Inhibitory effect of kinetin riboside in human heptamoa, HepG2
Jane
Cheong
a,
David
Goh
a,
Jean
Wan Hong Yong
b,
Swee
Ngin Tan
b and
Eng
Shi Ong
*c
aApplied Science School, Temasek Polytechnic, 21 Tampinese Avenue 1, Singapore, 529757, Republic of Singapore
bNatural Sciences and Science Education Academic Group, Nanyang Technological University, 1 Nanyang Walk, Singapore, 637616, Singapore
cDepartment of Community, Occupational and Family Medicine, National University of Singapore, 16, Medical Drive, Singapore, 117597. E-mail: cofoes@nus.edu.sg
First published on 25th November 2008
Abstract
Cytokinins ribosides such as kinetin riboside are a class of plant hormone that were first identified as factors that promote cell division and have since been implicated in many other aspects of plant growth and development. From the data obtained from cell cycle analysis with flow cytometry, the in vitro growth inhibition of human heptamoa, HepG2 cells with kinetin riboside was mediated by causing G2/M cell cycle arrest and cell death. At the same time, treatment with various doses of kinetin riboside in HepG2 cells did not result in a population of cells positive for the active caspase 3. Differentially expressed proteins in the mitochondria of HepG2 cells with cell death induced by kinetin riboside were investigated. Without the use of stable isotope labeling, the proposed method using LC/MSMS provided a rapid approach to study the differentially expressed proteins in the mitochondria due to the cell death induced by kinetin riboside in HepG2 cells. The ability of kinetin riboside to induce cell death and attenuate G1 to S transition is probably a consequence of its ability to interfere with several components in the mitochondria. Hence, it was proposed that the cell death caused by kinetin riboside in HepG2 cells affected a network of proteins involved in cell death and electron transport.
Introduction
Cytokinins, N6-substituted adenine derivatives, are a class of plant hormones that were first identified as factors that promoted cell division and have since been implicated in many other aspects of plant growth and development including shoot initiation and growth, apical dominance, senescence, and photomorphogenic development.1,2 The effects of various adenine analogues on the growth and differentiation of human myeloid leukemia HL-60 cells were examined. Kinetin riboside (Fig. 1A) was observed to be one of most potent cytokinins investigated for growth inhibition and apoptosis.3 Kinetin ribosides were shown to act on mouse and human tumor cells such as M4 Beu human and B16 murine melanoma cells at low concentration.4Kinetin (free base and riboside), which was assumed by many scientists to be a synthetic 3 cytokininplant growth hormone, has been detected for the first time in the endosperm 4 liquid of fresh young coconut fruits (“coconut water”) with a LC/MSMS method.5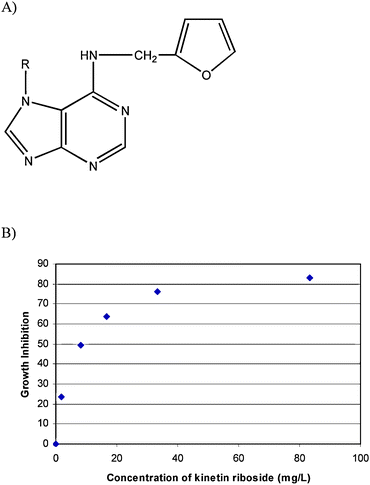 | ||
| Fig. 1 (A) Chemical structure of kinetin riboside, R: β-D-ribofuranosyl and (B) growth inhibitory effect of kinetin riboside on HepG2 human liver cancer cells. Cells were treated with different concentrations of kinetin riboside for 2 days when the cell viability was determined by the MTT assay. The growth inhibition was calculated as percentage of inhibition compared with the control. | ||
The mitochondria have long been considered to play a straightforward but critical role in the life of the cell and as a key regulator of mammalian apoptotic cell death. They carry out energy oxidative reactions that create the vast majority of ATP necessary to support all cellular functions. Interruption of this mitochondrial function in vivo leads to death. In the mitochondria two main pathways, the intrinsic and extrinsic pathways, can lead to apoptosis. The extrinsic pathway involves a death receptorprotein and an adapter protein. This in turn interacts with the cysteine aspartate protease pro-caspase 8 and often pro-caspase 10. Activation of caspase 8 culminates in the activation of other executioner caspases such as caspase 3, 6 and 7. The intrinsic pathway directly releases soluble proteins contained in the mitochondria inter-membrane space. These molecules include cytochrome c, apoptosis-inducing factors (AIF), Smac/DIABLO and endonuclease G (EndoG).6–9 Another apoptotic gene, death associated protein 3, localized to the mitochondria is involved in the process of mitochondrial fragmentation during cell death.10 With proteomic analysis, mitochondrial proteins that may hold the key to the mechanisms by which copper-zinc superoxide dismutase (SOD1) mutants cause mitochondrial dysfunction and neuronal death were investigated.11
