SAC1 lipid phosphatase and growth control of the secretory pathway
Anastasia
Blagoveshchenskaya
and
Peter
Mayinger
*
Division of Nephrology & Hypertension and Department of Cell & Developmental Biology, Oregon Health & Science University, Portland, Oregon, USA. E-mail: mayinger@ohsu.edu
First published on 24th November 2008
Abstract
Phosphoinositide lipids play a dual role in cell physiology. Specific sets of these molecules are short-lived downstream mediators of growth signals, regulating cell survival and differentiation. In addition, distinct classes of phosphoinositide lipids function as constitutive mediators of membrane traffic and organelle identity. Recent work has provided the first direct evidence that phosphoinositides also play a direct role in linking protein secretion with cell growth and proliferation. This review focuses on SAC1 lipid phosphatase and how this enzyme operates in an evolutionary conserved mechanism to coordinate the secretory capacity of ER and Golgi during cell growth.
 Anastasia Blagoveshchenskaya Anastasia Blagoveshchenskaya | Anastasia D. Blagoveshchenskaya received her PhD in Cell Biology in 1996 at the Institute of Cytology, Russian Academy of Sciences. Since 2005, she has been a postdoctoral fellow in the laboratory of Dr Peter Mayinger at the Division of Nephrology & Hypertension, OHSU, where she studies lipid phosphatase SAC1 and its role in cell proliferation. |
 Peter Mayinger Peter Mayinger | Peter Mayinger obtained his PhD in 1992 at the Ludwig-Maximilans University, Munich, Germany. In 1999 he became an independent group leader at the ZMBH, where he completed his Habilitation in Cell Biology in 2001. In 2003 he moved with his laboratory to Oregon Health & Science University, where he is currently an Assistant Professor in the Division of Nephrology and Hypertension. His research is centred on lipid signalling and membrane traffic with particular focus on the lipid phosphatases that regulate organelle function and how deficiencies in these enyzmes cause human disease. |
Introduction
Growth is essential for cell proliferation and differentiation in all organisms and causes a wide range of severe human diseases if misregulated. Most cells in multicellular organisms differentiate to perform specialized functions and then stop dividing. Under certain circumstances, such quiescent cells may re-enter the cell cycle in a highly regulated manner. Quiescent cells are not only present in multicellular organisms but are also found in stationary-phase yeast cultures when nutrients become limiting.1Growth control relies on the complex orchestration of a plethora of cellular activities. In higher eukaryotes, external stimuli, such as growth factors, induce intracellular signalling cascades that in turn regulate transcriptional programs, protein synthesis, metabolic rates, cytoskeletal dynamics and morphogenesis.2,3 Mitogenic stimulation of quiescent cells induces a general increase in protein synthesis, which allows the cell to double its protein content and size before mitosis.4 Active cell growth relies therefore on continuous delivery of exocytic transport vesicles to the cell periphery to ensure that average cell size is maintained during active proliferation. A landmark study in yeast three decades ago showed directly that secretion is essential for cell surface growth.5 Yet, how membrane traffic is coordinated with growth and proliferation remains largely unknown. The secretory pathway is coordinated via the interplay of different types of regulatory mechanisms. Organelle-specific small GTPases from the Arf and Rab families help defining the identity of individual compartments and function as important control element for membrane traffic.6Another important mechanism for regulating vesicular transport involves phosphoinositide lipids. Reversible phosphorylation of the inositol headgroup of phosphatidylinositol creates seven distinct phosphoinositide species, which have unique functions in cell physiology.7,8 These molecules serve as signal transducers at virtually every cellular membrane but have a particularly important role in controlling the function of secretory organelles.6,9 Though phosphatidylinositol is an abundant phospholipid present in all cellular membranes, organelle-specific biosynthesis and turnover by lipid kinases and phosphatases generates compartment-specific phosphoinositide pools.6,9 Generation of phosphoinositides by specific lipid kinases is essential for cell viability.10 However, the regulated dephosphorylation of these lipids by phosphatases is equally important and mutations in several lipid phosphatases were recently linked to a variety of specific human disorders.11 This review focuses on the lipid phosphatase SAC1 with particular emphasis on the novel roles this enzyme plays in the metabolic and growth control of membrane traffic within the biosynthetic pathway.
Cell growth and the secretory pathway
Cell proliferation is stimulated by a multitude of extracellular signals including growth factors, cytokines and hormones. These signals converge at several major downstream signalling pathways that act both antagonistically and in parallel to precisely control the diverse cellular functions required for growth and proliferation.Multicellular organisms have three well-characterized subfamilies of mitogen-activated protein kinases (MAPKs) that regulate complex programs such as proliferation, differentiation, embryogenesis and cell death.2 MAPKs form protein kinase cascades, which contain at least three enzymes acting in series.2,12 Another important signal transduction pathway activated by growth factors and cytokines is the phosphoinositide 3-kinase (PI3K) pathway. PI3K generates the short lived lipid signalling molecule PI(3,4,5)P2 that initiates a cascade of signalling events primarily via the induction of specific protein-serine/threonine kinases.13 Interest in the pathway has been driven by its frequent aberrant activation in disease and its impact on cell fate decisions owing to roles in survival signalling and metabolic control.14 Despite the progress in characterizing mitogen-dependent signalling cascades, it remains unresolved which one of these pathways regulates flux through the biosynthetic pathway in response to growth signals. Recent evidence indicates that growth factor signalling involves intracellular membranes including the ER and Golgi.15 Endogenous Ras and oncogenic H-Ras are activated in response to mitogens on the Golgi and endoplasmic reticulum (ER), respectively.15 In addition, the MEK/ERK MAP kinase modules are important regulators of Golgi fragmentation during mitosis.16,17 However, a direct functional link between mitogen stimulation of cell growth and constitutive secretion is missing.
A functional secretory pathway appears to be particularly important for cancer cell growth. Tumor cells interact with their environment through the secretion of soluble factors and malignant progression is thought to require substantial remodelling of the secretory machinery in cancer cells.18 Consistent with this idea, several Rab and Arf GTPases involved in the regulation of membrane traffic within the secretory pathway show upregulated expression levels in various liver, breast and ovarian tumors.19,20 In addition, chronic lymphocytic leukemia cells are highly sensitive to interruption of ER-to-Golgi traffic and studies are underway to target Arf proteins in cancer cells using Brefeldin A (BFA) ester derivatives.21,22 BFA binds to a complex of Arf with its guanine nucleotide exchange factor and thus inhibits Arf function. This blocks protein transport and results in redistribution of Golgi proteins to the ER.23,24 Unfortunately, BFA exhibits significant neurotoxicity and the use of its derivatives as anticancer agents remains problematic.25 The molecular analysis of the unknown mechanisms that regulate secretion in rapidly dividing cells is therefore of significant biomedical relevance.
Phosphoinositides and membrane dynamics
Phosphoinositides are phosphorylated derivatives of phosphatidylinositol (PI). The 3-, 4-, and 5-hydroxy positions of the inositol headgroup can be phosphorylated leading to 7 unique phosphoinositide species (Fig. 1A).8 Historically, phosphatidylinositol-4,5-bisphosphate PI(4,5)P2 has been considered the key player in phosphoinositide signalling because it is the precursor of various second messenger molecules such as Inositol-1,4,5-trisphosphate (IP3), diacylglycerol and PI(3,4,5)P3.26 IP3 modifies intracellular calcium levels, diacylglycerol regulates members of the protein kinase C family, and PI(3,4,5)P3 controls cell survival and propagation.26–28 However, recent work showed that the other phosphoinositide species have equally important functions acting as signal mediators in a spatially and temporally controlled manner.6,29 Localized production of the different phosphoinositide molecules is coupled to the recruitment of specific phosphoinositide-binding factors, which often interact additionally with other membrane-specific factors.9,30 The coincident detection of specific phosphoinositides and other membrane regulators by various effector proteins controls multiple lines of intracellular events such as gene expression, cytoskeletal organization, cell motility and vesicular membrane transport.7,31,32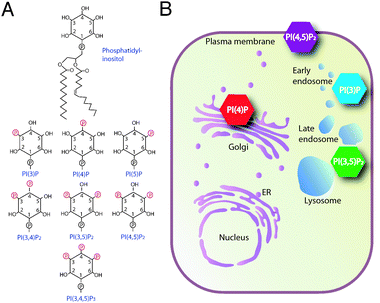 | ||
| Fig. 1 Organelle-specific distribution of phosphoinositide lipids. (A) Structure of phosphatidylinositol and its phosphorylated derivatives. The myo-inositol head group can be phosphorylated at D3-, D4- and D5-positions, resulting in 7 distinct phosphoinositide species. (B) Model illustrating spatially restricted pools of individual phosphoinositide species at cellular organelles. PI(4)P and PI(4,5)P2 play a role in anterograde trafficking and in recruiting clathrin to the sites of endocytosis. PI(3)P and PI(3,5)P2 are important for membrane dynamics along the endocytic pathway. | ||
A particularly important feature of phosphoinositide lipids is their restricted intracellular localization. With the exception of PI(3,4,5)P3 and PI(3,4)P2, which are generated upon stimulation with extracellular signals, most of the individual phosphoinositide species are constitutively concentrated at distinct organelles (Fig. 1B).6,9 However, the function of the various intracellular organelles and the rate of trafficking between them must be coupled to cell growth and cell division and there is increasing evidence that these compartment-specific phosphoinositide pools can be modulated in response to extracellular signals.33,34 Site-specific biosynthesis and turnover by lipid kinases and phosphatases are essential for dynamic regulation of the various phosphoinsositide pools.35 Yet, how the individual trafficking routes that are governed by phosphoinositides are synchronized in coordination with growth and proliferation is poorly understood. The phosphoinositides that control the organization of the secretory pathway are likely targets for growth regulation because the capacity of anterograde trafficking contributes to determining cell growth rates.
The importance of phosphorylated lipids in membrane trafficking was first recognized when the yeast VPS34gene, mutations in which cause defects in vacuolar protein sorting, was found to encode a PI3-kinase.36 Consistent with this result, 3-phosphorylated derivatives of PI were later found to be involved in endosomal membrane trafficking.37PI(3)P is enriched in endosomal membranes and recruits cytosolic effector proteins that are required for endosomal fusion reactions.38,39 Several other phosphoinositide species are also constitutively concentrated at particular organellar membranes, which is important for controlling compartment-specific membrane trafficking and maintaining organelle identity (Fig. 1B). A local pool of PI(3,5)P2 is believed to function in cargo selection at a late endosomal sorting compartment destined to eventually fuse with the vacuole.37
PI(4)P and PI(4,5)P2 are the most important lipid regulators of the biosynthetic secretory pathway (Fig. 1B). PI(4,5)P2 was reported to function in organizing Golgi structure by interacting with the cytoskeletal protein β-spectrin.40,41 The exact role of PI(4,5)P2 in trafficking is unclear and only little if any PI(4,5)P2 can be detected in membranes other than the plasma membrane.42 However, mutations in OCRL1, a PI(4,5)P2-specific 5-phosphatase localized to Golgi and endosomal membranes cause Lowe’s syndrome, a severe congenital disease in humans, probably because of defects in late Golgi and endosomal trafficking.43–45 A crucial role for PI(4)P in Golgi trafficking has been more firmly established. In both yeast and mammals, PI(4)P is important for forward trafficking from the Golgi to the cell periphery.46–48PI(4)P was shown to bind directly to the mammalian clathrin adaptor protein complex 1 (AP-1) and to both mammalian and yeast GGA proteins.49–51PI(4)P also associates with four-phosphate-adaptor protein 1 and 2 (FAPP1 and FAPP2), both of which form complexes with ADP-ribosylation-factor (ARF) at Golgi membranes.52 FAPP2 has recently been found to function as a glucosylceramide transfer protein required for glycosphingolipid biosynthesis, though the exact role of FAPP2 in this pathway is not entirely clear.53,54 In yeast, synthesis of PI(4)P at Golgi membranes is essential for anterograde trafficking of a variety of cargo proteins, such as invertase, Hsp150 and chitin synthases.46,55,56Localization studies using fluorescent lipid binding probes indicated that a substantial proportion of PI(4)P localizes to Golgi membranes.48,49,57 However, there is strong evidence that additional functionally distinct pools of PI(4)P exist58,59 and this phosphoinositide has also been implicated in ER-function, actin cytoskeletal dynamics, and vacuolar morphology.46,56,60,61
Sac1-domain lipid phosphatases
Members of the Sac1-domain subfamily of lipid phosphatases share a catalytically active domain, first identified in the yeast Sac1 protein.62Sac1 homology domain-containing phosphatases catalyze the hydrolysis of a variety of phosphoinositide species and are found from yeast to mammals.62,63 The Sac1 domain is a 400 amino acid region containing the motif CX5R(T/S), essential for catalytic activity, which is also found in many metal-independent protein and lipid phosphatases.64,65In vitro studies have shown that the Sac1 homology domain is capable of hydrolyzing phosphate from any of the three positions of inositol that may be phosphorylated (D-3, D-4 and D-5). However, inositol lipids with adjacent phosphates are resistant to hydrolysis by Sac1 domains and only PI(3)P, PI(4)P and PI(3,5)P2 can be dephosphorylated efficiently.64,66 In mammals, the Sac1 phosphatase domain is found in the transmembrane phosphatase SAC1, and in two additional genes present in the human genome termed SAC2 and SAC3, whose coding sequences are most closely related to the yeast Fig4 protein (Fig. 2). SAC3/FIG4 appears to be mainly a PI(3,5)P2-specific phosphatase in vivo and has recently been implicated in Charcot–Marie–Tooth peripheral neuropathies.67 A Sac1 domain is also present in the mammalian phosphoinositide 5-phosphatase synaptojanin and its various splice variants, all of which contain both Sac1 phosphatase and 5-phosphatase domains (Fig. 2).62,65,68 The synaptojanins play a role in clathrin-mediated synaptic vesicle endocytosis.68–70 In yeast, this protein family comprises five different members, Sac1p, Fig4p, and the three synaptojanin-like phosphatases Sjl1/Inp51p, Sjl2/Inp52p and Sjl3/Inp53p.57,62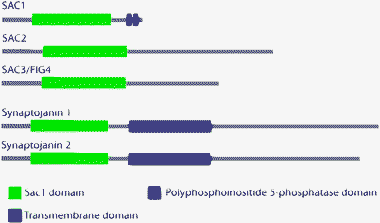 | ||
| Fig. 2 Domain structure of Sac1 domain-containing phosphatases. Sac1p- and Fig4p-like phosphatases have an N-terminal Sac1 domain. Synaptojanin-like phosphatases have an N-terminal Sac1 domain and a C-terminal 5-phosphatase domain. | ||
The Sac1 polyphosphoinositide phosphatase is the founding member of the Sac1 domain lipid phosphatase family. Yeast Sac1p was originally identified as a suppressor of actin mutations in yeast.71,72 Among the Sac1 domain-containing proteins, Sac1 itself is unique in that it is the only integral membrane lipid phosphatase in this protein family.73,74 Topology analysis showed that all Sac1 orthologs are type-II transmembrane protein containing two separate transmembrane segments (Fig. 3A).74,75 However, sequence comparison showed several species-specific features (Fig. 3B). All mammalian Sac1 orthologs contain a leucine zipper within their Sac1-domain, which is required for oligomerization, and a C-terminal di-lysine ER retrieval motif (Fig. 3A).76,77 Both of theses elements are absent in yeast Sac1p (Fig. 3B). Ablation of the yeast SAC1gene results in a dramatic increase of intracellular PI(4)P-levels and causes alteration in ER and Golgi function.61,74 Mutant mice lacking Sac1 are embryonically lethal and knock-down of Sac1 expression in mammalian tissue culture cells leads to changes in Golgi morphology and in mitotic spindle assembly.78 While both yeast and mammalian Sac1 orthologs can also dephosphorylate PI(3)P and PI(3,5)P2, Sac1 appears to act predominantly as a lipid 4-phosphatase in vivo.61,74,79
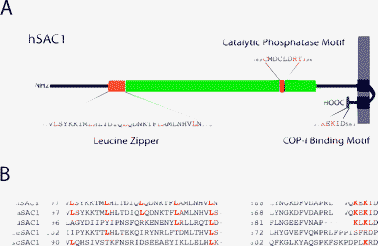 | ||
| Fig. 3 Structural model illustrating topology and important sequence elements of the Sac1 lipid phosphatase. (A) SAC1 is a type II transmembrane protein, with a large N-terminal cytosolic domain. The depicted topology of human SAC1 is based on predicted structural elements and on experiments in yeast.74 Regions that are important for catalytic activity, oligomerization and trafficking are highlighted. (B) Sequence comparison between various SAC1 orthologs. The leucine zipper motif and the C-terminal COP–I interacting motif are not conserved in all organisms. | ||
Yeast Sac1p: metabolic control of lipid signalling and secretion
Genetic and functional studies in yeast provided genetic and functional evidence that Sac1p stimulates ER function but antagonizes Golgi trafficking.72,74,75 These findings were surprising because the simultaneous execution of such strikingly opposing regulatory roles should lead to a severely compromised organization of the secretory pathway. The identification of the ER protein Dpm1p in a complex with Sac1p provided a first insight into the mechanism of this puzzling intracellular function of Sac1p.80 Dpm1p synthesizes dolicholphosphate-mannose, an important mannosyl donor for glycosylation reactions in the ER. By examining mutant forms of Dpm1p and Sac1p, it was established that the interaction with Dpm1p is essential for the ER localization of the lipid phosphatase.80 Interestingly, recruitment of Sac1p activity to the ER was required for efficient oligosaccharide biosynthesis but only during times of exponential growth. Glucose starvation-induced shut down of cell proliferation was accompanied by rapid and reversible translocation of Sac1p to Golgi membranes (Fig. 4A, upper panel).80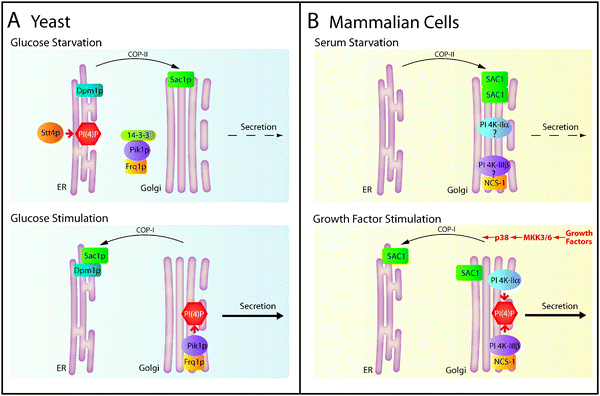 | ||
| Fig. 4 Regulation of Sac1 localization and secretion in yeast and mammals. (A) Metabolic control of Sac1p localization in yeast. Glucose starvation triggers translocation of Sac1p to the Golgi and release of Pik1p/Frq1p from Golgi membranes, which reverses the PI(4)P gradient and slows secretion. The cytosolic pool of Pik1p/Frq1p is stabilized by binding to 14-3-3 proteins. In the presence of glucose, Sac1p localizes to the ER in a complex with Dpm1p. During exponential growth, the PI 4-kinase complex Pik1p/Frq1p generates a pool of PI(4)P at the Golgi. The resulting steep PI(4)P gradient between ER and Golgi stimulates anterograde trafficking. (B) Mitogen-dependent regulation of SAC1 localization and secretion in mammals. In quiescent cells, SAC1 oligomerizes and accumulates in the Golgi, which in turn down-regulates Golgi PI(4)P and constitutive secretion. Whether resident PI 4-kinases are released from the Golgi under these conditions is unknown. After growth factor stimulation, p38 MAPK activity is required for dissociation of SAC1 complexes, which triggers retrograde traffic and redistribution of SAC1 to the ER. Reduction of SAC1 levels at the Golgi and biosynthesisviaPI 4-kinase IIα and PI 4-kinase IIIβ generates elevated concentration of PI(4)P at this organelle, thus accelerating constitutive secretion. | ||
PI(4)P is the major substrate for Sac1pin vivo. Consequently, glucose-dependent shuttling of Sac1p between ER and Golgi is responsible for reciprocal control of PI(4)P levels at these organelles. Anterograde trafficking from the Golgi requires sufficient levels of PI(4)P.81 If nutrients are abundant, the PI 4-kinase Pik1p generates a local pool of PI(4)P at Golgi membranes (Fig. 4A, lower panel).82,83 Under these conditions localization of Sac1 remains restricted to ER membranes, where it keeps PI(4)P levels low (Fig. 4A, lower panel).80,82 Pik1p association with the Golgi requires the non-catalytic cofactor Frq1p.84Glucose starvation induces the release of the Pik1p/Frq1p complex from the Golgi and leads to nuclear accumulation of Pik1p.82,83 In addition to its role in Golgi traffic, Pik1p plays an essential but as yet not completely understood role in nuclear lipid signalling.85 Nutrient-dependent binding of 14-3-3 proteins to Pik1p regulates the distribution of this protein between Golgi, nucleus and cytoplasm (Fig. 4A, upper panel).83 In the presence of glucose, the mutually exclusive intracellular localization of Sac1p and Pik1p creates a Golgi-restricted pool of PI(4)P, which is essential for efficient anterograde trafficking (Fig. 4A, lower panel).80,82 During glucose starvation, Sac1p translocation to the Golgi and the release of Pik1p/Frq1p reduces Golgi PI(4)P levels, thereby slowing anterograde trafficking (Fig. 4A, upper panel). Absence of Sac1p from the ER allows for increased levels of ER-localized PI(4)P, which appears to be synthesized by the PI 4-kinase Stt4p (Fig. 4A, upper panel).61,82 In turn, protein processing at the ER is reduced upon PI(4)P accumulation at this compartment.75,80 Sac1p and Pik1p are therefore elements of a unique metabolic control mechanism for synchronizing the secretory capacities of ER and Golgi membranes during cell growth.
Human SAC1: Connecting mitogen signalling, Golgi traffic and cell growth
Consistent with the studies in yeast, the human SAC1 also undergoes efficient shuttling between Golgi and ER. However, in mammalian cells this shuttling is triggered by mitogens rather than by glucose thus implicating SAC1 in the signal transduction cascades leading to cell growth and proliferation.77 The significance of phosphoinositides in the transmission of mitogenic stimuli has long been realized but this cross-talk between growth signalling and lipid signalling at the Golgi is novel. The cell growth-dependent shuttling of SAC1 has therefore important ramifications for re-evaluating the cellular function of this lipid phosphatase. Clearly, SAC1 is not just an enzyme involved in the constitutive spatial restriction of phosphoinositide pools but plays a major role in mitogen-induced signal transduction pathways.Although the membrane trafficking machinery and the molecular mechanisms involved in mitogen-induced SAC1 shuttling between the ER and Golgi are relatively well characterized, much less is understood about how SAC1 function is linked to mitogenic signal transduction.77 Recent data showed that SAC1 shuttling from the ER to Golgi is triggered when cells are deprived of growth factors. This translocation to the Golgi requires oligomerization of SAC1 via its leucine zipper (LZ) motif and subsequent binding of the coat protein complex II (COP-II), which facilitates ER exit (Fig. 4B, upper panel).77 Interestingly, a similar mechanism involving the interaction of LZ-mediated oligomers with components of the COP-II machinery has been documented for ER-to-Golgi trafficking of newly synthesized neurotransmitter transporters en route to the cell surface.86–89 Upon its arrival in the Golgi, SAC1 dephosphorylates Golgi PI(4)P which is accompanied by reduced constitutive secretion (Fig. 4B, upper panel).77 When quiescent cells are stimulated with mitogens, SAC1 oligomers dissociate and the monomers are retrieved back to the ER (Fig. 4B, lower panel). The retrograde traffic of SAC1 requires coatomer protein complex I (COP–I)-binding to its C-terminal double lysine ER-retrieval motif.76Growth-stimulated cells establish a concentrated pool of PI(4)P at the Golgi, which promotes anterograde traffic (Fig. 4B, lower panel). Whether any of the mammalian PI 4-kinases show growth-dependent rounds of Golgi association and release remains to be determined.77
The mammalian p38 mitogen-activated protein kinase (MAPK) and, to a lesser extent, the extracellular signal-regulated kinase 1/2 (ERK) are required for mitogen-dependent stimulation of SAC1 shuttling from the Golgi to the ER.77 Although it is unknown whether these kinases interact with SAC1 directly or exert their regulatory function via accessory proteins, SAC1 is an integral part of a regulatory pathway required for spatial organization of signal transduction at the Golgi. A large body of evidence documented a requirement for MAPKs in disassembly and fragmentation of the Golgi apparatus during mitosis.90,91 Only recently, a possible role for the Golgi in signal transduction during nonmitotic stages of the cell cycle has emerged. Recent studies using fluorescent probes in living cells detected Ras proteins and Ras-related GTPases at the Golgi.15,92 Ras proteins play a critical role in the control of normal and transformed cell growth and are among the most frequently affected genes in human cancer.93Mitogen-induced Ras signalling occurs via a downstream MAPK pathway with Raf, MEK and ERK being major downstream effectors, all of which have been detected at the Golgi.94
Recent evidence suggests that the p38 MAPK pathway may function as a tumor suppressor via inhibiting Ras-induced tumorigenesis.93 The role of the p38 MAPK pathway has been originally linked to transmission of inflammatory and stress responses but was later expanded with the findings that Ras activity is required for p38 MAPK activation by growth factors.95 It is now becoming apparent that the p38 MAPK pathway regulates Ras oncogenic activity by negative feedback. According to this mechanism, mitogenic stimulation with growth factors activates the components of the p38 MAPK pathway, such as MKK6, p38α and MK2/PRAK, which in turn suppress Ras activity.96 Interestingly, activation of p38 MAPKvia dominant active alleles of MKK6 and MKK3 was sufficient to trigger dissociation of SAC1 oligomers and induce retrograde traffic to the ER in the absence of mitogenic stimulation.77 There is therefore the intriguing possibility that SAC1 may play an important role as a downstream p38MAPK effector to modulate Ras-induced cell proliferation. Future work is needed to analyze this mechanistic link between growth signalling and ER/Golgi function and evaluate its possible implications in tumorigenesis.
Conclusion
Our recent work provided the first direct evidence that lipid signalling at the ER and Golgi is intimately connected to cell growth. Although this mechanism in yeast was largely dependent on the availability of glucose, the results in mammalian cells suggest that phosphoinositides play an evolutionary conserved role in integrating secretion and cell growth. Characterization of mitogen-responsive elements within the secretory pathway in mammalian cells will be critical for understanding growth control of secretion in molecular detail. This information may also shed new light on the cellular basis of human proliferative diseases such as fibrosis and cancer and may offer opportunities to identify novel drug targets to combat these diseases.Acknowledgements
We thank Jim McCormick for critical reading of the manuscript. We are also indebted to Vytas Bankaitis for discussing data prior to their publication.References
- C. Allen, S. Buttner, A. D. Aragon, J. A. Thomas, O. Meirelles, J. E. Jaetao, D. Benn, S. W. Ruby, M. Veenhuis, F. Madeo and M. Werner-Washburne, J. Cell Biol., 2006, 174, 89–100 CrossRef CAS.
- G. L. Johnson and R. Lapadat, Science, 2002, 298, 1911–1912 CrossRef CAS.
- X. Wang and C. G. Proud, Physiology, 2006, 21, 362–369 CrossRef CAS.
- K. A. Martin and J. Blenis, Adv. Cancer Res., 2002, 86, 1–39 CAS.
- P. Novick and R. Schekman, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1858–1862 CrossRef CAS.
- R. Behnia and S. Munro, Nature, 2005, 438, 597–604 CrossRef CAS.
- A. Toker, Cell. Mol. Life Sci., 2002, 59, 761–779 CrossRef CAS.
- K. F. Tolias and L. C. Cantley, Chem. Phys. Lipids, 1999, 98, 69–77 CrossRef CAS.
- G. Di Paolo and P. De Camilli, Nature, 2006, 443, 651–657 CrossRef CAS.
- D. A. Fruman, R. E. Meyers and L. C. Cantley, Annu. Rev. Biochem., 1998, 67, 481–507 CrossRef CAS.
- C. Erneux, C. Govaerts, D. Communi and X. Pesesse, Biochim. Biophys. Acta, 1998, 1436, 185–199 CAS.
- C. Widmann, S. Gibson, M. B. Jarpe and G. L. Johnson, Physiol. Rev., 1999, 79, 143–180 CAS.
- J. Downward, Semin. Cell Dev. Biol., 2004, 15, 177–182 CrossRef CAS.
- M. P. Wymann and R. Marone, Curr. Opin. Cell Biol., 2005, 17, 141–149 CrossRef CAS.
- V. K. Chiu, T. Bivona, A. Hach, J. B. Sajous, J. Silletti, H. Wiener, R. L. Johnson 2nd, A. D. Cox and M. R. Philips, Nat. Cell Biol., 2002, 4, 343–350 CAS.
- U. Acharya, A. Mallabiabarrena, J. K. Acharya and V. Malhotra, Cell, 1998, 92, 183–192 CrossRef CAS.
- Y. D. Shaul and R. Seger, J. Cell Biol., 2006, 172, 885–897 CrossRef CAS.
- S. Mkrtchian, M. Baryshev, E. Sargsyan, I. Chatzistamou, A. A. Volakaki, N. Chaviaras, A. Pafiti, A. Triantafyllou and H. Kiaris, Mol. Carcinog., 2008, 47, 886–892 CrossRef CAS.
- H. He, F. Dai, L. Yu, X. She, Y. Zhao, J. Jiang, X. Chen and S. Zhao, Gene Expression, 2002, 10, 231–242 Search PubMed.
- K. W. Cheng, J. P. Lahad, J. W. Gray and G. B. Mills, Cancer Res., 2005, 65, 2516–2519 CrossRef CAS.
- N. O. Anadu, V. J. Davisson and M. Cushman, J. Med. Chem., 2006, 49, 3897–3905 CrossRef CAS.
- J. S. Carew, S. T. Nawrocki, Y. V. Krupnik, K. Dunner Jr, D. J. McConkey, M. J. Keating and P. Huang, Blood, 2006, 107, 222–231 CrossRef CAS.
- S. Robineau, M. Chabre and B. Antonny, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 9913–9918 CrossRef CAS.
- J. Lippincott-Schwartz, L. C. Yuan, J. S. Bonifacino and R. D. Klausner, Cell, 1989, 56, 801–813 CrossRef CAS.
- S. Kikuchi, K. Shinpo, S. Tsuji, I. Yabe, M. Niino and K. Tashiro, J. Neurosci. Res., 2003, 71, 591–599 CrossRef CAS.
- S. B. Lee and S. G. Rhee, Curr. Opin. Cell Biol., 1995, 7, 183–189 CrossRef CAS.
- L. Combettes, B. Berthon and M. Claret, Biochimie, 1987, 69, 281–286 CrossRef CAS.
- R. Kapeller and L. C. Cantley, Bioessays, 1994, 16, 565–576 CAS.
- M. P. Czech, Annu. Rev. Physiol., 2003, 65, 791–815 CrossRef CAS.
- J. H. Hurley and T. Meyer, Curr. Opin. Cell Biol., 2001, 13, 146–152 CrossRef CAS.
- D. A. Cantrell, J. Cell Sci., 2001, 114, 1439–1445 CAS.
- A. Simonsen, A. E. Wurmser, S. D. Emr and H. Stenmark, Curr. Opin. Cell Biol., 2001, 13, 485–492 CrossRef CAS.
- M. Falasca and T. Maffucci, Arch. Physiol. Biochem., 2006, 112, 274–284 CrossRef CAS.
- K. K. Caldwell, M. Sosa and C. T. Buckley, Cell Commun. Signal, 2006, 4, 2 Search PubMed.
- M. Vicinanza, G. D'Angelo, A. Di Campli and M. A. De Matteis, EMBO J., 2008, 27, 2457–2470 CrossRef CAS.
- P. V. Schu, K. Takegawa, M. J. Fry, J. H. Stack, M. D. Waterfield and S. D. Emr, Science, 1993, 260, 88–91 CrossRef CAS.
- D. J. Katzmann, G. Odorizzi and S. D. Emr, Nat. Rev. Mol. Cell Biol., 2002, 3, 893–905 CrossRef CAS.
- A. E. Wurmser, J. D. Gary and S. D. Emr, J. Biol. Chem., 1999, 274, 9129–9132 CrossRef CAS.
- T. K. Sato, M. Overduin and S. D. Emr, Science, 2001, 294, 1881–1885 CrossRef CAS.
- D. A. Sweeney, A. Siddhanta and D. Shields, J. Biol. Chem., 2002, 277, 3030–3039 CrossRef CAS.
- A. Siddhanta, A. Radulescu, M. C. Stankewich, J. S. Morrow and D. Shields, J. Biol. Chem., 2003, 278, 1957–1965 CrossRef CAS.
- S. A. Watt, G. Kular, I. N. Fleming, C. P. Downes and J. M. Lucocq, Biochem. J., 2002, 363, 657–666 CrossRef CAS.
- M. A. Dressman, I. M. Olivos-Glander, R. L. Nussbaum and S. F. Suchy, J. Histochem. Cytochem., 2000, 48, 179–190 CAS.
- M. Lowe, Traffic, 2005, 6, 711–719 CrossRef CAS.
- M. Loi, Orphanet. J. Rare Dis., 2006, 1, 16 Search PubMed.
- C. Walch-Solimena and P. Novick, Nat. Cell Biol., 1999, 1, 523–525 CrossRef CAS.
- H. Hama, E. A. Schnieders, J. Thorner, J. Y. Takemoto and D. B. DeWald, J. Biol. Chem., 1999, 274, 34294–34300 CrossRef CAS.
- A. Godi, A. D. Campli, A. Konstantakopoulos, G. D. Tullio, D. R. Alessi, G. S. Kular, T. Daniele, P. Marra, J. M. Lucocq and M. A. Matteis, Nat. Cell Biol., 2004, 6, 393–404 CrossRef CAS.
- Y. J. Wang, J. Wang, H. Q. Sun, M. Martinez, Y. X. Sun, E. Macia, T. Kirchhausen, J. P. Albanesi, M. G. Roth and H. L. Yin, Cell, 2003, 114, 299–310 CrossRef CAS.
- J. Wang, H. Q. Sun, E. Macia, T. Kirchhausen, H. Watson, J. S. Bonifacino and H. L. Yin, Mol. Biol. Cell, 2007, 18, 2646–2655 CrossRef CAS.
- L. Demmel, M. Gravert, E. Ercan, B. Habermann, T. Muller-Reichert, V. Kukhtina, V. Haucke, T. Baust, M. Sohrmann, Y. Kalaidzidis, C. Klose, M. Beck, M. Peter and C. Walch-Solimena, Mol. Biol. Cell, 2008, 19, 1991–2002 CrossRef CAS.
- M. A. De Matteis and A. Godi, Nat. Cell Biol., 2004, 6, 487–492 CrossRef CAS.
- G. D’Angelo, E. Polishchuk, G. Di Tullio, M. Santoro, A. Di Campli, A. Godi, G. West, J. Bielawski, C. C. Chuang, A. C. van der Spoel, F. M. Platt, Y. A. Hannun, R. Polishchuk, P. Mattjus and M. A. De Matteis, Nature, 2007, 449, 62–67 CrossRef CAS.
- D. Halter, S. Neumann, S. M. van Dijk, J. Wolthoorn, A. M. de Maziere, O. V. Vieira, P. Mattjus, J. Klumperman, G. van Meer and H. Sprong, J. Cell Biol., 2007, 179, 101–115 CrossRef CAS.
- A. Audhya, M. Foti and S. D. Emr, Mol. Biol. Cell, 2000, 11, 2673–2689 CAS.
- M. Schorr, A. Then, S. Tahirovic, N. Hug and P. Mayinger, Curr. Biol., 2001, 11, 1421–1426 CrossRef CAS.
- C. J. Stefan, A. Audhya and S. D. Emr, Mol. Biol. Cell, 2002, 13, 542–557 CrossRef CAS.
- A. Roy and T. P. Levine, J. Biol. Chem., 2004, 279, 44683–44689 CrossRef CAS.
- S. Tahirovic, M. Schorr and P. Mayinger, Traffic, 2005, 6, 116–130 CrossRef CAS.
- H. Hama, E. A. Schnieders, J. Thorner, J. Y. Takemoto and D. B. DeWald, J. Biol. Chem., 1999, 274, 34294–34300 CrossRef CAS.
- M. Foti, A. Audhya and S. D. Emr, Mol. Biol. Cell, 2001, 12, 2396–2411 CAS.
- W. E. Hughes, F. T. Cooke and P. J. Parker, Biochem. J., 2000, 350 Pt 2, 337–352 CrossRef.
- B. Despres, F. Bouissonnie, H. J. Wu, V. Gomord, J. Guilleminot, F. Grellet, F. Berger, M. Delseny and M. Devic, Plant J., 2003, 34, 293–306 CrossRef CAS.
- S. Guo, L. E. Stolz, S. M. Lemrow and J. D. York, J. Biol. Chem., 1999, 274, 12990–12995 CrossRef CAS.
- J. C. Whisstock, F. Wiradjaja, J. E. Waters and R. Gurung, IUBMB Life, 2002, 53, 15–23 Search PubMed.
- W. E. Hughes, R. Woscholski, F. T. Cooke, R. S. Patrick, S. K. Dove, N. Q. McDonald and P. J. Parker, J. Biol. Chem., 2000, 275, 801–808 CrossRef CAS.
- C. Y. Chow, Y. Zhang, J. J. Dowling, N. Jin, M. Adamska, K. Shiga, K. Szigeti, M. E. Shy, J. Li, X. Zhang, J. R. Lupski, L. S. Weisman and M. H. Meisler, Nature, 2007, 448, 68–72 CrossRef CAS.
- O. Cremona, G. Di Paolo, M. R. Wenk, A. Luthi, W. T. Kim, K. Takei, L. Daniell, Y. Nemoto, S. B. Shears, R. A. Flavell, D. A. McCormick and P. De Camilli, Cell, 1999, 99, 179–188 CrossRef CAS.
- R. M. Perera, R. Zoncu, L. Lucast, P. De Camilli and D. Toomre, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19332–19337 CrossRef CAS.
- S. Y. Lee, M. R. Wenk, Y. Kim, A. C. Nairn and P. De Camilli, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 546–551 CrossRef CAS.
- P. Novick, B. C. Osmond and D. Botstein, Genetics, 1989, 121, 659–674 CAS.
- A. E. Cleves, P. J. Novick and V. A. Bankaitis, J. Cell Biol., 1989, 109, 2939–2950 CrossRef CAS.
- E. A. Whitters, A. E. Cleves, T. P. McGee, H. B. Skinner and V. A. Bankaitis, J. Cell Biol., 1993, 122, 79–94 CrossRef CAS.
- G. Konrad, T. Schlecker, F. Faulhammer and P. Mayinger, J. Biol. Chem., 2002, 277, 10547–10554 CrossRef CAS.
- K. U. Kochendorfer, A. R. Then, B. G. Kearns, V. A. Bankaitis and P. Mayinger, EMBO J., 1999, 18, 1506–1515 CrossRef CAS.
- H. M. Rohde, F. Y. Cheong, G. Konrad, K. Paiha, P. Mayinger and G. Boehmelt, J. Biol. Chem., 2003, 278, 52689–52699 CrossRef CAS.
- A. Blagoveshchenskaya, F. Y. Cheong, H. M. Rohde, G. Glover, A. Knodler, T. Nicolson, G. Boehmelt and P. Mayinger, J. Cell Biol., 2008, 180, 803–812 CrossRef CAS.
- Y. Liu, M. Boukhelifa, E. Tribble, E. Morin-Kensicki, A. Uetrecht, J. E. Bear and V. A. Bankaitis, Mol. Biol. Cell, 2008, 19, 3080–3096 CrossRef CAS.
- Y. Nemoto, B. G. Kearns, M. R. Wenk, H. Chen, K. Mori, J. G. Alb Jr, P. De Camilli and V. A. Bankaitis, J. Biol. Chem., 2000, 275, 34293–34305 CrossRef CAS.
- F. Faulhammer, G. Konrad, B. Brankatschk, S. Tahirovic, A. Knodler and P. Mayinger, J. Cell Biol., 2005, 168, 185–191 CrossRef CAS.
- M. A. De Matteis, A. Di Campli and A. Godi, Biochim. Biophys. Acta, 2005, 1744, 396–405 CAS.
- F. Faulhammer, S. Kanjilal-Kolar, A. Knodler, J. Lo, Y. Lee, G. Konrad and P. Mayinger, Traffic, 2007, 8, 1554–1567 CrossRef CAS.
- L. Demmel, M. Beck, C. Klose, A. L. Schlaitz, Y. Gloor, P. P. Hsu, J. Havlis, A. Shevchenko, E. Krause, Y. Kalaidzidis and C. Walch-Solimena, Mol. Biol. Cell, 2008, 19, 1046–1061 CAS.
- K. B. Hendricks, B. Q. Wang, E. A. Schnieders and J. Thorner, Nat. Cell Biol., 1999, 1, 234–241 CrossRef CAS.
- T. Strahl, H. Hama, D. B. DeWald and J. Thorner, J. Cell Biol., 2005, 171, 967–979 CrossRef CAS.
- H. H. Sitte, H. Farhan and J. A. Javitch, Mol. Interventions, 2004, 4, 38–47 CrossRef CAS.
- E. Fernandez-Sanchez, F. J. Diez-Guerra, B. Cubelos, C. Gimenez and F. Zafra, Biochem. J., 2008, 409, 669–681 CrossRef CAS.
- H. Farhan, V. Reiterer, V. M. Korkhov, J. A. Schmid, M. Freissmuth and H. H. Sitte, J. Biol. Chem., 2007, 282, 7679–7689 CAS.
- H. Farhan, M. Freissmuth and H. H. Sitte, Handb. Exp. Pharmacol., 2006, 233–249 Search PubMed.
- H. Cha and P. Shapiro, J. Cell Biol., 2001, 153, 1355–1367 CrossRef CAS.
- A. Colanzi, C. Sutterlin and V. Malhotra, J. Cell Biol., 2003, 161, 27–32 CrossRef CAS.
- T. G. Bivona and M. R. Philips, Curr. Opin. Cell Biol., 2003, 15, 136–142 CrossRef CAS.
- M. Loesch and G. Chen, Front. Biosci., 2008, 13, 3581–3593 CrossRef CAS.
- S. Torii, M. Kusakabe, T. Yamamoto, M. Maekawa and E. Nishida, Dev. Cell, 2004, 7, 33–44 CrossRef CAS.
- O. Rausch and C. J. Marshall, J. Biol. Chem., 1999, 274, 4096–4105 CrossRef CAS.
- G. Chen, M. Hitomi, J. Han and D. W. Stacey, J. Biol. Chem., 2000, 275, 38973–38980 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |