing 0.2% (m/v) DTT. Then, protein concentration was determined according to the Bradford method using bovine serum albumin as standard.12 For that, the protein pellets were re-dissolved employing a solution containing 1 mol L−1Tris-HCl (pH 6.8).2-DE was carried out following the manufacturer’s (GE Healthcare, Uppsala, Sweden) recommendations.13 A mass of 2.4 mg protein was suspended into 300 μL of lysis buffer (7 mol L−1urea, 2 mol L−1thiourea, 2% m/v 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 0.002% m/v bromophenol blue, 0.5% (v/v) carrier ampholytes (pH 3–10) and 1% m/v DTT) at room temperature (25 °C) for 5 min. Immobilized gradient pH strips (13 cm, pH 3–10, linear) were loaded with sample proteins during rehydration overnight at room temperature. Isoelectric focusing was carried out in a Multiphore II system (GE Healthcare), totalizing 16000 V h.
The second dimension separation was carried out at 25 mA per gel and 100 W during ca. 5.5 h in an Ettan DALT System (GE Healthcare) with lab cast 1 mm SDS polyacrylamide gels having a 12.5% (m/v) acrylamide concentration. After separation, the protein spots were visualized using 0.12% (m/v) colloidal Coomassie Brilliant Blue G-250.14 At least three gels of each treatment were obtained in independent experimental days.
The gels were scanned using an ImageScanner II (GE Healthcare) with the densitometer operating at 10000 dpi resolution. ImageMaster 2D Platinum 6.0 software (GeneBio, Geneva, Switzerland) was used to analyze the gel images. Spot detections were done using default detection parameters and without manual editing. Spot volume was used instead of %volume (commonly employed to compare differential protein expression between gels) because the latter is more appropriate for gels with similar spot patterns.
Protein characterization by MALDI-QTOF MS
Protein spots that showed changes in expression due to different environmental conditions, as well as some others that were chosen randomly were characterized by MALDI-QTOF MS. For this task, an in-gel digestion of protein spots was performed using the Montage In-Gel DigestZP Kit (Millipore, Bedford, MA, USA) according to the manufacturer’s recommendations.All samples obtained by tryptic digestion were analyzed using the dried droplet method.15 The sample was acidified by adding a volume of 0.1% (v/v) trifluoroacetic acid (TFA) to the digested samples in a ratio 2 (TFA) to 1 (sample). The acidified sample (1 μL) was spotted on the MALDI plate for protein characterization, and kept at room temperature until complete solvent evaporation. MALDI matrix (1 μL) was then added to the sample, which also allowed drying at room temperature. This latter matrix was prepared from 1% (m/v) α-cyano-4-hydroxycinnamic acid, which was dissolved in a 1:1 (v/v) acetonitrile–H2O solution containing 0.1% (v/v) TFA.16
MALDI-QTOF mass spectra were acquired in a MALDI-QTOF Premier mass spectrometer (Waters–Micromass, Manchester, UK). The mass spectra were obtained in the positive mode (LDI+) with a fixed nitrogen ion source and LockMass correction with 0.1% (v/v) phosphoric acid using the following main parameters: mass range from 880.0 to 3000.0 Da, peak detection threshold for MS/MS of 1500.0, mass threshold of 200.0 Da, scan time of 2 s, resolution of 10000 in “V” mode, trigger threshold of 700 mV, signal sensitivity of 80 mV, and microchannel-plate photomultiplier set to 2100 V. Each spectrum was collected over a 1 s scan, and the spectra were accumulated over ca. 2 min. The instrument was controlled by MassLynx 4.1v software. All mass spectra were processed into peak list files with a *.pkl extension using ProteinLynxGlobalServer 2.2.5v (Waters, UK).
Protein identification was achieved by searching a database using the peptide peak list (*.pkl file) masses and intensities files generated by MALDI-QTOF pos-processing mass spectra through ProteinLynxGlobalServer. Identification of proteins was performed using ProteinLynxGlobalServer 2.2.5v (Waters, UK). The Expressed Sequence Tag databank was obtained through The Institute of Genomic Research file transfer protocol download (ftp://ftp.tigr.org/pub/data/plantta/Helianthus_annuus)17 which was added and compiled into PLGS 2.2.5v to generate a concise open reading-frame of proteinamino acids. Monoisotopic peak lists were processed with the following search parameters: HELIANTHUS-1.0 databank field input file, one missed cleavage, tryptic digestion, carbamidomethylation as a cysteine modification. The search error tolerance was set at 5 ppm with a [M+H]+ charge state.
Results and discussion
Influence of metal-ion contamination in the protein profile
In an earlier work11 we used one dimensional electrophoresis separations to evaluate the effect of metal-ion contamination on sunflower development as related to height, mass production, nutrition status, and protein level. Significant differences were observed for protein expression in sunflower leaves. Inspired by this result, we performed a series of new experiments using 2-DE and mass spectrometry to elucidate in more detail which sunflower proteins would be up- or down-regulated according to some specific metal-ion contamination; hence, those proteins of potential biotechnological interest for metal-ion detoxification and accumulation.Using control sunflower leaves, more than 270 spots (Fig. 1A) were reproducibly detected in the 3–10 pH range (n = 3, 70% match). But when sunflower plants were irrigated with a zinc solution (Fig. 1B) or with a mixed ions solution (Fig. 1C) only ca. 105 (n = 3, 58% match) and 135 spots (n = 3, 55% match) were detected, respectively. Then, the presence of different metal-ions was found to result in considerable alterations of sunflower leaf protein compositions. Additionally, the comparison between proteomes was based on the expression of those more affected proteins so as to evaluate the sunflower plant development responses obtained after each metal treatment.
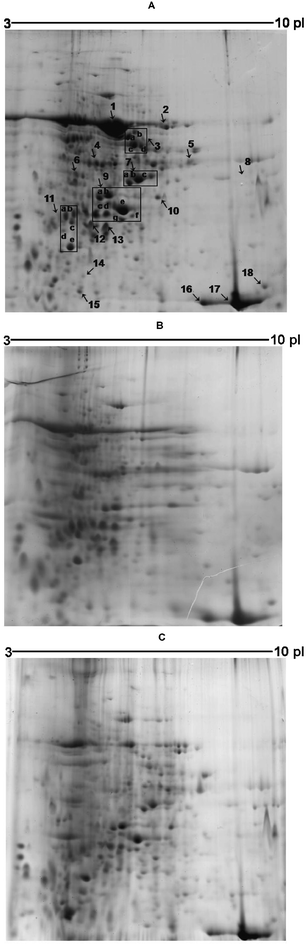 | ||
| Fig. 1 Representative 2-DE gel for proteins for sunflower leaves. 2.4 mg of protein was loaded and run on 13 cm IPG strips (pH 3–10, linear). SDS-PAGE gels were used in the second dimension. The protein spots were visualized by Coomassie Brilliant Blue. (A) Control, (B) zinc contamination and (C) mixed-ion contamination. Arrows indicate the protein spots/regions analyzed, which were designated as numbers 1–18. The arrow 8 indicates proteins that were expressed only in the conditions specified in Fig. 1C. | ||
The proteins with the most remarkable differences in expression are identified in Fig. 1A. Representative examples of four different protein profiles are also highlighted in Fig. 2. The proteins depicted in Fig. 2 and some others also presented in the Fig. 1 were quantified (in terms of absolute volume). The absolute volume of the spot was determined using ImageMaster 2D Platinum 6.0 software (GeneBio, Geneva, Switzerland). This software calculates spot volume by multiplication of spot area (expressed in mm2) and spot intensity (based on the highest calibrated pixel intensities in the spot from which the background has been withdrawn). The same procedure to proteins from contamination treatments was done. These volumes were then compared to those from the control (Table 1). Proteins were considered as up- or down-regulated when the spot volume changed at least 1.8-fold after contamination. This cutoff is the average value found in the literature concerning comparative studies on protein expression.18,19
 | ||
| Fig. 2 Different gel regions were magnified using a 3D map to emphasize different expression of protein spots (numbered according to Fig. 1A). | ||
| Spot/Gel Region | Volume (104)a | Situation | |||
|---|---|---|---|---|---|
| Control | Zinc contamination (1) | Mixed ions contamination (2) | (1) | (2) | |
| a Volume calculated using ImageMaster 2D Platinum 6.0 software. b Spot not determined using the ImageMaster 2D Platinum 6.0 software. | |||||
| 1 | 276.0 | b | 19.6 | Down | Down |
| 2 | 13.0 | 11.7 | 8.5 | Same | Same |
| 3a | 11.2 | b | 3.7 | Down | Down |
| 3b | 8.5 | 8.9 | 1.8 | Same | Down |
| 3c | 24.5 | b | 3.2 | Down | Down |
| 3d | 12.4 | b | 4.0 | Down | Down |
| 4 | 11.9 | 14.0 | 5.3 | Same | Down |
| 5 | 11.1 | b | 23.7 | Down | Up |
| 6 | 8.0 | 8.9 | b | Same | Down |
| 7a | 16.5 | b | 14.1 | Down | Same |
| 7b | 55.0 | b | 28.2 | Down | Down |
| 7c | 18.7 | 8.9 | 19.8 | Down | Same |
| 7d | b | b | 5.0 | Same | Up |
| 8a | b | b | 4.2 | Same | Up |
| 8b | b | b | 3.2 | Same | Up |
| 8c | b | b | 4.5 | Same | Up |
| 8d | b | b | 2.5 | Same | Up |
| 9a | 14.6 | 17.4 | 4.4 | Same | Down |
| 9b | 34.5 | 43.0 | 15.0 | Same | Down |
| 9c | 21.0 | 31.6 | 15.9 | Same | Same |
| 9d | 15.5 | 31.9 | 9.8 | Up | Same |
| 9e | 63.1 | 23.2 | 36.6 | Down | Down |
| 9f | 4.4 | 6.4 | 5.3 | Same | Same |
| 9g | 9.5 | b | 7.0 | Down | Same |
| 9h | b | b | 8.7 | Same | Up |
| 10 | 6.5 | 7.1 | 15.5 | Same | Up |
| 11a | 12.8 | 20.9 | b | Same | Same |
| 11b | 11.5 | 17.4 | b | Same | Down |
| 11c | 11.5 | 19.0 | 7.7 | Same | Same |
| 11d | 5.2 | 1.1 | b | Down | Down |
| 11e | 18.1 | 26.5 | 26.5 | Same | Same |
| 12 | 11.9 | 6.8 | 13.3 | Down | Same |
| 13 | 18.2 | 28.6 | 9.6 | Same | Down |
| 14 | 3.2 | b | b | Down | Down |
| 15 | 5.6 | 16.5 | 9.1 | Up | Same |
| 16 | 42.5 | 89.9 | 27.3 | Up | Same |
| 17 | 94.1 | 236.4 | 98.4 | Up | Same |
| 18 | 8.7 | 20.2 | 6.9 | Up | Same |
For the zinc and mixed ions treatments, 9 spots displayed significant changes in volume. For at least one treatment, 23 other protein spots also showed significant variation (see also Fig. 2 for some examples). The number of protein spots differentially regulated was more evident when the zinc solution was used for plant irrigation. Thirteen protein spot expressions were markedly decreased (1, 5, 7a–c, 9e and 11d, Table 1) and another 5 protein spots were up-regulated (9d and 15–18, Table 1). For the treatment with the mixed ions solution, 15 protein spots were down-regulated and 8 protein spots were up-regulated. Among the up-regulated proteins, 6 were detected only under this condition (7d, 8a–d and 9h, see Fig. 1C).
These alterations in protein expression could be explained based on the zinc and cadmium phytotoxic levels found in sunflower leaves after growth: Zn(II) > 500 μg g−1 in both treatments and Cd > 5 μg g−1 in the mixed ions treatment. To obtain these results, leaves samples were decomposed using microwave-assisted procedure with nitric acid and the metal ions were quantified by electrothermal atomic absorption spectrometer (for more details, see ref. 11). Additionally, the presence of reactive oxygen species was verified in our earlier work11 through enzymatic analyses. Changes on glutathione reductase and superoxide dismutase activities were observed in sunflower leaves due to metal-ion contamination.11
Characterization of proteins by MALDI-QTOF MS
Protein regulation as a response to metal-ion contamination is important to understand the role of the altered proteins in sunflower adaptation. Among 9 protein spots (that showed differences in expression when taken into account both metal treatments, see also Table 1), 3 of them (identified as 1, 5 and 14—Fig. 1A) were successfully characterized by MALDI-QTOF MS. Some protein spots were also included for characterization to add new sunflower leaf information to the protein databank. Since the Helianthus annuus L. genome has not yet been completely sequenced, few proteins entries (1129) are available in SwissProt and TREMBL, mainly when it is compared to Arabidopsis thaliana (51528) and Oryza sativa (142893), which already have their genome sequence elucidated. Despite this difficulty for Helianthus annuus L. protein identification, we succeeded in the characterization of 12 proteins (Table 2) and a good correlation (SCORE higher than 10.4 and COVERAGE between 69 to 88%) was obtained. Among the 12 proteins identified, six of them have been characterized for the first time for the Helianthus annuus L. species, whereas two other proteins showed new sequences that were also inserted in the protein databank.| Spot | Protein | Accession number |
|---|---|---|
| a New sequences inserted in the Expasy Proteomics Server | ||
| 1 | Ribulose bisphosphate carboxylase large chain | AAB01594 |
| 2 | Putative receptor protein kinasea | P85193 |
| 4 | Cytochrome P450 a | P85191 |
| 5 | Delta-12 oleate desaturase | AAL68982 |
| 6 | Oxygen-evolving enhancer protein 1a | P85194 |
| 9a | F6F9.12 proteina | P85200 |
| 9b | DEAD-box ATP-dependent RNA helicase 3a | P85199 |
| 12 | Unconventional myosin [Fragment] | AAB71528 |
| 13 | Oxygen-evolving enhancer protein 2, chloroplast precursora | P85189 |
| 14 | Cell division protease ftsH homologa | P85190 |
| 17 | Hypothetical protein 1a | P85192 |
| 18 | CC-NBS-LRR-like protein [Fragment] | AAT08958 |
Spot 1 was identified as ribulose bisphosphate carboxylase large chain (the major leaf protein—called RuBisCO). This protein participates in the Calvin cycle (CO2 fixation) during photosynthesis processes as well as the oxidative fragmentation of the pentose substrate in the photorespiration process.11
A significant down-regulation of ribulose bisphosphate carboxylase was induced for both treatments (zinc and mixed ions), but it was more pronounced when the zinc solution alone was used for plant irrigation. Kim et al.20 reported that other stresses such as salt, drought, high temperature and ozone contribute to the ribulose bisphosphate carboxylase large chain accumulation. This protein was also up-regulated in Arabidopsis thaliana cells when they were exposed for 24 h to 200 μmol L−1CdCl2.21 Tuomainen et al.22 reported, however, that cadmium contamination causes RuBisCO degradation in the Thlaspi caerulescens species (plant employed for bioremediation purposes).
Spot 5 was attributed to delta-12 oleate desaturase. This protein participates in oxidoreductase and phosphatidylcholine desaturase, and it can promote electron pair donors, resulting in molecular oxygen reduction and the production of two water molecules. The delta-12 oleate desaturase also participates in the fatty acid biosynthetic process.23 Here, we found that changes in the expression of this protein is metal-ion dependent since down- and up- protein expression were observed when zinc and mixed ions solution were used, respectively.
The unconventional myosin [Fragment] was identified in spot 12, which has the molecular function of ATP binding and motor activity. The function of the CC-NBS-LRR-like protein [Fragment] (spot 18) is up to now unknown.23 These last two proteins showed modification in their expression due to zinc stress. In this condition, unconventional myosin and CC-NBS-LRR-like proteins were down- and up-regulated, respectively.
Some proteins shown in Table 2 are being reported for the first time, hence their functions are putatively described based on homologies to others plants. Putative receptor protein kinase (spot 2) is responsible for ATP binding, protein binding, protein serine/threonine kinase activityand receptor activity as well as protein amino acid phosphorylation.23 This protein abundance was not affected by metal-ion stress.
Cytochrome P450 (spot 4) participates in electron transport, promotes iron ion and heme binding as well as monooxygenase activity, which is responsible for the insertion of one atom of oxygen into an organic compound, whereas the other oxygen atoms are reduced to water.23 This protein showed down-expression when the plants were irrigated with the mixed ions solution. According to Aina et al.,24cytochrome P450 expression was also inhibited in Oryza sativa L. roots due to cadmium toxicity.
Oxygen-evolving enhancer protein 1 and oxygen-evolving enhancer protein 2 (spots 6 and 13, respectively) were associated to the photosystem II complex. The first one stabilizes the manganese cluster, which is the primary site of water splitting. The second one may be involved in photosystem II regulation.23 According to Tuomainen et al.,22 these proteins (involved in the energy metabolism) were up-regulated in Thlaspi caerulescens exposed to 500 mmol L−1ZnSO4 for three weeks. However, we observed that these proteins were down-regulated due to the mixed ions treatment.
DEAD-box ATP-dependent RNA helicase 3 (present in spot 9b, see also Fig. 2), which is responsible for ATP-binding and hydrolysis and also shows helicase activity(catalyzes the unwinding of double-stranded nucleic acids). According to Roth et al.,25 the exposure of Arabidopsis thaliana roots to cadmium contamination showed significant changes in terms of helicase family abundance (it was down-regulated). We observed similar behavior for mixed ions stress.
Cell division protease ftsH homolog (spot 14) seems to act as an ATP-dependent zinc metallopeptidase and also to be related to metalloprotease and metal-binding.24 This protein was not expressed when either zinc or the mixed ions solution were used for inducing stress.
Hypothetical protein 1 (spot 17) shows the function of peptidyl–prolyl cis–trans isomerase activity. There is no information about the F6F9.12 protein (spot 9a) function in the literature.23
Other substantial changes were observed in protein expression (spots 5, 9a, 12 and 17, Table 1), which have not been previously reported. This contrast could be due to the different systems (cell cultures, roots, and leaves) or plant species as well as the different conditions investigated, making any comparative evaluation difficult.
Conclusions
The evaluation of metal-ion stress was successfully carried out in this work, inside the experimental domain. Metal-ion stress affected a significant number of proteins once that there was a decrease on the quantity of proteins in ca. 61% and 50% when the protein map from control sunflower leaves was compared with those obtained from plants irrigated with a zinc solution and with mixed ions solution, respectively. Additionally, metal-ion stress did affect a significant number of proteins, which showed up- or down-regulation in sunflower leaves. These alterations were more significant for those proteins related to energy metabolism (e.g. ribulose bisphosphate carboxylase large chain, putative receptor protein kinase, oxygen-evolving enhancer protein 1, oxygen-evolving enhancer protein 2 and DEAD-box ATP-dependent RNA helicase 3).In order to improve proteomic information related to sunflower, some proteins were also successfully identified. Eight new proteins sequences for Helianthus annuus L. were characterized. These proteins, reported for the first time, were appropriately inserted in the Expasy Proteomics Server.
Finally, we consider that the combination of “omics” approaches would be expected to generate new information not only related to sunflower responses to metal-ion contamination but also related to other plants that show the capability of metal-ion accumulation, which could be used in the phytoremediation process.
Acknowledgements
We thank the Fundação de Amparo a Pesquisa do Estado de São Paulo (FAPESP), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Financiadora de Estudos e Projetos (FINEP), and Proteomic Network of the São Paulo state for financial support and for fellowships to J.S.G. (FAPESP), G.H.M.F.S. (CAPES) and M.A.Z.A and M.N.E (CNPq). The authors also thank Fabio C. Gozzo for use of Mass Spectrometry Laboratory at the Brazilian Synchrotron Light Laboratory facilities, and Carol H. Collins for language assistance.References
- R. Gopal and A. H. Rizvi, Chemosphere, 2008, 70, 1539–1544 CrossRef CAS.
- J. S. Garcia, C. S. de Magalhães and M. A. Z. Arruda, Talanta, 2006, 69, 1–15 CrossRef CAS.
- A. I. Sousa, I. Caçador, A. I. Lillebø and M. A. Pardal, Chemosphere, 2008, 70, 850–857 CAS.
- M. P. Benavides, S. M. Gallego and M. L. Tomaro, Braz. J. Plant Physiol., 2005, 17, 21–34 Search PubMed.
- P. L. Gratão, A. Polle, P. J. Lea and R. A. Azevedo, Funct. Plant Biol., 2005, 32, 481–494 CrossRef CAS.
- A. Arora, R. K. Sairam and G. C. Srivastava, Curr. Sci. India, 2002, 82, 1227–1238 Search PubMed.
- N. Ahsan, D. G. Lee, S. H. Lee, K. Y. Kang, J. J. Lee, P. J. Kim, H. S. Yoon, J. S. Kim and B. H. Lee, Chemosphere, 2007, 67, 1182–1193 CrossRef CAS.
- G. Caruso, C. Cavaliere, C. Guarino, R. Gubbiotti, P. Foglia and A. Lagana, Anal. Bioanal. Chem., 2008, 391, 381–391 CrossRef CAS.
- M. Labra, E. Gianazza, R. Waitt, I. Eberini, A. Sozzi, S. Regondi, F. Grassi and E. Agradi, Chemosphere, 2006, 62, 1234–1244 CrossRef CAS.
- A. Sussulini, G. H. M. F. Souza, M. N. Eberlin and M. A. Z. Arruda, J. Anal. At. Spectrom., 2007, 22, 1501–1506 RSC.
- J. S. Garcia, P. L. Gratão, R. A. Azevedo and M. A. Z. Arruda, J. Agric. Food Chem., 2006, 54, 8623–8630 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- T. Berkelman and T. Stenstedt, 2-D Electrophoresis—Principles and Methods, Amersham Biosciences, Uppsala, 1998 Search PubMed.
- G. Candiano, M. Bruschi, L. Musante, L. Santucci, G. M. Ghiggeri, B. Carnemolla, P. Orecchia, L. Zardi and P. G. Righetti, Electrophoresis, 2004, 25, 1327–1333 CrossRef CAS.
- P. Onnerfjord, S. Ekstrom, J. Bergquist, J. Nilsson, T. Laurell and G. Marko-Varga, Rapid Commun. Mass Spectrom., 1999, 13, 315–322 CrossRef CAS.
- B. Granvogl, M. Plöscher and L. A. Eichacker, Anal. Bioanal. Chem., 2007, 389, 991–1002 CrossRef CAS.
- K. L. Childs, J. P. Hamilton, W. Zhu, E. Ly, F. Cheung, H. Wu, P. D. Rabinowicz, C. D. Town, C. R. Buell and A. P. Chan, Nucleic Acids Res., 2007, 35, D846–D85 CrossRef CAS.
- R. Parker, T. J. Flowers, A. L. Moore and N. V. J. Harpham, J. Exp. Bot., 2006, 57, 1109–1118 CrossRef CAS.
- M. Eravci, S. Fuxius, O. Broedel, S. Weist, S. Eravci, U. Mansmann, H. Schuter, J. Tiemann and A. Baumgartner, Proteomics, 2007, 7, 513–523 CrossRef CAS.
- Dea-Wook Kim, R. Rakwai, G. K. Agrawal, Young-Ho Jung, J. Shibato, Nam-Soo Jwa, Y. Iwahashi, H. Iwahashi, D. H. Kim, Ie-Sung Shim and K. Usui, Electrophoresis, 2005, 26, 4521–4539 CrossRef CAS.
- J. E. Sarry, L. Kuhn, C. Ducruix, A. Lafaye, C. Junot, V. Hugouvieux, A. Jourdain, O. Bastien, J. B. Fievet, D. Vailhen, B. Amekraz, C. Moulin, E. Ezan, J. Garin and J. Bourguignon, Proteomics, 2006, 6, 2180–2198 CrossRef CAS.
- M. H. Tuomainen, N. Nunan, S. J. Lehesranta, A. I. Tervahauta, V. H. Hassinen, H. Schat, K. M. Koistinen, S. Auriola, J. McNicol and S. O. Kärenlampi, Proteomics, 2006, 6, 3696–3706 CrossRef CAS.
- Expasy Proteomics Server-http://www.expasy.org, accessed in January, 2007.
- R. Aina, M. Labra, P. Fumagalli, C. Vannini, M. Marsoni, U. Cucchi, M. Bracale, S. Sgorbati and S. Citterio, Environ. Exp. Bot., 2007, 59, 381–392 CrossRef CAS.
- U. Roth, E. von Roepenack-Lahaye and S. Clemens, J. Exp. Bot., 2006, 57, 4003–4010 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |