 Open Access Article
Open Access ArticleGenome-wide survey of microRNA–transcription factor feed-forward regulatory circuits in human†
Angela
Re‡
a,
Davide
Corá‡
bd,
Daniela
Taverna
cd and
Michele
Caselle
*bd
aCIBIO-Centre for Integrative Biology, University of Trento, Via delle Regole 101, I-38100 Trento, Italy. E-mail: re@science.unitn.it
bDepartment of Theoretical Physics, University of Torino and INFN, Via Pietro Giuria 1, I-10125 Torino, Italy. E-mail: cora@to.infn.it; caselle@to.infn.it; Fax: +39 011-6707214; Tel: +39 011-6707205
cDepartment of Oncological Sciences, University of Torino and Molecular Biotechnology Center, Via Nizza 52, I-10126 Torino, Italy. E-mail: daniela.taverna@unito.it
dCenter for Complex Systems in Molecular Biology and Medicine, University of Torino, Via Accademia Albertina 13, I-10100 Torino, Italy
First published on 19th June 2009
Abstract
In this work, we describe a computational framework for the genome-wide identification and characterization of mixed transcriptional/post-transcriptional regulatory circuits in humans. We concentrated in particular on feed-forward loops (FFL), in which a master transcription factor regulates a microRNA, and together with it, a set of joint target protein coding genes. The circuits were assembled with a two step procedure. We first constructed separately the transcriptional and post-transcriptional components of the human regulatory network by looking for conserved over-represented motifs in human and mouse promoters, and 3′-UTRs. Then, we combined the two subnetworks looking for mixed feed-forward regulatory interactions, finding a total of 638 putative (merged) FFLs. In order to investigate their biological relevance, we filtered these circuits using three selection criteria: (I) GeneOntology enrichment among the joint targets of the FFL, (II) independent computational evidence for the regulatory interactions of the FFL, extracted from external databases, and (III) relevance of the FFL in cancer. Most of the selected FFLs seem to be involved in various aspects of organism development and differentiation. We finally discuss a few of the most interesting cases in detail.
Background
A basic notion of modern systems biology is that biological functions are performed by groups of genes that act in an interdependent and synergic way. This is particularly true for regulatory processes for which it is by now mandatory to assume a “network” point of view.Among the various important consequences of this approach, a prominent role is played by the notion of “network motifs”. The idea is that a complex network (say a regulatory network) can be divided into simpler, distinct regulatory patterns called network motifs, typically composed of three or four interacting components that are able to perform elementary signal processing functions. Network motifs can be thought of as the smallest functional modules of the network and, by suitably combining them, the whole complexity of the original network can be recovered.
In this paper we shall be interested in “mixed” network motifs involving both transcriptional (T) and post-transcriptional (PT) regulatory interactions, and in particular we shall especially focus our attention on the mixed feed-forward loops. Feed-forward loops (FFLs) have been shown to be one of the most important classes of transcriptional network motifs.1,2 The major goal of our work is to extend them to those also including post-transcriptional regulatory interactions.
Indeed, in the last few years it has become more and more evident that post-transcriptional processes play a much more important role than previously expected in the regulation of gene expression.
Among the various mechanisms of post-transcriptional regulation, a prominent role is played by a class of small RNAs called microRNAs (miRNAs), reviewed in refs. 3 and 4. miRNAs are a family of ∼22 nt small non-coding RNAs, which negatively regulate gene expression at the post-transcriptional level in a wide range of organisms. They are involved in different biological functions, including developmental timing, pattern formation, embryogenesis, differentiation, organogenesis, growth control and cell death. They certainly play a major role in human diseases as well.5,6
Mature miRNAs are produced from longer precursors, which in some cases cluster together in so-called miRNA “transcriptional units” (TU),7 and their expression is regulated by the same molecular mechanisms that control protein-coding gene expression. Even though the precise mechanism of action of the miRNAs is not well understood, the current paradigm is that in animals, miRNAs are able to repress the translation of target genes by binding, in general, in a Watson–Crick complementary manner to 7 nucleotides (nts) long sequences present at the 3′-untranslated region (3′-UTR) of the regulated genes. The binding usually involves nts 2–8 of the miRNA, the so-called “seed”. Often, the miRNA binding sites at the 3′-UTR of the target genes are over-represented.8–14
All these findings, in addition to the large amount of work related to the discovery of transcription factor binding sites (for a recent review, see for instance ref. 15), suggest that both transcriptional and post-transcriptional regulatory interactions could be predicted in silico by searching over-represented short sequences of nts present in promoters or 3′-UTRs, and by filtering the results with suitable evolutionary or functional constraints.
Stemming from these observations, the aim of our work was to use computational tools to generate a list of feed-forward loops in which a master transcription factor (TF) regulated a miRNA, and together with it, a set of target genes (see Fig. 1a). We performed a genome wide “ab initio” search, and we found in this way a total of 638 putative (merged) FFLs. In order to investigate their biological relevance, we then filtered these circuits using three selection criteria: (I) GeneOntology enrichment among the joint targets of the FFL, (II) independent computational evidence for the regolatory interactions of the FFL, extracted from the ECRbase, miRBase, PicTar end TargetScan databases, and (III) relevance to cancer of the FFL as deduced from their intersection with the Oncomir and Cancer gene census databases.
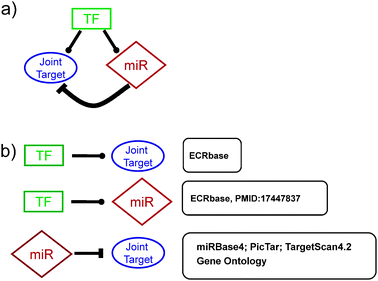 | ||
Fig. 1
Feed-forward loops. (a) Representation of a typical mixed feed-forward loop (FFL) analyzed in this work. In the square box, TF is the master transcription factor; in the diamond-shaped box miR represents the microRNA involved in the circuit, while in the round box, the Joint Target is the joint protein-coding target gene (JT). Inside each circuit, –• indicates transcriptional activation/repression, whilst  indicates post-transcriptional repression. (b) Flow-chart of the annotation strategies for the feed-forward circuits. After building the catalogue of closed FFLs (see Fig. 2), each side of the circuit was expanded and analyzed using external support databases and functional annotations. Beside each circuit link the source used for its annotation is reported; see Materials and Methods for details. indicates post-transcriptional repression. (b) Flow-chart of the annotation strategies for the feed-forward circuits. After building the catalogue of closed FFLs (see Fig. 2), each side of the circuit was expanded and analyzed using external support databases and functional annotations. Beside each circuit link the source used for its annotation is reported; see Materials and Methods for details. | ||
In a few cases some (or all) of the regulatory interactions that composed the feed-forward loop were found to be already known in the literature, with their interplay in a closed regulatory circuit not noticed, thus representing an important validation of our approach. However, for several loops we predicted new regulatory interactions, which represent reliable targets for experimental validation.
Let us finally notice that in this work we only discuss the simplest non-trivial regulatory circuits (feed-forward loops). However, our raw data could be easily used to construct more complex network motifs. For this reason, we make them accessible to the interested investigators as ESI.†
Results
Here we provide a collection of circuits that explicitly link a transcription factor (TF) and a microRNA (miRNA), which both regulate a set of common target genes (Fig. 1a). To this end we (1) constructed a transcriptional regulatory network, (2) defined a miRNA-mediated post-transcriptional regulatory network, (3) merged the two networks, and (4) filtered the results with various selection criteria (Fig. 2). In the next section we shall then discuss a few cases in more detail.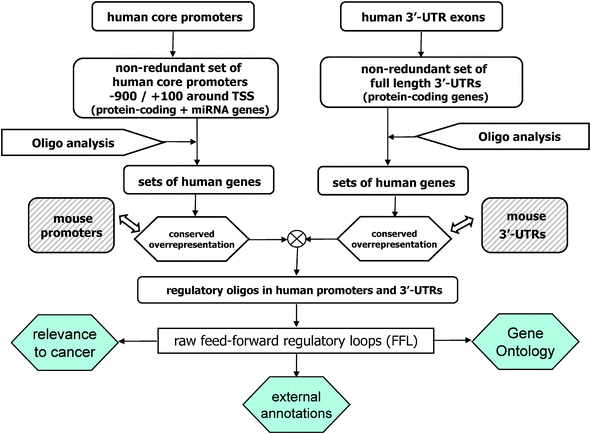 | ||
| Fig. 2 Flow-chart of our pipeline for the identification of the mixed feed-forward regulatory loops. We built two independent but symmetrical pipelines for the construction of a transcriptional and, separately, a post-transcriptional regulatory network in humans. On the left: we defined a catalogue of core promoter regions around the transcription start sites (TSS) for protein-coding and miRNA genes in the human genome. We then applied a genome-wide sequence analysis strategy in order to identify a catalogue of human putative transcriptional regulatory motifs and the corresponding regulated genes. In so doing, the key ingredients used were statistical properties of short DNA words (oligo analysis) and conservation to mouse, implemented in an alignment-free manner (conserved over-representation). On the right: a similar strategy was used, starting from a catalogue of 3′-UTRs in humans, to obtain a catalogue of human post-transcriptional regulated genes, with a focus for miRNA-mediated interactions. We fixed 0.1 as the false discovery rate (FDR) level for both the two motifs discovery pipelines. At the end, the two regulatory networks were merged to extract the complete dataset of closed mixed feed-forward loops (FFLs), as defined in Fig. 1a, and the results were filtered according to three different procedures: by looking for (I) significant functional (Gene Ontology) annotations between the joint targets of the FFLs, (II) independent computational evidences for the regulatory interactions of the FFLs, and (III) relevance to cancer. See Materials and Methods for details. | ||
Circuits identification
Construction of a human transcriptional regulatory network
The starting point of our analysis was the construction of a database of promoter regions for both protein-coding and miRNA genes for human and mouse. Details of this construction are reported in the Materials and Methods section. Here we only stress our main choices. For protein-coding genes we selected the core promoter region near the transcription start site (TSS), whereas for the miRNA promoters, we chose to merge together all the miRNAs present in the so called “transcriptional units” (TUs) proposed in ref. 7, kept only the conserved TUs (human and mouse) and then selected the putative core promoter regions (see the ESI, supplementary files S1 and S2†).We then identified, separately for humans and mice, sets of genes (protein-coding plus miRNAs) sharing over-represented oligonucleotides (oligos), 6–9 nts long, in their associated promoter regions. Next, we selected the oligos for which the human and mouse sets contained a statistically significant fraction of orthologous genes. In doing so, we used a binomial model for the assessment of over-representation and an alignment-free evolutionary methodology for the identification of conserved oligos, as previously used in refs. 16 and 17. This approach was also extended to the putative promoters of miRNA genes. All the sequences were repeat-masked, and we took into account either redundancy due to superposition of the same genomic areas or protein-coding exons, or correction for CG content of the sequences themselves. As a final result, we ended up with a catalogue of cis-regulatory motifs conserved in the core promoter regions of human and mouse protein-coding or miRNA genes, each endowed with a score (the p-value of the evolutionary conservation test, described in the Materials and Methods). We then applied corrections for multiple testing and ranking, setting 0.1 as the false discovery rate (FDR).
The last step was the association of the serving motifs with known transcription factor binding sequences (TFBSs), where possible, to obtain a list of putative TF–target gene interactions. To this end we used the TRANSFAC18 database and the list of consensus motifs reported in ref. 13.
Fixing 0.1 as the FDR level, we obtained a catalogue of 2031 oligos that could be associated to known TFBSs for a total of 115 different TFs. These 2031 oligos targeted a total of 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 159 genes (20972 protein-coding and 187 miRNAs), and almost every gene in the Ensembl19 database was present at least once in our network. In parallel to that, our motif discovery procedure further identified 20216 significant motifs but for which we were not able to make any strong association with known TFBSs consensus.
159 genes (20972 protein-coding and 187 miRNAs), and almost every gene in the Ensembl19 database was present at least once in our network. In parallel to that, our motif discovery procedure further identified 20216 significant motifs but for which we were not able to make any strong association with known TFBSs consensus.
The dataset of associations between motifs and genes represents our transcriptional regulatory network and was the starting point for the circuits identification (see the ESI, supplementary files S3 and S4†). A relevant role in the following will be played by the subnetwork describing the transcriptional regulation of miRNAs. This subnetwork involves 110 TFs (out of 115 of the whole network) targeting a total of 187 miRNAs (see the ESI, supplementary file S4†).
Construction of a human post-transcriptional regulatory network
We used a very similar approach for the construction of the post-transcriptional regulatory network and used a dataset of 3′-UTRs for all the protein-coding genes in the human and mouse genomes. We ended up with a catalogue of 3989 short oligos (in this case 7-mers) over-represented and conserved in humans and mice after corrections for multiple testing and ranking, again setting 0.1 as the FDR threshold in our motifs discovery pipeline. Although the ab initio unbiased procedure that we used could discover different kinds of post-transcriptional regulatory motif,17 we kept only those motifs that could be associated with “seeds” of our known mature miRNAs (193 in total). 182 out of 3989 motifs turned out to match with at least one seed present in 140 out of 193 mature miRNAs (in some cases the motif could be associated to more than one miRNA). These motifs targeted a total of 17266 protein-coding genes, which represented our post-transcriptional regulatory networks reported in the ESI, supplementary file S5.†
Construction of the human mixed feed-forward loops catalogue
Once equipped with these two regulatory networks, we could, in principle, integrate their complementary information in various different ways. Here, we concentrated on the class of mixed FFLs discussed for instance in refs. 20–22, because biologically important and relatively simple to relate to experimental evidences and validations. We integrated the two networks, looking for all possible cases in which a master TF regulates a miRNA, and together with it, a set of protein-coding joint targets (JT). Notice that, as mentioned above, for each TF we associated all the motifs compatible with its binding site and its variants as they are reported in the TRANSFAC18 and in the ref. 13 collections. In this way, the intrinsic variability of regulatory binding sites, apparently neglected by our method, since we used fixed motifs, was restored in the final results.We were able to obtain a list of 5030 different “single target circuits”, each of them defined by a single TF as master regulator, a single mature miRNA and a single protein-coding joint target. We then grouped together all the single target circuits sharing the same pair of TF and miRNA and obtained as final result, 638 “merged” circuits, each composed by a known TF acting as master regulator, a mature miRNA and a list of protein-coding joint targets (see Fig. 1a). These 638 circuits involved a total of 2625 joint target genes, 101 transcription factors and 133 miRNAs. The number of joint targets in these circuits ranged from 1–38 and 74% of the circuits targeted up to 10 genes.
The raw data relative to these circuits can be found in the ESI, supplementary file S6.†
Besides the motifs used to build the above described circuits, we have several other cis-regulatory upstream motifs in our transcriptional networks that could not be related to a known TFBS. These motifs can be considered as new, putative, regulatory sequences16 and, even if we are not able to associate a precise TF (or any other kind of regulatory mechanism) to them, we decided to extend the above construction to these sequences as well. In these cases it would be too difficult to reconstruct the variability of the binding site for the corresponding putative unknown TF, so we decided to construct only the FFL in which the exact same unidentified and fixed motif was present in the upstream region of both the target protein-coding gene and the co-regulating miRNA and, as above, closed the loop only if the target gene was also a target of the considered miRNA.
In this way, we obtained 4035 different circuits, which included various motifs with different sizes on the promoter regions: 170, 6 nts long; 128, 7 nts long; 440, 8 nts long; 3297, 9 nts long. The number of joint targets in these circuits, after merging on the same cis-regulatory motifs, ranged from 1–5 and 79% of the circuits targeted one single gene.
All the raw data concerning these fixed-motif circuits can be found in the ESI, supplementary file S7.†
Circuits assessment I: functional analysis
As a first way to select biologically relevant FFLs among our results, we analyzed each one of the 638 merged circuits looking for an enrichment in Gene Ontology categories in the set of their joint targets. To assess this enrichment, we used the standard exact Fisher test with a p-value threshold p < 10−4. Previous experience on similar enrichment tests16,23 shows that this is a rather robust way to keep into account multiple testing of GO categories, which, being highly correlated, cannot be treated with a standard Bonferroni correction. Details regarding this analysis are available in Materials and Methods.As a final result of this analysis, we end with a list of 32 merged mixed feed-forward loops (corresponding to 380 single-target FFLs). These circuits involve a total of 344 joint target protein-coding genes, 24 TFs and 25 mature miRNAs. We report in Table 1 a selected list of such loops with a subset of the most representative Gene Ontology enriched annotations; the complete list of results is available in the ESI, supplementary file S8.†
| FFL id | JTs | Fisher test p-value | Gene Ontology characterization |
|---|---|---|---|
| AP-4|hsa-miR-133b | ADORA1 AP1GBP1 | 7.42e-5 | endocytosis (P) |
| AREB6|hsa-miR-126 | STRBP HERPUD1 CARD14 TRIM4 NP_995324.1 | 4.01e-6 | cellular developmental process (P) |
| EGFL7 PIK3R1 WFDC12 CDKN2A KLF10 C17orf70 | 3.63e-5 | regulation of osteoclast differentiation (P) | |
| RORB FBXL2 PPP3CB | 6.20e-5 | leukocyte differentiation (P) | |
| AREB6|hsa-miR-375 | PCSK6 LRP5 HABP2 USP6 GUF1 CNN3 PTPN4 | 1.94e-5 | anterior/posterior pattern formation (P) |
| XR_017284.1 ATPAF1 LCN1L1 NLGN3 LRFN1 AQP4 | 7.86e-5 | regionalization (P) | |
| TCF2 | |||
| C-REL|hsa-miR-126 | ARHGAP22 DSCR1 EGFR PIK3R2 Q96N05_HUMAN | 2.64e-6 | regulation of cell migration (P) |
| TOX2 PIK3R1 PARP16 ADAMTS9 EGFL7 | 2.97e-6 | phosphoinositide 3-kinase regulator activity (F) | |
| 4.00e-6 | regulation of cell motility (P) | ||
| 4.74e-6 | regulation of locomotion(P) | ||
| C-REL|hsa-miR-199a | ENO3 DDR1 SP2 CCNL1 PALLD | 9.10e-5 | transmembrane receptor protein tyrosine kinase activity(F) |
| ELF-1|hsa-miR-342 | C22orf15 ADAMTS5 CCDC32 IBRDC2 C5orf24 UBE4B CCR2 RPE PHB Q6PK04_HUMAN | 2.97e-6 | protein ubiquitination during ubiquitin-dependent protein catabolic process (P) |
| ER|hsa-miR-135b | GBE1 HCN2 CD99L2 TTC21A BSN RNASE11 NP_787078.1 PRLR | 4.11e-5 | cellular protein complex assembly(P) |
| ANGPT2 Q49AQ9_HUMAN | |||
| ZNF69 FAM129A FMOD IL11 ISCA1 PR285_HUMAN | |||
| CITED1 TGM2 MUSK DEFB123 MFSD3 C17orf28 | |||
| NP_057628.1 LZTS2 | |||
| HMGIY|hsa-miR-152 | EDG1 Q86V52_HUMAN DMRTA2 SLC25A32 FGF1 ITGA5 MEOX2 EPAS1 ZNF33A ADAM17 MAPK6 RNF182 | 6.48e-5 | angiogenesis (P) |
| ICSBP|hsa-miR-223 | ADM GAST PRL GTDC1 FOXO3A | 1.40e-6 | hormone activity (F) reproductive process (P) multicellular organism reproduction (P) |
| 2.18e-5 | |||
| 7.49e-5 | |||
| IRF1|hsa-miR-126 | EGFR EGFL7 GOLPH3 BDH2 ZADH2 | 8.01e-5 | regulation of cell migration (P) |
| IRF-7|hsa-miR-26a | VAX1 GALNT10 CA3 EIF2S1 NDUFA4 | 8.01e-5 | regulation of cell migration (P) |
| ARP19_HUMAN FBXO42 RPIA FBXL19 ALS2CR2 | 6.25e-5 | cellular response to stress (P) | |
| XR_017723.1 GSK3B DBR1 TTC13 NT5DC1 | |||
| MYC|hsa-miR-17-5p | BICC1 STK33 VSX1 EDD1 SLC24A4 NFAT5 E2F1 | 9.40e-0 | cellular metabolic process (P) primary metabolic process (P) |
| C21orf25 C9orf117 MYNN MAPK1 | 9.56e-5 | ||
| MYOD|hsa-miR-140 | ANK2 TSSK2 EIF2AK1 HMX2 THY1 ALAS2 UROC1 | 7.20e-6 | hemoglobin metabolic process (P) organ development (P) |
| CDKL4 PPARA CYBB PPL CDS2 ZIC3 | 6.61e-5 | ||
| SRY|hsa-miR-26a | FANCA GSK3B RPIA Q6ZQV3_HUMAN ALS2CR2 | 2.68e-5 | protein export from nucleus (P) |
| KIF1C RG9MTD2 CDS1 BAG4 PPP2R3C | 5.64e-5 | anti-apoptosis (P) |
Circuits assessment II: comparison with existing computational databases
To further assess the relevance of the circuits that we identified, we developed an annotation scheme based on the existence of additional computational evidences for each circuit link. To this end we used ECRbase24 and the data collected in ref. 25 for the transcriptional links, and the miRBase,26 PicTar11 and TargetScan9 databases for the post-transcriptional ones. Let us see in more detail how we used these sources of information:• The Evolutionary Conserved Regions database (ECRbase24) is a collection of evolutionary conserved regions, promoters and TFBSs in vertebrate genomes, based on genome-wide alignments created mainly with the Blastz program. Even if both our pipeline and ECRbase are based on evolutionary conservation, this ingredient is implemented in a very different way in the two approaches. ECRbase looks for conserved blocks identified via whole-genome alignments, while we implemented evolutionary conservation using an alignment-free approach. In this way, we were able to validate 216 TF–target gene links and 98 TF–miRNA links.
•Ref. 25 is a computational study of miRNA biogenesis. The regulatory interactions reported in ref. 25 are of particular interest for our assessment procedure since their pipeline is very different from ours. With this tool, we were able to validate 64 TF–miRNA links. It is interesting to notice that these 64 miRNAs were controlled by only nine transcription factors (the important role of these “hub” TFs was already noticed in ref. 25)
• The miRBase,26 PicTar11 and TargetScan9 databases are by now an accepted standard in the miRNA literature. They are based on strategies that are definitely different from our pipeline and are somehow complementary in their approaches. In this way, we were able to validate the miRNA–target gene link for 607 circuits (503 by miRBase, 343 by PicTar and 560 by TargetScan).
The results of these comparisons are summarized in Table 2a, while Table 3 reports the top ten TFs ranked by out-degree and the top ten miRNAs scored by in-degree.
| (a) | General view: | |
|---|---|---|
| Number of annotated links | Number of circuits | |
| 3 | 75 | |
| 2 | 207 | |
| 1 | 334 | |
| Detailed view: | ||
|---|---|---|
| Link type | Number of circuits | |
| TF -> miR: | 150 | |
| ECRbase: 98 | ||
| PMID 17447837: 64 | ||
| TF -> JT: | 216 | |
| ECRbase: 216 | ||
| miR -> JT: | 607 | |
| miRBase: 503 | ||
| PicTar: 343 | ||
| TargetScan: 560 | ||
| (b) | FFL id | JTs |
|---|---|---|
| AML1|has-miR-223 | RHOB DNAJB13 NDUFA3 TBC1D17 NP_001007596.1 IGSF21 SPTLC2 WNT2B RIPK3 ELF5 SLC2A11 C13orf31 FOXO3A | |
| LEF1|hsa-miR-138 | MYO3A NP_775790.1 RNMTL1 ZNF704 GPR124 NOTUM KRT83 FGF6 ITK | |
| MAZ|hsa-miR-34a | CA9 AKTIP SLC6A3 | |
| MEF-2|has-miR-133a | BRUNOL4 PLCL2 | |
| SMAD-3|hsa-miR-200b | MAP3K3 MAGED4 MAGED4B BAZ2A EBAG9 ZNF323 SCN5A WBP1 | |
| SOX5|hsa-miR-302d,c,c*,b,b* | TSPAN6 E2F2 WWC3 SIDT1 NFX1 C20orf7 GSPT1 ACO1 CHD6 GLT25D1 C19orf40 CLEC10A TNS3 PI15 ZNF291 NP_060887.1 ATP6V0D2 HTR3B LATS2 MAT1A FAM128B CDCP2 GNPDA2 SRGAP2 MON1A C10orf28 HNRPUL2 PBK NP_001034885.1 ZFP42 C9orf31 LRRIQ2 FAM22A TMCO2 HLA-DOA C4A C4B C6orf15 | |
| YY1|hsa-miR-101 | Q9HCM6_HUMAN NACA3P PRKD3 PFDN6 RAB15 ARID1A LRRC4 RAB5A FGD6 ARHGAP1 C17orf39 RBM25 NP_060164.3 STC1 FAM114A1 RNF213 Q96NB8_HUMAN |
| TF | Out-degree | miRNA | In-degree |
|---|---|---|---|
| MEIS1 | 31 | hsa-mir-148b | 15 |
| ER | 30 | hsa-mir-203 | 14 |
| SRY | 29 | hsa-mir-181d | 13 |
| HNF-1 | 27 | hsa-mir-99a | 12 |
| SOX-5 | 27 | hsa-mir-125b-2 | 12 |
| LEF1 | 23 | hsa-mir-423 | 11 |
| AREB6 | 22 | hsa-mir-129-2 | 11 |
| NCX | 18 | hsa-mir-149 | 11 |
| SRF | 18 | hsa-mir-214 | 11 |
| C-REL | 17 | hsa-mir-296 | 11 |
In Table 2b we report a selection of a few circuits which turned out to be validated by the above tests. In the ESI, supplementary file S8,† one can find the complete list of results.
Circuits assessment III: looking for cancer related FFLs
In these last few years it has become increasingly clear that miRNAs play a central role in cancer development. About half of the human miRNAs are located in cancer-related chromosomal regions and miRNA expression profiling correlates with various cancers and it is used to improve cancer diagnosis. This supports the definition of a subset of miRNAs as “oncomiRs”.27We filtered our results looking for circuits containing at least one cancer-related miRNA or target gene. To identify cancer related genes, we used the list of oncomiRs reported in ref. 27 and 28, while for the protein-coding target genes we compiled a list of genes showing mutations in cancer based on the Cancer Gene Census catalogue.
In particular we found 24 circuits in which at least two cancer-related genes (e.g. an oncomiR and a target or a TF and an oncomiR) were present (see Table 4). The full list of cancer-related circuits is available in the ESI, supplementary files S9 and S10.†
| FFL id | TF | miRNA | JTs |
|---|---|---|---|
| AP-1|hsa-miR-142-3p | hsa-miR-142-3p | DDIT3 | |
| ATF-1|hsa-miR-199a* | hsa-miR-199a* | MTCP1 | |
| ATF6|hsa-miR-199a* | hsa-miR-199a* | MTCP1 | |
| ER|hsa-miR-375 | TPR, USP6 | ||
| HIF-1|hsa-miR-199a* | hsa-miR-199a* | MTCP1 | |
| HNF-3|hsa-let-7a | hsa-let-7a | CCND2 | |
| HNF-3|hsa-let-7f | hsa-let-7f | CCND2 | |
| HNF-3|hsa-miR-30a-5p | MYH11, BCL9 | ||
| HNF-3|hsa-miR-30c | MYH11, BCL9 | ||
| HSF2|hsa-let-7a | hsa-let-7a | MYCN | |
| HSF2|hsa-let-7f | hsa-let-7f | MYCN | |
| HSF2|hsa-miR-199a* | hsa-miR-199a* | MYCN | |
| IRF|hsa-miR-125b | hsa-miR-125b | BCL2 | |
| IY|hsa-miR-296 | RPL22, BCL2 | ||
| MYC|hsa-miR-17-5p | MYC | hsa-miR-17-5p | |
| MYC|hsa-miR-19a | MYC | hsa-miR-19a | |
| MYC|hsa-miR-20a | MYC | hsa-miR-20a | |
| NF-Y|hsa-miR-223 | APC, ATF1 | ||
| OCTAMER|hsa-miR-125b | hsa-miR-125b | IRF4 | |
| PAX-4|hsa-miR-125b | hsa-miR-125b | IRF4 | |
| SOX-5|hsa-miR-125b | hsa-miR-125b | SS18 | |
| SOX-5|hsa-miR-29a | EXT1,COL1A1 | ||
| SRY|hsa-miR-221 | hsa-miR-221 | CCND2 | |
| SRY|hsa-miR-412 | BRAF, ATIC |
| CAGACAATG|hsa-miR-125b | hsa-miR-125b | IRF4 | |
| GGACTGCAA|hsa-miR-200c | hsa-miR-200c | MTCP1 | |
| GCCAACTGA|hsa-miR-199a* | hsa-miR-199a* | MTCP1 | |
| GCCCCCC|hsa-miR-200a | hsa-miR-200a | TFRC | |
| ACTTCACCC|hsa-miR-125b | hsa-miR-125b | BRD4 | |
| CGGGAAAAG|hsa-miR-125b | hsa-miR-125b | BRD4 | |
| GGCAATTTA|hsa-miR-19a | hsa-miR-19a | CCND1 | |
| AGAACTAAT|hsa-miR-19a | hsa-miR-19a | CCND1 | |
| CAGGTTGCA|hsa-miR-200c | hsa-miR-200c | MTCP1 | |
| AATTAGTTC|hsa-miR-19a | hsa-miR-19a | CCND1 | |
| ATCATTTTA|hsa-miR-125b | hsa-miR-125b | IRF4 | |
| AACCAGACA|hsa-let-7e | hsa-let-7e | SDHC | |
| GGATCTTAA|hsa-let-7a | hsa-let-7a | CCND2 |
Discussion
Potential function of mixed feed-forward circuits
Depending on the type of transcriptional regulation (excitatory or inhibitory) exerted by the master TF on the miRNA and on the targets, the FFLs that we study in this paper may be classified (following ref. 20) as coherent, if the master TF and the miRNA act in a coherent way on the target, or incoherent in the opposite case. A similar classification can be found in ref. 21 where the two classes of FFL were named as Type II or Type I, respectively (see Fig. 3, in which we chose to follow the same notations as ref. 21). Due to the computational procedure that we adopted to identify the FFLs, based on sequence analysis only, we were not able to recognize if the action of the master TF was excitatory or inhibitory, and thus if the FFL that we obtained was of Type I or Type II. Accordingly, in Fig. 1 and 4 we avoided identifying the links that connect the master TF to its targets as excitatory or inhibitory and used a different notation. Obviously the two types of circuits may lead to very different behaviours.20,21 Type II (coherent) circuits lead to a reinforcement of the transcriptional regulation at the post-transcriptional level and might be important to eliminate the already transcribed mRNAs when the transcription of a target gene is switched off. Type I can be used to stabilize the steady state production of a protein by dumping transcriptional fluctuations. In a simple TF–target interaction, any fluctuation of master TF could induce a non-linear increase in the amount of its target products. The presence, among the targets, of a miRNA that down-regulates the other targets might represent a simple and effective way to control these fluctuations. Another interesting possibility (discussed for instance in ref. 22) occurs if a temporal gap exists between the activation of the target gene and the miRNA repressor. This could be the case, for instance, if the binding sequences of the master TF in the two promoters have different affinities, or if there is a delay in the miRNA maturation process. In this case, the Type I circuit could be used to express the target protein within a well defined time window. In this respect, it is interesting to observe that one of the most studied mixed FFLs is Type I-like: here the role of the master TF is played by c-Myc, which induces the expression of miR-17-5p and miR-20a, and also of the joint target, E2F1, which, in turn, is repressed by the same miRNAs.29 Needless to say, these elementary FFLs, when embedded in more complex circuits, can lead to more sophisticated behaviours (see for instance the discussion in refs. 21, 22, and in particular, 30). We shall see below an example of this type of construction.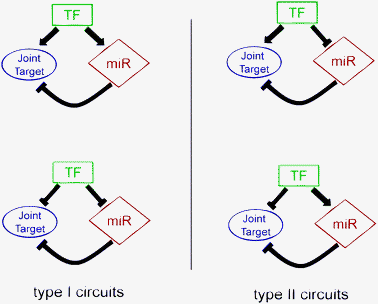 | ||
Fig. 3
Graphical representation of Type I and Type II circuits.
TF is the master transcription factor, miR represents the microRNA involved in the circuit and Joint Target is the joint target gene. Inside each circuit, → indicates transcription activation, whilst  indicates transcription or post-transcriptional repression. In representing Type I and Type II circuits, we followed the nomenclature used in ref. 21. indicates transcription or post-transcriptional repression. In representing Type I and Type II circuits, we followed the nomenclature used in ref. 21. | ||
 | ||
Fig. 4
Graphical representation of the c-Myc|E2F1|hsa-miR-20a circuit, with its extension to E2F2. The c-Myc|E2F1|hsa-miR-20a is the only feed-forward circuit already validated experimentally, as stated in the literature. Its components are embedded in a more sophisticated network, in particular, when mining our database we recognized the interplay with E2F2. E2F2 is down-regulated by hsa-miR-20a at the post-transcriptional level, and it is a direct transcriptional target of E2F1 itself. –• indicates transcriptional activation/repression, whilst 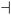 post-transcriptional repression. Mature microRNA ids are written according to the standard nomenclature of miRBase,47 for the TF and JT protein-coding genes, we used the standard HGNC ids. post-transcriptional repression. Mature microRNA ids are written according to the standard nomenclature of miRBase,47 for the TF and JT protein-coding genes, we used the standard HGNC ids. | ||
Analysis of the mixed feed-forward circuits in terms of network motifs
Elementary regulatory circuits (the so called “network motifs”) were shown to be over-represented in transcriptional networks.1,2 This very interesting observation led a few authors to conjecture that functionally important network motifs should always be over-represented and to use this criterion as a tool to identify them. This assumption is somewhat controversial and is currently challenged by some other authors.30,31 Our data represent a perfect setting to test this over-representation conjecture.In order to quantify the over-representation we performed a set of randomization tests. The results are reported in the ESI, Fig. S1 and details are available in the supporting text.† Briefly, we carried out three types of randomizations:
•Random reshuffling of miRNA promoters and seeds. We rebuilt the entire database of mixed feed-forward circuits (i.e. the entire pipeline designed in Fig. 2), but using randomly shuffled versions of the miRNA promoters and random sets of 7-mers as miRNA seeds. The principle of this procedure was to perform the same analysis of correlation between transcriptional and post-transcriptional regulatory networks, but considering the connection between the two regulatory layers a randomized version of the real known miRNAs, in terms of their in-degree (the miRNA promoter) and out-degree (the miRNA seed).
•Edge switching. We applied a randomization strategy on the real transcriptional and post-transcriptional regulatory network obtained with our pipeline, similar to the one used in ref. 32. The edge switching strategy is able to randomize the real network, preserving the individual degree of each node in the network.
•Complete node replacement. We applied a second, more drastic, randomization strategy on the real transcriptional and post-transcriptional regulatory network obtained with our pipeline, in this case with no constraint on the randomization procedure.32
The results reported in the ESI, Fig. S1 (panel A)† show that for the three randomization strategies, the number of circuits recognized in the real regulatory network is statistically higher than the one found in the random versions (random reshuffling of miRNA promoters and seeds: Z = 3.5; edge switching: Z = 8.3; complete node replacement: Z = 8.9). However, it is important to notice that the actual number of mixed feed-forward loops identified in the randomized versions of the regulatory network is always rather large. Thus, even if the over-representation is statistically significant, it would be very inefficient (i.e. it would lead to a large number of false positive identifications) to use it as the only tool to identify functionally relevant mixed FFLs. Interestingly, our results are in good agreement with a similar analysis reported in ref. 32. This is particularly significant since our approach and that of ref. 32 for the identification of TF and miRNA regulatory interactions are totally different. In ref. 32, the authors presented the first genome-scale Caenorhabditis elegansmiRNA regulatory network that contains experimentally mapped transcriptional TF → miRNA interactions, as well as computationally predicted post-transcriptional miRNA → TF connections. They then looked at the properties of mixed feedback loops, comparing their findings with network randomizations: the average number of loops in randomized networks was always about half the number of real loops they identified.
Analysis of the gene ontology enrichment results
In the ESI, supplementary file S8† we report a detailed view of the GO enrichment results at the level of joint target sets and of single gene analysis. Besides the intrinsic interest of several of these annotations, it is interesting to observe that the set of GO categories enriched in our circuits somehow shows a general trend.We observe over-representation of GO terms describing several aspects of organism development such as differentiation, proliferation, apoptosis, programmed cell death and cellular migration. These results are in good agreement with the predictions about the biological meanings of the FFLs reported in ref. 20. Specifically, our data provide evidence for functions of several circuits in the cardiac and skeletal, neural and hematopoietic cell lineages.
A similar pattern emerges if we look at the single-gene enrichment analysis. Multi-cellular organisms development, cell differentiation, cell proliferation and apoptosis directly annotate, respectively, 108, 56 and 48 target genes included in the annotated circuits.
Finally it is interesting to notice that several circuits seem to be involved, according to the GO analysis, in basal mechanisms of post-translational regulation such as protein amino acid phosphorylation and in the ubiquitin cycle (with as much as 57 annotated genes).
All these observations agree with the idea that the mixed (T-PT) motifs and in particular the feed-forward loops that we discuss in this paper play a fundamental role in all those processes (like tissue development and cell differentiation), which are characterized by a high degree of complexity and require the simultaneous fine tuning of several different players. Strikingly, it is worth noting that this result was obtained here with a completely ab initio bioinformatics sequence analysis strategy.
Comparison of our results with the database33 of chip-pet c-Myc targets
Besides the above tests, in order to evaluate the reliability of our transcriptional regulatory network we compared our results with a set of c-Myc targets reported in ref. 33. This database contains a genome-wide, unbiased characterization of direct Myc binding targets in a model of human B lymphoid tumor using chromatin immunoprecipitation coupled with pair-end ditag sequencing analysis (ChIP–PET), and reports a total of 2088 targets.The choice of the c-Myc TF is not random. Besides being a very interesting TF, it is present in several of our FFLs and as such, it plays a central role in the transcriptional side of our regulatory networks. In particular, the first example that we shall discuss below contains c-Myc as master TF.
Looking at the intersection between the 2088 targets of ref. 33, and the 1979 predicted by our analysis, we found 253 targets in common, corresponding to a p-value of 1.1 × 10−6 (Fisher test). This result is even more impressive if compared with the number of intersections of the Zeller dataset with the list of c-Myc targets reported in the TRANSFAC database.18 Out of 235 TRANSFAC targets, only 27 were present in Zeller’s dataset, corresponding to a p value of 0.21.
As a further test, we performed the same comparison for the transcriptional network obtained choosing as promoter the (−500/+100) region around the TSS as promoter. In this case we found 1612 putative c-Myc targets, in which 203 were in common with the dataset of ref. 33, corresponding to a slightly higher p-value p = 8.4 × 10−5.
Dependence of our results on the choice of the promoter’s region
In the construction of the transcriptional regulatory network, we chose to consider the interval (−900/+100) around the TSS for the promoter regions. In order to test the dependence of our results on this choice, we performed the same analysis choosing as promoter region the interval (−500/+100) around the TSS. This is somehow an extreme choice and represents a very stringent test of the robustness of our network. Looking at the mixed FFL, we found a total of 6682 “single target” FFLs (to be compared with the 5030 of the (−900/+100) case), of which 1769 were in common with the (−900/+100) run. Remarkably enough, all the circuits that we discussed in the text (and more generally most of the circuits surviving our assessment tests) turned out to be present in both releases. The complete list of circuits obtained in the (−500/+100) run is reported in the ESI, supplementary file S11.†In order to complete this analysis we also performed the randomization tests and the comparison with the c-Myc database discussed above for the (−500/+100) FFLs. We found comparable results with those obtained in the (−900/+100) case: the number of circuits of the real regulatory network turned out to be statistically higher than the ones found in the random simulations. In particular, for the first two tests, we found an improvement of the Z values, while for the third one, we found slightly worse values of Z. All these results are reported in the second panel of ESI, Fig. S1.† Also for the c-Myc analysis, we found results comparable with those obtained in the (−900/+100) case, with a slight worsening of the p-value of the intersection. More precisely the c-Myc targets in the (−500/+100) transcriptional network turned out to be 1612, of which 203 were in common with the Zeller c-Myc dataset, corresponding to a p-value of 8 × 10−5.
We consider all these findings as an indication of the overall robustness of our results.
Comparison with related works
Mixed T-PT regulatory circuits have been recently studied in two interesting papers.21,22 It is worthwhile to compare their results with our analysis, which is similar in spirit, but slightly more complete in the final results.In ref. 21, the authors studied various types of feed-forward and feedback loops involving miRNAs, their target genes and transcriptional regulators as a tool to explain the (anti-)correlations between the expression levels of miRNAs and of their target genes. This study relied on a predicted miRNA-mediated network and did not use the transcriptional regulatory network of miRNAs that was unavailable at that time. Hence, to the best of our knowledge, no actual explicit loops were identified (see also ref. 32).
In ref. 22, the authors used pre-compiled TF- and miRNA-mediated networks, and studied global and local properties of the two networks separately. Additionally, they provided a catalogue of network designs in the co-regulated network, including feed-forward loops. Both the TF- and the miRNA-mediated networks in ref. 22 were obtained from sequence-based identification of regulatory features in promoters and 3′-UTRs. This makes the study in ref. 22 more comparable to ours than that in ref. 21. For this reason, we decided to perform a more detailed comparison with our results. Unfortunately, this study did not report explicitly the circuits (including joint target genes) but only provided a list of 16 pairs of co-regulating TFs and miRNAs involved in feed-forward loop. We obtained these pairs using as input the PSSMs (position specific scoring matrices) and microRNAs listed in the supplementary Table S2 of ref. 22 and then mapping the PSSMs to the corresponding transcription factors. We compared this list with our results. It turns out that none of these predictions are contained in our dataset. A detailed comparison of the two pipelines shows that there are a few important reasons behind this disagreement:
• Different annotation for mature miRNA identifiers due to the older miRBase release used in ref. 22 (8.2 vs. 9.2): e.g. pairs involve miR-10 in ref. 22, while miRBase 9.2 reports miR-10a and miR-10b; similarly for miR-142 and miR-142-5p,-3p.
• Different assignment of mature miRNAs to pre-miRNAs: e.g. in ref. 22 the authors assign miR-7 to mir-7-1, while miRBase 9.2 assigns miR-7 to mir-7-3.
• Different organization of pre-miRNAs in transcriptional units: in ref. 22, miRNAs are clustered in precursors according to physical proximity, while we relied on human/mouse conserved transcriptional units reported in ref. 7.
• Different definition of miRNA promoters: ref. 22 uses 10 kb upstream of the 5′-most pre-miRNA for each cluster, while we used 1 kb upstream of the 5′-most pre-miRNA for each transcriptional unit.
• Different solutions for predicted transcription factor binding sites: ref. 22 uses PSSMs from TRANSFAC release 8.3, and using pre-compiled lists of interactions available in the UCSC hg17 genome assembly, while we mainly mapped ab initio conserved and over-represented motifs to transcription factor binding sites.
• Different solutions for predicted mature miRNA binding sites: ref. 22 uses TargetScan (release 3.0) and PicTar (picTarMiRNA4Way track in the UCSC genome browser) while we mapped conserved and over-represented motifs in 3′-UTRs to mature miRNAs by means of miRBase release 9.2.
As a final comment on this comparison, let us stress that probably one of the major novelties of the present analysis with respect to existing works is the particular attention we paid to the definition of miRNA promoters and in the search of their putative binding sequences. Accordingly, besides the final list of FFLs, we consider as one of our most interesting results the subset of our transcriptional regulatory network involving miRNAs as targets. This subnetwork includes a total of 110 TFs targeting 187 miRNAs and is reported in the ESI, supplementary file S4.†
Description of a few interesting circuits
As a final part of this section, let us discuss in more detail the biological relevance of a few of our results. We have chosen to discuss a few examples for each of the three assessment pipelines.We first present a case in which our pipeline is able to predict circuits already known in the literature and for which all the links are experimentally validated: this is the case of the circuits involving c-Myc as master TF, and hsa-miR-17-5p and hsa-miR-20a as post-transcriptional regulators. In particular, one of the predicted joint target genes results in being the E2F1 gene, in this way closing the circuit exactly on the target gene experimentally assessed and used as a major example in the discussion of ref. 20.
In the remaining examples some (or all) of the genes embedded in the circuits were already annotated to related functions in the literature but their combination in a closed FFL was not noticed. We consider these cases as further successful validations of our approach.
•The c-Myc, hsa-miR-20a/miR-17-5p circuit
In this circuit, c-Myc is the master TF and hsa-miR-20a the post-transcriptional regulator. This circuit contains eleven joint targets, among which is E2F1. The complete list of joint targets is reported in Table 1. The FFL involving E2F1 is well known in the literature. It was discussed for the first time in ref. 29 and is expected to play a role in the control of cell proliferation, growth and apoptosis. With our analysis, we could identify several other genes sharing the same regulatory pattern of E2F1 and we expect that at least some of them could be involved in the same biological processes. In this respect, it is interesting to find among the other targets NFAT5, which is known to play a critical role in heart, vasculature, muscle and nervous tissue development. Similarly, it seems interesting to find MAPK1, which, like E2F1, is an anti-apoptotic gene. These observations could suggest a similar functional role also for the remaining joint targets.
This circuit also allows us to discuss how our data could be used to obtain more complex regulatory motifs. Combining different entries of our databases, it is easy to find a circuit involving, besides c-Myc, hsa-miR-20a and E2F1, also E2F2, which turns out to be simultaneously targeted by E2F1 and by hsa-miR-20a (see Fig. 3). This is a rather non-trivial result, since it is well known that different TFs of the E2F family tend to act together in a concerted way. We see in this example a simple network motif in which this cooperative action is present and is tightly regulated.
•The AREB6, hsa-miR-375 circuit
One of the most interesting entries of Table 1 is the feed-forward loop that involves the transcriptional repressor zinc-finger E-box binding homeobox 1 AREB6 (also known as ZEB1), hsa-miR-375 and a set of 14 joint target genes. Owing to the following observations, we surmise its function in embryonic development and the physiology of the pancreas. ZEB1 is a crucial inducer of the embryonic program ‘epithelial-mesenchymal transition’ (EMT) that facilitates tissue remodelling during embryonic development. miR-375 is essential for embryonic pancreatic islet development, as well as for endocrine pancreas function, where it was demonstrated to regulate the process of exocytosis of insulin during glucose-stimulated insulin release.34 Notably GO analysis globally annotates the set of target genes to patterning in embryonic development, which is consistent with the regulatory roles of ZEB-1 and miR-375. Moreover, the hypothesis of a function in insulin secretion is strengthened by the following observations:35 reports of strong evidence that EMT can provide cells for replacement therapy in diabetes; among the target genes, HNF1β (also known as TCF2) is responsible for MODY,36 a form of diabetes characterized by defective insulin secretion of pancreatic β-cells.
•The MEF-2, hsa-miR-133a circuit
This is one of the entries of Table 2. It contains only two joint targets: BRUNOL4 and PLP2, but the presence of BRUNOL4 turns out to be highly non-trivial. In fact, the myocyte enhancing factor-2 (MEF-2), hsa-miR-133a and the RNA-binding protein BRUNOL4 have been shown to altogether control cardiomyocyte hypertrophy. In this case, it is also possible to envisage a feedback effect, because cardiac repression of BRUONOL4 activity disrupts alternative splicing of MEF-2 and leads to cardiac hypertrophy.37 Finally, it is important to stress that the regulatory interaction between MEF-2 and hsa-mir-133a, which we predicted with our in silico analysis, was indeed observed experimentally in ref. 38.
•The C-REL, hsa-mir-199a circuit
Another interesting circuit relates C-REL, a member of the NFKB family, and miR-199a. MiR-199a has been identified as a miRNA signature in human ovarian cancer. miR-199a down-modulation in epithelial ovarian cells is reported in ref. 39 and, interestingly, miR-199a has lately been shown to affect NFKB activity in ovarian cancer cells.40 Among the joint targets for this circuit, let us mention: DDR1, a receptortyrosine kinase, whose expression is restricted to epithelial cells and significantly high in epithelial ovarian cells, and Sp2, which is a transcriptional repressor of the tumor suppressor gene CEACAM1 in epithelial cells.41
•The HSF2, hsa-let-7f circuit
Looking at the cancer-related list, one of the most interesting entries is the one which relates the transcription factor, HSF2, and the hsa-let-7f miRNA. The DNA-binding protein heat shock factor-2 (HSF2) and hsa-let-7f jointly regulate a number of target genes such as MYCN, ESPL1, PLSCR3, PDCD4, MTO1 and FMO2. Several observations point to an involvement of this circuit in cell cycle progression with relevant implications in cancer. HSF2’s role in cancer is being elucidated42 by the observation that it functions as a bookmarking factor, not only for heat shock responsive genes, but also for genes that are involved in the regulation of cell apoptosis and proliferation (such as Hsp90, Hsp27 and c-Fos). Among the target genes, the MYCN oncogene is crucial in neuronal development, and its amplification is currently the only molecular marker adopted in neuroblastoma clinical treatments. The MYC family oncogenes are known to deregulate cell cycle progression, apoptosis and genomic instability. In neuroblastoma cell lines, N-Myc can induce genomic instability by centrosome amplification. Interestingly, HSF2 and hsa-let-7f regulate the extra spindle poles like-1 (ESPL1) that mediates mitotic sister chromatid segregation. The programmed cell death-4 (PDCD4) is also linked to progression through the cell cycle by mediating MAPK kinase activity and JNK activity. The phospholipid scramblase-3 (PLSCR3) is a mitochondrial integrator of apoptotic signals. Interestingly, also the mitochondrial translation optimization-1 homolog (MTO1) and the flavin containing monooxygenase-2 (FMO2) promote local effects on mitochondria. Finally, MYCN has recently been reported as a direct target of miR-34a. Here we add that let-7f targets MYCN. Notably let-7f belongs to the let-7 family of oncomiRs and, in particular, let-7f has been found to be involved in cell aging.43
As a final remark, we would like to stress that interesting convergence of cooperative biological functions can also be observed in circuits in which we were not able to identify a putative master TF, and therefore were not processed with our assessment pipeline. As an example let us mention the UST gene (Ensembl id: ENSG00000111962), which is involved in heparan sulfate-dependent growth factor signaling during myogenesis and in ion buffering; UST linked to hsa-miR-1 (see the ESI, supplementary file S7†), which in turn promotes skeletal muscle proliferation and differentiation, and is involved in heart electrical conductions as well.44
Conclusions
The main purpose of this work was to systematically investigate connections between transcriptional and post-transcriptional network interactions in the human genome. To this end, we designed a bioinformatic pipeline, mainly based on sequence analysis of human and mouse genomes, which is able to construct, in particular, a catalogue of mixed feed-forward loops (FFLs) in which a master transcription factor regulates a miRNA and, together with it, a set of joint target protein-coding genes. These circuits were then prioritized based on various selection criteria. We also analyzed a few of them in detail looking for a possible biological role. The lists of FFLs selected in this way are the major results of our work, and our findings demonstrate in particular a connection between such loops and aspects of organisms’ development and differentiation. Moreover, one of the outcomes resulting from our study is the design of a putative TF regulatory network of human miRNA genes.As a concluding remark it is important to stress that we consider the present work only as a first step along this research line. For both technical and biological reasons, it is likely that we missed several regulatory circuits in our network. We discussed in detail the technical issues and the related problems. Let us comment here on one of the main biological issues, which should certainly be addressed in future works. One of our main assumptions is that we can associate a well defined promoter to a well defined gene. However several recent studies on the widespread presence of alternative splicing and transcription start sites (TSS) (see for instance ref. 45) show that this is probably a restrictive choice. Moreover, alternatively spliced isoforms of the same gene may have completely different functions and play different roles in the regulatory network. More generally the notion of “gene” by itself is experiencing a deep redefinition in the last few years.46 Notwithstanding this, the good agreement that we found with some existing experimental data suggests that our approach may represent a reliable step toward a better understanding of gene regulatory networks, and in particular, it could give some useful insight on the complex interplay of their transcriptional and post-transcriptional layers.
Materials and methods
miRNA transcriptional units
We obtained genomic coordinates of human and mouse pre-miRNA hairpins from the miRBase47miRNA sequence database (release 9.2). Consistently, human and mouse protein-coding genes and annotations were obtained from the Ensembl database,19 release 46, corresponding to the human genome assembly hg18 and to the mouse genome assembly mmu8. Mapping of pre-miRNAs to overlapping protein-coding genes was performed using the mirGen database (http://www.diana.pcbi.upenn.edu/miRGen/v3/), which provided us with a list of all the pre-miRNA hairpins that overlapped to annotated genes and gave the precise location of the pre-miRNA hairpin within the gene. In this study, from the Ensembl database we selected only protein-coding genes labelled as “KNOWN”, for both human and mouse. Pre-miRNAs were defined as genic if they were located within annotated exons, introns or flanking untranslated regions. miRNA hairpins were retained in our study only if they had an orthologous copy in mice. This selection was performed using the human-to-mouse orthology table compiled by ref. 7 and provided as their supplementary table S15.An important role in our analysis is played by the notion of “transcriptional units” (TU), which are clusters of miRNA hairpins located in nearby positions along the DNA, and supposed to be transcribed together in a single poly-miRNA precursor.7 Both cDNA and EST expression data7,48 support the idea that miRNAs belonging to the same TU are co-transcribed. For this reason, we shall treat them as a unique (miRNA) gene and associate the same promoter (the one corresponding to the transcriptional start site (TSS) of the transcriptional unit) to all the miRNAs belonging to the TU.
Taking together isolated miRNAs and TUs, we were able to identify a total of 130 miRNA precursors for the human genome and the corresponding 130 orthologues for the mouse genome. 68 out of 130 were non-genic and 62 were located within a KNOWN gene. A direct inspection showed that 53 of these genic pre-miRNAs shared the same orientation with the host gene, while the remaining nine had the opposite orientation. These 130 precursors corresponded to a total of 193 mature miRNAs. These mature miRNAs and their “seeds” represented the list of input motifs for the target search algorithms and the bases of our discussions.
The list of TUs, their most 5′-upstream members, their genomic coordinates, their locations relative to protein-coding genes and additional orthology annotations can be found in the ESI, supplementary file S1,† for humans and mice. We then provide the corresponding mature miRNAs used in this study in supplementary file S2,† for humans and mice.
Definition of promoter regions
For the analysis of promoter regions, we prepared two distinct datasets, one for protein-coding genes and one for miRNA genes. All the sequences and annotations used were extracted from the Ensembl database, version 46.316 promoters for human and 21814 for mouse protein-coding genes, respectively.
• If the pre-miRNA was non-genic, we selected the region ranging from nt − 900 upstream to nt + 100 downstream of the 5′-start of the pre-miRNA.
• If the pre-miRNA was genic, with the same orientation of the host gene, we used the promoter region selected for the host gene.
• If the pre-miRNA was genic, but with opposite orientation with respect to the host gene, we again selected the region ranging from nt − 900 upstream to nt + 100 downstream of the 5′-start of the pre-miRNA.
In all these cases, we then repeat-masked and exon-masked the sequences as we did for the protein-coding genes discussed above. Repeat-masking was performed with the default values provided by Ensembl.
Merging together protein-coding and miRNA promoters, we ended up with a collection of 21446 human and 21944 mouse regulatory sequences.
Definition of 3′-UTR regions
For the analysis of post-transcriptional regulation, we downloaded the complete 3′-UTR sequences for all protein-coding genes from the Ensembl database, version 46. Similarly to the promoters, we retained only those genes labeled as “KNOWN”. Then we selected only the longest transcript, again among those labeled as “KNOWN”. Since in the Ensembl database not all the genes have defined 3′-UTR regions, we ended up with only 17486 human and 15921 mouse genes. We then repeat-masked these sequences using the default values provided by Ensembl as masking parameters.
It is worth noticing that, differently from the promoter case, the 3′-UTR sequences have different sizes. The average length of human or mouse 3′-UTR regions was ∼1157 nts or ∼982 nts, respectively.
Oligos analysis
All the details relevant to the oligos analysis are described in the supporting text of the ESI.† The promoter and 3′-UTR sequences used as input, and the software described in the text are available upon request from the authors.TF–miR pairs and their joint target genes
By crossing the lists of putative TF and miRNA targets obtained above we constructed all possible feed-forward circuits composed by a transcription factor, which regulates a miRNA with which it co-regulates a set of target genes. In some cases in which a mature miRNA is transcribed from more than one genomic locus, all possible promoters were taken into account.Assessment of miRNA targets using existing databases
In silico predicted targets were obtained from the following three resources: TargetScan, PicTar and miRBase. These three algorithms predict and assign target genes to miRNAs essentially based on sequence multi-species conservation. TargetScan targets were obtained from miRGen Release 3 (http://www.diana.pcbi.upenn.edu/miRGen/v3/) where human miRNA family targets predicted by TargetScanS were downloaded from the TargetScan Release 4.2 download site (http://www.targetscan.org/) and miRNA family names were expanded to include all family members. We downloaded PicTar targets from the UCSC hg17 database where they were presented as the picTarMiRNA4Way track. miRBase predicted targets were downloaded from http://microRNA.sanger.ac.uk/targets/v4/. Since different resources use different genomic annotation sets, we maintained Ensembl as main namespace and mapped both Gene Symbol IDs and RefSeq IDs to Ensembl Gene IDs.Comparison with ECRbase
From ECRbase (http://ecrbase.dcode.org/) we downloaded the complete dataset of transcription factor binding sites (TFBSs) predictions in the CoreECR regions (al least 355 nts long with 77% indentity) from the tfbs_correEcrs.hg18mm8.v94.txt file.We mapped the predicted TFBSs stored in those databases onto our promoter regions according to genomic coordinates, for protein-coding and miRNA genes. To avoid mismatches due to different masking and/or misannotations, we assigned the binding of ECRbase TF to our gene only if the complete sequence contained in the ECRbase was present in our promoter sequence.
Gene Ontology analysis
We downloaded the Gene Ontology (GO) annotation DAGs from the GO website (http://www.geneontology.org) and gene product annotations from the Ensembl database, version 46. We always considered a gene annotated to a GO term if it was directly annotated to it or to any of its descendants in the GO graph. We implemented an exact Fisher’s test to assess whether a certain set of genes could be enriched in a certain GO category as done in our previous studies.16,23 The Fisher’s test gave us the probability p of obtaining an equal or greater number of genes annotated to the term in a set made of the same number of genes, but randomly selected. To account for multiple testing, in this work, only p-values <10−4 were reported.Identification of cancer related genes
OncomiRs were obtained from ref. 27 and 28. We obtained the complete working list of mutated genes causally implicated in cancer from the Cancer Gene Census catalogue (http://www.sanger.ac.uk/genetics/CGP/Census/). The list was annotated with information concerning chromosomal location, tumour types in which mutations were found, classes of mutation that contributed to oncogenesis and other genetic properties. We considered as cancer-related a circuit if it included at least one oncomiR or one gene listed in the Cancer Gene Census catalogue. The full lists of these circuits, provided with detailed properties on cancer-related genes, are available in the ESI, supplementary files S9 and S10.†Supporting information
All the supplementary files and raw data are available upon request from the authors.Acknowledgements
We thank Paolo Provero, Ferdinando Di Cunto, Francesca Orso, Paolo Macchi and Alessandro Quattrone for useful suggestions and discussions. We also thank Marco Consentino-Lagomarsino for discussions about network motifs. This work was partially supported by the Fund for Investments of Basic Research (FIRB) from the Italian Ministry of the University and Scientific Research, No. RBNE03B8KK-006.References
- R. Milo, S. Shen-Orr, S. Itzkovitz, N. Kashtan, D. Chklovskii and U. Alon, Network motifs: simple building blocks of complex networks, Science, 2002, 298, 824–827 CrossRef CAS.
- S. Shen-Orr, R. Milo, S. Mangan and U. Alon, Network motifs in the transcriptional regulation network of escherichia coli, Nat. Genet., 2002, 31, 64–68 CrossRef CAS.
- L. He and G. Hannon, MicroRNA: small rnas with a big role in gene regulation, Nat. Rev. Genet., 2004, 5, 522–531 CrossRef CAS.
- W. Filipowicz, S. Bhattacharyya and N. Sonenberg, Mechanisms of post-transcriptional regulation by micrornas: are the answers in sight?, Nat. Rev. Genet., 2008, 9, 102–114 CrossRef CAS.
- I. Alvarez-Garcia and E. Miska, MicroRNA function in animal development and human disease, Development, 2005, 132, 4653–4662 CrossRef CAS.
- G. Calin and C. Croce, MicroRNA-cancer connection: the beginning of a new tale, Cancer Res., 2006, 66, 7390–7394 CrossRef CAS.
- P. Landgraf, M. Rusu, R. Sheridan, A. Sewer and N. Iovino et al., A mammalian microRNA expression atlas based on small rna library sequencing, Cell, 2007, 129, 1401–1414 CrossRef CAS.
- E. Lai, Micro rnas are complementary to 3′ utr sequence motifs that mediate negative post-transcriptional regulation, Nat. Genet., 2002, 30, 363–364 CrossRef CAS.
- B. Lewis, I. Shih, M. Jones-Rhoades, D. Bartel and C. Burge, Prediction of mammalian microRNA targets, Cell, 2003, 115, 787–798 CrossRef CAS.
- C. Nielsen, N. Shomron, R. Sandberg, E. Hornstein, J. Kitzman and C. B. Burge, Determinants of targeting by endogenous and exogenous microrRNA and sirnas, RNA, 2007, 13, 1894–1910 CrossRef CAS.
- A. Krek, D. Grun, M. Poy, R. Wolf, L. Rosenberg, E. J. Epstein, P. MacMenamin, I. da Piedade, KC. Gunsalus, M. Stoffel and N. Rajewsky, Combinatorial microRNA target predictions, Nat. Genet., 2005, 37, 495–500 CrossRef CAS.
- B. Lewis, C. Burge and D. Bartel, Conserved seed pairing, often flanked by adenosine, indicates that thousands of human genes are microRNA targets, Cell, 2005, 120, 15–20 CrossRef CAS.
- X. Xie, J. Lu, E. Kulbokas, T. Golub, V. Mootha, K. Lindblad-Toh, E. S. Lander and M. Kellis, Systematic discovery of regulatory motifs in human promoters and 3′ utrs by comparison of several mammals, Nature, 2005, 434, 338–345 CrossRef CAS.
- C. Chan, O. Elemento and S. Tavazoie, Revealing posttranscriptional regulatory elements through network-level conservation, PLoS Comput. Biol., 2005, 1, e69 Search PubMed.
- L. Elnitski, V. X. Jin, P. J. Farnham and S. J. Jones, Locating mammalian transcription factor binding sites: A survey of computational and experimental techniques, Genome Res., 2006, 16, 1455–1464 CrossRef CAS.
- D. Corà, C. Herrmann, C. Dieterich, F. Di Cunto, P. Provero and M. Caselle, Ab initio identification of putative human transcription factor binding sites by comparative genomics, BMC Bioinformatics, 2005, 6, 110 CrossRef CAS.
- D. Corà, F. Di Cunto, M. Caselle and P. Provero, Identification of candidate regulatory sequences in mammalian 3′ utrs by statistical analysis of oligonucleotide distributions, BMC Bioinformatics, 2007, 8, 174 CrossRef.
- V. Matys, O. Kel-Margoulis, E. Fricke, I. Liebich, S. Land, A. Barre-Dirrie, I. Reuter, D. Chekmenev, M. Krull, K. Hornischer, N. Voss, P. Stegmaier, B. Lewicki-Potapov, H. Saxel, A. E. Kel and E. Wingender, Transfac and its module transcompel: transcriptional gene regulation in eukaryotes, Nucleic Acids Res., 2006, 34, D108–D110 CrossRef CAS.
- T. Hubbard, B. Aken, K. Beal, B. Ballester and M. Caccamo et al., Ensembl 2007, Nucleic Acids Res., 2007, 35, D610–D617 CrossRef CAS.
- E. Hornstein and N. Shomron, Canalization of development by micrornas, Nat. Genet., 2006, 38(6s), S20 CrossRef CAS.
- J. Tsang, J. Zhu and A. van Oudenaarden, MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals, Mol. Cell, 2007, 26, 753–67 CrossRef CAS.
- R. Shalgi, D. Lieber, M. Oren and Y. Pilpel, Global and local architecture of the mammalian microRNA-transcription factor regulatory network, PLoS Comput. Biol., 2007, 3, e131 Search PubMed.
- D. Corà, F. Di Cunto, P. Provero, L. Silengo and M. Caselle, Computational identification of transcription factor binding sites by functional analysis of set of genes sharing overrepresented uptream motifs, BMC Bioinformatics, 2004, 5, 57 CrossRef.
- G. Loots and I. Ovcharenko, Ecrbase: database of evolutionary conserved regions, promoters, and transcription factor binding sites in vertebrate genomes, Bioinformatics, 2006, 23, 122 CrossRef.
- J. Lee, Z. Li, R. Brower-Sinning and B. John, Regulatory circuit of human microRNA biogenesis, PLoS Comput. Biol., 2007, 3, e67 Search PubMed.
- B. John, A. Enright, A. Aravin, T. Tuschl, C. Sander and D. S. Marks, Human microRNA targets, PLoS Biol., 2004, 2, e363 CrossRef.
- A. Esquela-Kerscher and F. Slack, Oncomirs-micrornas with a role in cancer, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS.
- B. Zhang, X. Pan, G. Cobb and T. Anderson, Micrornas as oncogenes and tumor suppressors, Dev. Biol., 2007, 302, 1–12 CrossRef CAS.
- K. O’Donnell, E. Wentzel, K. Zeller, C. Dang and J. Mendell, c-myc-regulated micrornas modulate e2f1 expression, Nature, 2005, 435, 839–843 CrossRef CAS.
- A. Mazurie, S. Bottani and M. Vergassola, An evolutionary and functional assessment of regulatory network motifs, Genome Biol., 2005, 6, R35 CrossRef.
- A. Konagurthu and A. Lesk, On the origin of distribution patterns of motifs in biological networks, BMC Syst. Biol., 2008, 2, 73 CrossRef.
- N. Martinez, M. Ow, M. Barrasa, M. Hammell, R. Sequerra, L. Doucette-Stamm, F. P. Roth, V. R. Ambros and A. J. Walhou, A c. elegans genome-scale microRNA network contains composite feedback motifs with high flux capacity, Genes Dev., 2008, 22, 2535–2549 CrossRef CAS.
- K. Zeller, X. Zhao, C. Lee, K. Chiu, F. Yao, , J. T. Yustein, H. S. Ooi, Y. L. Orlov, A. Shahab, H. C. Yong, Y. Fu, Z. Weng, V. A. Kuznetsov, W. K. Sung, Y. Ruan, C. V. Dang and C. L. Wei, Global mapping of c-myc binding sites and target gene networks in human b cells, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17834–17839 CrossRef CAS.
- M. Joglekar, V. Parekh and A. Hardikar, Pancreas from old: microregulators of pancreas regeneration, Trends Endocrinol. Metab., 2007, 18, 393–400 CrossRef CAS.
- M. Gershengom, A. Hardikar, C. Wei, E. Geras-Raaka, B. Marcus-Samuels and B. M. Raaka, Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells, Science, 2004, 306, 2261–2264 CrossRef.
- J. Gudmundsson, P. Sulem, V. Steinthorsdottir, J. Bergthorsson and G. Thorleifsson et al., Two variants on chromosome 17 confer prostate cancer risk, and the one in tcf2 protects against type 2 diabetes, Nat. Genet., 2007, 39, 977–983 CrossRef CAS.
- A. Ladd, G. Taffet, C. Hartley, D. Kearney and T. Cooper, Cardiac tissue-specific repression of celf activity disrupts alternative splicing and causes cardiomyopathy, Mol. Cell. Biol., 2005, 25, 6267 CrossRef CAS.
- N. Liu, A. Williams, Y. Kim, J. McAnally and S. Bezprozvannaya et al., An intragenic mef2-dependent enhancer directs muscle-specific expression of micrornas 1 and 133, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20844–20849 CrossRef CAS.
- M. Iorio, R. Visone, G. Di Leva, V. Donati, F. Petrocca, P. Casalini, C. Taccioli, S. Volinia, C. G. Liu, C. G. Liu, H. Alder, G. A. Calin, S. Ménard and C. M. Croce, MicroRNA signatures in human ovarian cancer, Cancer Res., 2007, 67, 8699–8707 CrossRef CAS.
- R. Chen, A. Alvero, D. Silasi, M. Kelly, S. Fest, I. Visintin, A. Leiser, P. E. Schwartz, T. Rutherford and G. Mor, Regulation of ikkbeta by mir-199a affects nf-kappab activity in ovarian cancer cells, Oncogene, 2008, 27, 4712–4723 CrossRef CAS.
- D. Phan, C. Cheng, M. Galfione, F. Vakar-Lopez, J. Tunstead, N. E. Thompson, R. R. Burgess, S. M. Najjar, L. Y. Yu-Lee and S. H. Lin, Identification of sp2 as a transcriptional repressor of carcinoembryonic antigen-related cell adhesion molecule 1 in tumorigenesis, Cancer Res., 2004, 64, 3072–3078 CrossRef CAS.
- D. Wilkerson, H. Skaggs and K. Sarge, Hsf2 binds to the hsp90, hsp27, and c-fos promoters constitutively and modulates their expression, Cell Stress Chaperones, 2007, 12, 283–290 Search PubMed.
- W. Wagner, P. Horn, M. Castoldi, A. Diehlmann, S. Bork, R. Saffrich, V. Benes, J. Blake, S. Pfister, V. Eckstein and A. D. Ho, Replicative senescence of mesenchymal stem cells; a continuous and organized process, PLoS ONE, 2008, 3, e2213 CrossRef.
- Y. Zhao, J. Ransom, A. Li, V. Vedantham, M. von Drehle, A. N. Muth, T. Tsuchihashi, M. T. McManus, R. J. Schwartz and D. Srivastava, Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking mirna-1-2, Cell, 2007, 129, 303–317 CrossRef CAS.
- Q. Pan, O. Shai, L. Lee, B. Frey and B. Blencowe, Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing, Nat. Genet., 2008, 40, 1413–1415 CrossRef CAS.
- G. Pesole, What is a gene? an updated operational definition, Gene, 2008, 417, 1–4 CrossRef CAS.
- S. Griffiths-Jones, mirbase: the microRNA sequence database, Methods Mol. Biol., 2006, 342, 129–138 Search PubMed.
- H. Saini, S. Griffiths-Jones and A. Enright, Genomic analysis of human microRNA transcripts, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17719–17724 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Description of oligo analysis and randomizations for network motifs analysis, randomization results for the network motifs analysis of mixed feed-forward loops, and supplementary files S1–S11. See DOI: 10.1039/b900177h |
| ‡ Equal contribution |
| This journal is © The Royal Society of Chemistry 2009 |