Proteomic analysis often involves the identification and quantification of expressed protein components in cells, tissue and organisms. It is a useful tool in investigating biological events as it provides significant information about the relevant gene products and how their levels and modifications change in response to the effects of various internal and external factors. The quantitative profiling of tryptic digest of proteins in complex mixtures without isotope labeling using liquid chromatography and mass spectrometry had been reported. The expected and calculated protein ratios differed no more than 16%.12 An approach using proteolytic digest with two dimensional liquid chromatography nanospray mass spectrometry without the use of stable isotope was applied for the study of differential protein expression in epidermal cell lines grown in the presence or absence of epidermal growth factor.13 In our laboratory, a method using proteolytic digest with two-dimensional liquid chromatography with tandem mass spectrometry without the use of stable isotope was used to identify differentially expressed proteins in human liver cancer cell lines (HepG2) and colon cancer cell lines, LoVo in response to the standardized extract from Scutellariae radix14 and Scutellaria barbata.15 At the same time, the method was applied for the profiling of differentially expressed proteins of mouse liver in the control group and groups treated with standardized extract from Scutellariae radix.16 The proposed method was able to identify changes at the molecular level and have satisfactory level of reproducibility.14–17 Currently, other label free protein quantitation with LC/MSMS from complex proteomes had been reported.18–20
In this work, we will study the inhibitory effects of kinetin riboside in human heptamoa, HepG2. The mitochondrial proteins obtained from the control and treated cells were digested with trypsin and the peptides were separated by reversed phase liquid chromatography tandem mass spectrometry (LC/MSMS). For the current method, labeling of cells with stable isotope would not used. The method developed allowed the molecular mechanism due to the inhibitory effects of the kinetin riboside to be investigated. The differentially expressed proteins identified using the current method will be used to correlate with cell viability assay, cell cycle analysis and activation of caspase 3 with flow cytometry.
Experimental
Chemicals and reagents
Dulbecco’s modified Eagles medium (DMEM), penicillin, streptomycin, trypsin–EDTA were obtained from Hyclone (Logan, Utah, USA). Fetal bovine serum (FBS) was obtained from Biological Industries (Israel). Dimethyl sulfoxide (DMSO), methanol and acetonitrile of HPLC grade were purchased from APS (NSW, Australia). Ultra pure water was obtained from Millipore Alpha-Q water system (Bedford, MA, USA). Sequencing grade modified trypsin was purchased from Promega (Madison, WI). Formic acid and ammonium acetate were purchased from Merck (Darmstadt, Germany). Kinetin riboside (>99.0% purity, MW: 347.33) was purchased from Olchemim (Olomouc, Czech).Cell cultures
All cell lines were obtained from ATCC. Human Hepatoma HepG2 was maintained with Eagle’s minimum essential medium with Earle’s BSS and 2 mM L-glutamine (EMEM) that was modified to contain 1 mM sodium pyruvate, 0.1 mM nonessential amino acids and 1.5 g l−1sodium bicarbonate and supplemented with 10% fetal bovine serum (FBS), 100 U ml−1 Penicillin, 100 μg ml−1 Stretomycin, incubated at 37 °C, and 5% CO2 atmosphere. Initially, 10 mg of kinetin riboside was dissolved in 10 ml of water to form stock solution A.Cell viability with MTT assay
HepG2 cells were seeded at a concentration of 5 × 104 cells ml−1 in a 96 well plate. After overnight incubation, serial concentrations of kinetin riboside were added. Each concentration was repeated three times. These cells were incubated in a humidified atmosphere with 5% CO2 for 2 days. Then, 20 μl MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) (Sigma) solution (4.14 mg ml−1) was added to each well and incubated at 37 °C for 4 h. The medium was removed and formazan was dissolved in DMSO and the optical density was measured at 590 nm using a Bio-assay reader (BioRad, USA). The growth inhibition was determined using: growth inhibition = (control O.D. − sample O.D.)/control O.D.Flow cytometry analysis
A total of 1 × 106 cells were plated on 75 cm2 tissue culture flask and incubated for 24 h at 37 °C. Cells were treated with extracts from kinetin riboside or control with medium for another 48 h. Cells were then washed, pelleted, fixed with cold 70% ethanol for at least 30 min. Before analysis, the 70% ethanol was removed by spinning at 2000 rpm and washed with phosphate saline buffer (PBS). The resulting solution was incubated with 100 μg ml−1Rnase A and 50 μg ml−1 of propidium iodide at room temperature for 30 min. Samples were immediately analyzed by flow cytometry (Becton Dickinson, San Jose, CA). Cell cycle distribution was determined using Modfit software (Verify Software House, Topsham, ME).Flow cytometric analysis of active caspase 3
A total of 1 × 105 of HepG2 cells were plated on 25 cm2 tissue culture flask and incubated for 24 h at 37 °C. Cells were treated with kinetin riboside or control with medium for another 48 h. The study of the activation of caspase 3 was done using a kit available from BD Biosciences (Becton Dickinson, San Jose, CA). Briefly, the cells were washed, pelleted, thoroughly resuspended with 250 μl of BD Cytofix/Cytoperm solution and incubated for 20 min at 4 °C. The cells (1 × 106 cells) were then washed 2 times in a buffer containing a cell permeabilizing agent such as saponin (BD Perm/Wash™ buffer). Finally, the cells were thoroughly resuspend fixed/permeabilized in 50 μl of BD Perm/Wash™ buffer containing the anti active caspase 3 monoclonal antibodies mAb (C92-605). The cells were allowed to incubate at room temperature for 60 min in the dark. Following incubation, cells were washed with BD Perm/Wash™ buffer, resuspended in BD Perm/Wash™ buffer and analyzed by flow cytometry. For positive control, HepG2 cells were treated with 1 μM staurosporine and 2 μM of camptothecin for 48 h.Mitochondrial protein extraction and protein analysis
A total of 1 × 105 cells were plated on a 75 cm2 tissue culture flasks and incubated for 24 h at 37 °C under 5% CO2. Cells were treated with kinetin riboside (8.33 and 16.67 mg l−1) or control with medium for another 48 h. The mitochondriaproteins were isolated using a kit from Pierce (Rockford, IL). Cells were trypsinized and pelleted at 2000 rpm for 5 min. The supernatant was discarded and 1 ml phosphate buffered saline (PBS) was used to reconstitute the pellet. It was centrifuged at 2000 rpm for another 5 min and the supernatant was removed. Proteins were extracted with 800 μl reagent A and vortexed at medium speed for 5 s. It was incubated on ice for 2 min. 10 μl reagent B was added and vortexed at maximum speed for 5 s. The tube was incubated on ice for 5 min and vortexed at maximum speed every minute. 800 μl Reagent C was added and the tube was inverted several times to mix. It was then centrifuged at 2500 rpm for 10 min at 4 °C. The supernatant was pelleted at 9500 rpm for 15 min at 4 °C. 500 μl Reagent C was added to the pellet and centrifuged at 9500 rpm for another 5 min. The supernatant was discarded and 0.5 ml of M-Per (Pierce, Rockford, IL) was added to reconstitute the pellet. Protein concentration was assayed using Bradford assay reagent (Pierce, Rockford, IL).The proteins were reduced with DTT (3 μl of 1000 mM in water). The mixture was incubated at 37 °C for 30 min. To alkylate the protein, iodoacetamide (7 μl of 1000 mM in 0.1 M KOH) was added and the mixture was incubated at room temperature for an additional 30 min in the dark. DTT (13 μl of 1000 mM in water) was added to react with excess iodoacetamide. The reduced and alkylated proteins were digested with sequencing grade trypsin (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) for 18 h.
:50) for 18 h.
For C-18 SPE, each 500 mg Strata (Phenomenex, USA) C-18 SPE column was conditioned with 10 ml of methanol, water and water with 0.1% acetic acid before loading the enzymatically digested samples. The loaded SPE columns were washed with 5 ml of water with 0.1% formic acid before eluting with 0.5 ml acetonitrile. Excess solvent for each of the fractions collected was evaporated in a speedvac. All fractions were reconstituted in 100 μl of water with 0.1% acetic acid prior to analysis by LC/MSMS.
Reversed phase HPLC/MSMS analysis for protein expression
For LC/MSMS experiments, an Agilent 1100 series (Waldbronn, Germany) equipped with a quaternary gradient pump, autosampler with sample cooler, column oven and diode array detector was coupled with a Agilent LC/MSD Trap VL ion trap mass spectrometer (Waldbronn, Germany). The gradient elution involved a mobile phase consisting of (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid. The initial condition was set at 5% of (B), gradient up to 40% in 70 min, up to 90% in the next 5 min and returning to initial condition for 15 min. Oven temperature was set at 40 °C and flow rate was set at 200 μl min−1. For all experiments, 30 μl of sample was injected. The column used for the separation was a reversed phase C18 Jupiter, 150 × 2.0 mm, 5 μ, 300 Å (Phenomenex, USA). The ESI-MS was acquired in the positive ion mode. The scanning mass range was from 400 to 1500. The heated capillary temperature was maintained at 350 °C, the drying gas and nebulizer nitrogen gas flow rates were 10 l min−1 and 50 psi, respectively. Data were acquired using automated MSMS. The target was set at 30000, maximum accumulation time: 300 ms, the number of average scans was 5 and SmartSelect™ was used.Database searching procedure of MS/MS data for protein identification
Mass data collected during a LC/MSMS run were submitted to the search software Mascot (http://www.matrixscience.com). Preliminary protein identifications were obtained by comparing experimental data to NCBInr database and Swiss-prot. The taxonomy was set to Homo sapien (human), 1 missed cleavage was allowed and carbamidomethyl was selected for fixed modifications. Peptide charge was set at 2+ and 3+. Searches were done with a tolerance on mass measurement of 1.0 Da in MS mode. From the searches obtained from Mascot, only the first five hits were considered. For MS/MS ion search, proteins with one peptide ion scoring higher than 45 or two peptide ions scoring higher than 30 were considered an unambiguous identification without manual inspection.A sequence tag of several continuous amino acids (5–20 residues) and the peptide mass were generally sufficient to identify the protein of a peptide. The sub-cellular location and tissue specificity of the proteins identified was examined using Swiss-prot. The raw data were inspected manually for confirmation prior to acceptance.
Results
Growth inhibition of kinetin ribosides on human liver cancer cell lines
The effect of different doses of kinetin riboside on cell growth was examined on HepG2 cell lines. Under the experimental conditions used with 48 h treatment, kinetin riboside exhibited a marked growth inhibitory effect on HepG2 in a dose-dependent manner (Fig. 1B). The IC50 was approximately 8.33 ± 0.73 mg l−1 for HepG2. A significant cell shrinkage and change in cell shape was observed in HepG2 cells treated with a higher concentration of kinetin riboside.The effects of kinetin riboside on cell proliferation may be due to cell cycle regulation, we examined the effect on cell cycle perturbation using flow cytometric analysis. It was observed that kinetin ribosides at 1.67 mg l−1 did not induce a mark increase sub-G0 phase in HepG2 cells. However, different doses of kinetin riboside were observed to decrease the population of G1 phase and an increased in the G2/M phase after 48 h. At the same time, a marked increase in sub-G0 phase was observed in a dose dependent manner (Fig. 2). The results were repeated on a different day and a similar trend was observed. The results showed that kinetin riboside induced in vitro growth inhibition of HepG2 cells was mediated by causing G2/M cell cycle arrest and cell death.
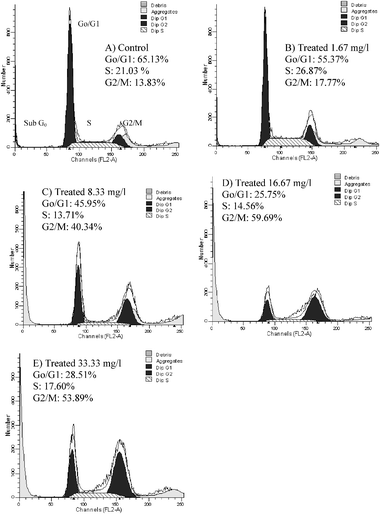 | ||
| Fig. 2 Cell cycle analysis by flow cytometry. The HepG2 cells lines were (A) treated with medium for control, (B) treated with 1.67 mg l−1 of kinetin riboside (C) treated with 8.33 mg l−1 of kinetin riboside, (D) treated with 16.67 mg l−1 of kinetin riboside and (E) treated with 33.33 mg l−1 of kinetin riboside for 48 h. The significant increase in the sub G0 phase in C, D and E is indicative of cell death. | ||
Treatment with different doses of kinetin riboside for 48 h did not result in induced capase-3 activity in HepG2 cells (Fig. 3). From Fig. 3A, the untreated HepG2 cells were primarily negative for active caspase 3. The different doses of kinetin riboside did not result in any activation of active caspase 3 (Fig. 3B, C, D and E). As a positive control, human liver cells treated with 1 μM staurosporine that will result in the activation of active caspase 3 was used.21 The determination of caspase 3/7 activities in HepG2 cells treated for 24 h with butyric acid, carbonyl cyanide 4 (trifluoromethoxy) phenylhydrazone and camptothecin was reported.22 By comparing the results obtained with the positive control, the different doses of kinetin ribosides did not result in a significant activation of active caspase 3. For the positive control (Fig. 3F), activation of caspase 3 can be seen in the changes of the profile of the stained and unstained HepG2 cells. We propose that HepG2 cells treated with different doses of kinetin ribosides had resulted in a significant degree of DNA fragmentation and cell death. At the same time, the various doses used did not result in a population of cells positive for the active caspase 3.
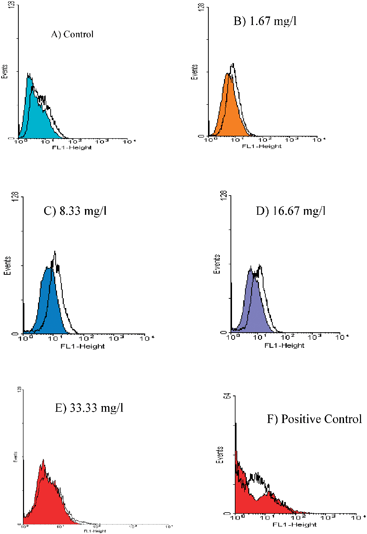 | ||
| Fig. 3 Flow cytometric analysis of active caspase 3 in HepG2 cells using the anti-active caspase 3 mAb (clone C92-605). Stained and unstained HepG2 cells were (A) left untreated (control), (B) treated with 1.67 mg l−1 of kinetin riboside, (C) treated with 8.33 mg l−1 of kinetin riboside, (D) treated with 16.67 mg l−1 of kinetin riboside, (E) treated with 33.33 mg l−1 of kinetin riboside and (F) stained and unstained HepG2 cells treated with 1 μM of staurosporine for 48 h (positive control). The untreated HepG2 cells were primarily negative for active caspase 3. The higher doses kinetin riboside (4.0 and 24.0 mg l−1) did not result in a significant activation of active caspase 3. | ||
Effect of kinetin riboside on cell mitochondria protein expression by HPLC/MSMS
Using reversed phase LC/MSMS, we next assessed the effect of kinetin riboside on the expression of mitochondria proteins that may be involved in the control of cellular proliferation. The reproducibility of the proposed method was validated by profiling the mitochondriaproteins from 3 different untreated HepG2 cells (Fig. 4A, B and C). By superimposing the chromatograms (TIC) from the all three different untreated groups, it was found that good reproducibility was obtained from cell lysates from 3 different flasks. The experiments were repeated on a different day with a different analyst and a similar reproducibility was observed. Using the current LC/MSMS approach, however, higher degree of variation of the chromatographic profile was observed from cell lysates obtained from different days. The method variation on the same day was estimated to be less than 20% and our data showed the feasibility of profiling the tryptic digest of the mitochondriaproteins without the use of isotope labeling. From the data obtained in Fig. 4, it was clear that by superimposing the different chromatograms obtained, it was possible to identify differentially expressed peptides from the mitochondriaproteins.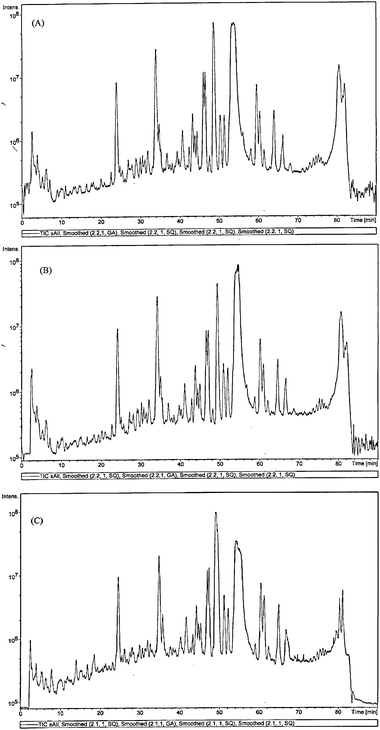 | ||
| Fig. 4 Chromatograms (TIC) from LCMSMS of three different cell lysates of the mitochondriaproteins from untreated Hep G2 cell line . With the assistance of the software, by superimposing the chromatograms (TIC) from the all the different untreated groups it was found that good reproducibility was obtained from cell lysates from 3 different flasks. | ||
The chromatograms in Fig. 5A and B showed the tryptic digest of two different cell lysates with reversed phase HPLC/MSMS from the HepG2 cell lines for the control and cell line treated with 16.67 mg l−1 of kinetin riboside, respectively. By superimposing the chromatograms (TICs) from Fig. 5A and B, a number of peptides that remained unchanged in the control and treated group were obtained. It was proposed that they were likely to be house keeping proteins as observed in our earlier works.14–16 With the assistance of peptides from house keeping proteins, the identification of peptides from proteins where their expression had been modified can be obtained. Hence, a list of mitochondria proteins (Table 1) where the expression were changed by more than 2 fold after treatment with 16.67 mg l−1 of kinetin riboside was identified using tandem mass spectrometry. At the same time, we did not observe significant changes in the mitochondriaprotein expression for HepG2 cells treated with 8.33 mg l−1 of kinetin riboside. The criteria was selected as it was observed that the proposed method would not induce variation higher than 2 fold (peak reduction or increased by two times).
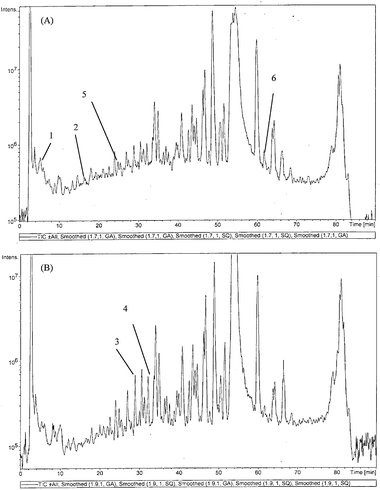 | ||
| Fig. 5 Chromatograms (TIC) from LCMSMS of mitochondria proteins from (A) HepG2 cell line , control with medium, (B) HepG2 cell line , treated with 16.67 mg l−1 of kinetin riboside for 48 h. The box regions are where differential expressed peptides (up-regulated or down-regulated more than two times) were observed. Peak 1–6 is as labeled in Table 1. | ||
| Accession | Mass/Da | Description | Up/down regulated | Proposed functions | |
|---|---|---|---|---|---|
| 1 | P49448 | 61434 |
Glutamate dehydrogenase [NAD (P)] | Down regulated | Molecular function: electron transporter activity; glutamate dehydrogenase activity. |
| Biological process: glutamate catabolism. | |||||
| 2 | Q92934 | 18392 |
Bcl2-antagonist of cell death | Down regulated | Molecular function: protein binding. |
| Biological process: apoptotic program; and induction of apoptosis | |||||
| 3 | Q9UNM1 | 10295 |
Chaperonin 10-related protein [Fragment] | Up regulated | Molecular function: ATP binding, unfolded protein binding. |
| Biological process: protein folding. | |||||
| 4 | Q5KTR4 | 111651 |
Flavoprotein oxidoreductase | Up regulated | Molecular function: oxidoreductase activity; zinc ion binding |
| Biological processs: electron transport; and metabolism | |||||
| 5 | P40939 | 83000 |
Trifunctional enzyme alpha subunit, mitochondrial [Precursor] | Down regulated | Molecular function: 3-hydroxylacyl-CoA dehydrogenase activity; acetyl-CoA C-acetyltransferase activity; and enoyl-CoA hydratase activity. |
| Biological function: lipid metabolism, metabolism and fatty acid metabolism | |||||
| 6 | Q5SZ02 | 19762 |
Mitochondrial ribosomal protein L24 [Fragment] | Down regulated | Molecular function: structural constituent of ribosome |
| Biological process: proteinbiosynthesis. |
The current approach of identification of proteins was based on our earlier works14–17 and other reports.11,13,23–28 The potential for false positive identifications from large databases through tandem mass spectra data had been discussed.28,29 Hence, for the searches obtained from Mascot, websites such as Swiss-prot (http://us.expasy.org/sprot) and NCBInr database (http://www.ncbi.nlm.nih.gov) were used to examine domains and motifs present in identified proteins. In this work, the proteins identified must be present in the mitochondria and the tissue specificity was examined using Swiss-prot.
Discussion
Cytokinin ribosides such as kinetin riboside, isopentenyladenosine and benzyl-aminopurine were found to be the most potent for growth inhibition and apoptosis in human myeloid leukemia cells (HL-60). Cytokinin ribosides greatly reduced the intracellularATP content and disturbed the mitochondrial membrane potential and accumulation of reactive oxygen species, whereas cytokinin did not.3 The growth inhibition and induction of cell death of kinetin ribosides in HepG2 in this study was consistent with other works on other cell lines.3,4Active caspase 3 is a marker for cells undergoing apoptosis.30,31 Caspases were originally identified as the mediators of apoptosis where it was hypothesized that many of their substrates were essential proteins whose destruction ensured the inevitability of cell death. However, caspase-independent cell death is observed in many systems indicating that cells still die even if the executioner is absent.30,32–35Cell death induced by 5-hydroxyphenyl butanoate retinamide in human cancer cell lines such as HCT116 and MCF7 did not result in cleavages of caspase 3 and 8. However, cleavages of caspases 3 and 8 were evident in PLC/PRF/5 and CaSki cells.36Activation of caspase 3 was observed within 6 h after treatment of HL-60 cells with kinetin riboside.3 For HepG2 cells, treatment with different doses of kinetin riboside had resulted in cell death but not the activation of active caspase 3.
Mitochondria are an important part of the apoptotic machinery. The mechanisms of action of chemically diverse small molecules on specific mitochondria loci such as respiratory chain, DNA biogenesis, potassium channels, the Bcl-2 family proteins and the permeability transition pores were well reported.9,37 Mitochondrial damage promotes apoptosis in two ways. On the one hand, it leads to the release of apoptogenic factors and on the other hand, it disrupts energy production of the cell. The ability of kinetin riboside to induce cell death and attenuate G1 to S transition is probably a consequence of its ability to interfere with several components in the mitochondria.
Genetic and biochemical studies have demonstrated that Bcl-2 family proteins are central to the regulation of mitochondria membrane permeabilization. The Bcl-2 family proteins localize or can be targeted to the mitochondria and regulate the permeability of the outer membrane to various apoptotic factors.8,9,35 The identification of the Bcl2-antagonist of cell deathprotein with the current method was consistent with all the data obtained from cell viability assay and cell cycle analysis. It is located at the outer mitochondrial membrane, upon phosphorylation, it locates to the cytoplasm. The Bcl2-antagonist of cell deathprotein encoded by this gene is a member of the Bcl-2 family. This protein positively regulates cell apoptosis by forming heterodimers with Bcl-xL and Bcl-2, and reversing their death repressor activity. Proapoptotic activity of this protein is regulated through its phosphorylation. Protein kinases AKT and MAP kinase, as well as protein phosphatase calcineurin were found to be involved in the regulation of this protein.38
Heat shock proteins (HSP) are the products of several distinct gene families that are required for cell survival during stress.39 The significances of chaperonin 10-mediated inhibition of ATP hydrolysis by chaperonin 60 was reported.40 Recent works have proposed the anti-apoptotic role of two heat shock proteins, Hsp10 and Hsp60 in various cells. These two proteins can be induced when cells are under stress. An over-expression of Hsp10 and Hsp60 differentially modulated the Bcl-2 family and in turn attenuate doxorubicin induced apoptosis in primary cardiomyocytes.41 Over-expression of Hsp10 by adenoviral infection decreased myocyte death induced by hydrogen peroxide, sodium cyanide, simulated ischemia and reoxygenation.42 The identification of Chaperonin 10-related protein in HepG2 cells treated with kinetin riboside was consistent with the proposition of cell death. The up-regulation of Chaperonin 10-related protein and down-regulation of Bcl2-antagonist of cell death suggested a possible link between the two proteins.
Electron transport is carried out in the mitochondrial inner membrane through a series of membrane-embedded proteins that communicate via several smaller molecules, the lipid-soluble ubiquinone and the water soluble proteincytochrome c.6 In our current work, the growth inhibition of HepG2 cells with kinetin riboside resulted in proteins such as glutamate dehydrogenase, flavoproteinoxidoreductase and trifunctional enzyme alpha subunit, mitochondrial [Precursor] that were involved in electron transport and metabolism to be differentially expressed. For trifunctional enzyme alpha subunit, mitochondrial [Precursor], it encodes the alpha and beta subunits of the mitochondrial trifunctional protein, respectively. The heterocomplex contains 4 alpha and 4 beta subunits and catalyzes 3 steps in mitochondrial beta-oxidation of fatty acids, including the long-chain 3-hydroxyl-CoA dehydrogenase (LCHAD) step. The alpha subunit harbors the 3-hydroxyacyl-CoA dehydrogenase and enoyl-CoA hydratase activities.38 Lastly, cell death induced by kinetin riboside had affected the mitochondrial ribosomal protein L24 [Fragment] that was involved in proteinbiosynthesis.
Conclusions
This paper reports our current data on the inhibitory effects of kinetin ribosides in human cancer cell lines. Together with other techniques, the current method allows us to study the differentially expressed proteins in the mitochondria due to the inhibitory effects of different doses of kinetin riboside in human liver cancer cells, HepG2. The data obtained from cell cycle analysis with flow cytometry provides some insights for the proteins that may be differentially expressed in HepG2 cell lines treated with kinetin riboside.13,14 In our earlier work, we did not identify any differentially expressed proteins that were associated with signal transduction, cell cycle and cell death when significant changes in sub G0, G0/G1, S and G2/M phases were not observed in cell cycle analysis with flow cytometry.13Without the use of stable isotope labeling, the proposed method provided a rapid approach to study the molecular mechanism due to the inhibitory effects of different doses of kinetin riboside on HepG2 cell lines. The differentially expressed proteins identified in the mitochondria were consistent with what was obtained from cell cycle analysis with flow cytometry. It provided further information on how the mitochondria play a critical role in the life of the cell and as a key regulator of mammalian apoptotic cell death. The cell death caused by kinetin riboside in HepG2 cells did not result in an activation of active caspase 3 and affected a network of proteins involved in cell death and electron transport.
References
- W. C. Evans, Trease and Evans’ Pharmacognosy, Harcourt Publisher, London, 15th edn, 2002 Search PubMed.
- I. B. D’Agostino and J. J. Kieber, Curr. Opin. Plant Biol., 1999, 2, 359–364 CrossRef CAS.
- Y. Ishii, Y. Hori, S. Sakai and Y. Honma, Cell Growth Diff., 2002, 13, 19–26 Search PubMed.
- B. Griffaut, R. Bos, J.-D. Maurizis, J.-D. Madelmont and G. Ledoigt, Int. J. Biol. Macromol., 2004, 34, 271–275 CrossRef CAS.
- L. Ge, J. W. H. Yong, N. K. Goh, L. S. Chia, S. N. Tan and E. S. Ong, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 829, 26–34 CrossRef CAS.
- D. D. Newmeyer and S. Ferguson-Miller, Cell, 2003, 112, 481–490 CrossRef CAS.
- L. E. Broker, F. A. E. Kruyt and G. Giaccone, Clin. Cancer Res., 2005, 11(9), 3155–3162 CrossRef.
- D. G. Breckenridge and D. Xue, Curr. Opin. Cell Biol., 2004, 16, 647–652 CrossRef CAS.
- N. N. Danial and S. J. Korsmeyer, Cell, 2004, 116, 205–219 CrossRef CAS.
- Z. Mukamel and A. Kimchi, J. Biol. Chem., 2004, 279(35), 36732–36738 CrossRef CAS.
- K. Fukada, F. Zhang, A. Vien, N. R. Cashman and H. Zhu, Mol. Cell. Proteomics, 2004, 3, 1211–1223 CrossRef CAS.
- D. Chelius and P. V. Bondarenko, J. Proteome Res., 2002, 1, 317–323 CrossRef CAS.
- D. Chelius, T. Zhang, G. Wang and R. F. Shen, Anal. Chem., 2003, 75, 6658–6665 CrossRef CAS.
- E. S. Ong, S. M. Len and A. C. H. Lee, J. Agric. Food Chem., 2005, 3, 8–16 CrossRef.
- D. Goh, Y. H. Lee and E. S. Ong, J. Agric. Food Chem., 2005, 53, 8197–8204 CrossRef CAS.
- E. S. Ong, S. M. Len, A. C. H. Lee, P. Chui and K. F. Chooi, Rapid Commun. Mass Spectrom., 2004, 18, 2522–2530 CrossRef CAS.
- Y. L. Tan, D. Goh and E. S. Ong, Mol. Biosyst., 2006, 2, 250–258 RSC.
- J. Gao, M. S. Friedrichs, A. R. Dongre and G. J. Opiteck, J. Am. Soc. Mass Spectrom., 2005, 16, 1231–1238 CrossRef CAS.
- G. Wang, W. W. Wu, W. Zeng, C. L. Chou and R. F. Shen, J. Proteome Res., 2006, 5, 1214–1223 CrossRef CAS.
- P. R. Cutillas and B. Vanhaesebroeck, Mol. Cell. Proteomics, 2007, 6(9), 1560–1573 CrossRef CAS.
- M. Giuliano, G. Bellavia, M. Lauricella, A. D’Anneo, B. Vassallo, R. Vento and G. Tesoriere, Int. J. Mol. Med., 2004, 13(4), 565–71 Search PubMed.
- S. Miret, E. M. de Groene and W. Klaffke, J. Biomol. Screening, 2006, 11, 184–93 CAS.
- L. J. Foster, C. L. de Hoog and M. Mann, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5813–5818 CrossRef CAS.
- L. Xiong, D. Andrews and F. Regnier, J. Proteome Res., 2003, 2, 618–625 CrossRef CAS.
- T. Natsume, Y. Yaamuchi, H. Nakayama, T. Shinkawa, M. Yanagida, N. Takahsahi and T. Isobe, Anal. Chem., 2002, 74, 4725–4733 CrossRef CAS.
- H. T. Tang, B. R. Halpern, I. V. Shilov, S. L. Seymour, S. P. Keating, A. Loboda, A. A. Patel, D. A. Schaeffer and L. M. Nuwaysir, Anal. Chem., 2005, 77, 3931–3946 CrossRef.
- D. Tsur, S. Tanner, E. Zandi, V. Bafna and P. A. Pevzner, Nat. Biotechnol., 2005, 23, 1562–1567 CrossRef CAS.
- D. J. States, G. S. Omenn, T. W. Blackwell, D. Fermin, J. Eng, D. W. Speicher and S. M. Hanash, Nat. Biotechnol., 2006, 24, 333–338 CrossRef CAS.
- B. J. Cargile, J. L. Bundy and J. L. Stephenson Jr, J. Proteome Res., 2004, 3, 1082–1085 CrossRef CAS.
- N. A. Thornberry and Y. Lazebnik, Science, 1998, 281, 1312–1316 CrossRef CAS.
- C. Dai and S. B. Krantz, Blood, 1999, 33, 3309–3316.
- R. A. Lockshin and Z. Zakeri, Oncogene, 2004, 23, 2766–2773 CrossRef CAS.
- L. L. Broker, F. A. E. Kruyt and G. Giaccone, Clin. Cancer Res., 2005, 11(9), 3155–3162 CrossRef.
- S. Y. Park, S. J. Cho, H. C. Kwon, K. R. Lee, D. K. Rhee and S. Pyo, Cancer Lett., 2005, 224, 123–132 CAS.
- C. Garrido and G. Kroemer, Curr. Opin. Cell Biol., 2004, 16, 639–646 CrossRef CAS.
- H. S. Han, Y. J. Kwon, S. H. Park, E. J. Kim, Y. S. Rho, H. S. Sin and S. J. Um, Int. J. Cancer, 2004, 109, 58–64 CrossRef CAS.
- N. Dias and C. Bailly, Biochem. Pharmacol., 2005, 70, 1–12 CrossRef CAS.
- Swiss-prot, (http://us.expasy.org/sprot/).
- S. K. Calderwood, M. Abdul Khaleque, D. B. Sawyer and D. R. Ciocca, Trends Biochem. Sci., 2006, 31, 164–171 CrossRef CAS.
- Y. Dubaquie, R. Looser and S. Rospert, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(17), 9011–9016 CrossRef CAS.
- K. M. Lin, J. M. Hollander, V. Y. Kao, B. Lin, L. Macpherson and W. H. Dillmann, FASEB J., 2004, 18(9), 1004–1006 CAS.
- Y. X. Shan, T. J. Liu, H. F. Su, A. Samsamshariat, R. Mestril and P. H. Wang, J. Mol. Cell. Cardiol., 2003, 35, 1135–1143 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |