MAC-CCD system: a novel lymphocyte microwell-array chip system equipped with CCD scanner to generate human monoclonal antibodies against influenza virus†
T.
Ozawa
a,
K.
Kinoshita
a,
S.
Kadowaki
a,
K.
Tajiri
ab,
S.
Kondo
a,
R.
Honda
a,
M.
Ikemoto
c,
L.
Piao
c,
A.
Morisato
c,
K.
Fukurotani
d,
H.
Kishi
*a and
A.
Muraguchi
a
aDepartment of Immunology; Graduate School of Medicine and Pharmaceutical Sciences; University of Toyama, 2630 Sugitani, Toyama, 930-0194, Japan. E-mail: immkishi@med.u-toyama.ac.jp; Tel: +81-76-434-7251; Fax: +81-76-434-5019
b3rd Department of Internal Medicine; Graduate School of Medicine and Pharmaceutical Sciences; University of Toyama, 2630 Sugitani, Toyama, 930-0194, Japan
cSC World Inc, 529 Takata, Toyama, 930-0866, Japan
dDepartment of Neural Computation; Graduate School of Science and Engineering; University of Toyama, 3190 Gofuku, Toyama, 930-8555, Japan
First published on 22nd October 2008
Abstract
We previously developed a lymphocyte microwell-array system, which effectively detects antigen-specific B-cells by monitoring intracellular Ca2+ mobilization at the single-cell level with a fluorescent Ca2+ indicator, fluo-4. However, it is difficult for the system to perform time-lapse monitoring. Here, we developed a novel method, a lymphocyte microwell-array chip system equipped with a charge-coupled device (CCD) time-lapse scanner (MAC-CCD system), for monitoring intracellular Ca2+ mobilization. The MAC-CCD system is able to monitor intracellular Ca2+ mobilization of more than 15,000–20,000 individual live B-cells every 10 s. In addition, we adopted a correlation method in a MAC-CCD system, which enabled us to detect B-cells with a frequency of as few as 0.046%. Furthermore, we succeeded in obtaining six influenza nucleoprotein-specific human monoclonal antibodies from the peripheral blood of influenza-vaccinated volunteers. These results demonstrate that the MAC-CCD system with a correlation method could detect very rare antigen-specific B-cells.
Introduction
Following DNA and protein microarrays, cell microarray technology has been emerging for investigating gene expression, cell-surface interactions, extracellular matrix composition, cell migration and proliferation, the effects of drugs on cellular activity, and many other areas.1 In the early studies, adherent cells, but not non-adherent cells, were used because it is difficult to place non-adherent cells such as blood cells at fixed positions on a chip. Recently, others and we have developed a microwell-array for analyzing non-adherent cells on a single-cell basis.2–10 These microwells were designed to capture single cells, thus enabling to track and analyze cellular functions at the single-cell level. Single-cell analysis with a microwell-array is especially useful in studying cells consisting of heterogeneous cell populations, such as blood cells that contain various cell lineages derived from hematopoietic progenitor cells or cells derived from tumor biopsies that consist of normal cells as well as tumor cells.B-cells play an important role in the immune response. Each B-cell clone expresses an antibody as an antigen receptor (B-cell receptor, BCR) with a unique antigen-binding structure on the cell surface. When an individual encounters microbes, B-cells that express antibodies specific to the microbes capture them with BCR. Engagement of BCR with the microbes triggers intracellular signal transduction, which finally initiates the proliferation and differentiation of the B-cells into antibody-secreting cells whose antibodies capture the microbes to get rid of them from the bodies.11,12 In any individual, it is estimated that 107 or more different B-cell clones exist.13 Thus, the B-cell population consists of tremendously heterogeneous cells.
Calcium signaling is important for signal transduction in the growth, death, differentiation, and function of immune cells.11,12,14–17 Measurements of intracellular Ca2+ concentration ([Ca2+]i) are significant for the immunological, physiological, and biological research of immune cells, and can also be used for detecting antigen-specific B-cells.3 In order to detect antigen-specific B-cells in a large number of B-cells with diverse antigen specificity by analyzing [Ca2+]i, it is necessary to measure it on a single-cell basis. To date, intracellular Ca2+ mobilizations in individual cells are monitored by flow cytometry or fluorescence microscopy using a fluorescent Ca2+ indicator. Flow cytometry can rapidly analyze intracellular Ca2+ mobilizations in a large number of cells, but cannot track the time course of Ca2+ mobilizations in individual cells. In contrast, fluorescence microscopy can track the time course of Ca2+ mobilizations in up to 5,000 cells at single cell levels,18 but it is difficult to monitor fluorescence signals of a large number of cells. We have previously reported that microwell-array chips that have an array of 45,000 to 234,000 microwells each of which has a size and shape that just fit a single cell.3,8 We could track the time course of Ca2+ mobilization of a large number of individual cells that were arrayed on a microwell-array chip using a cell scanner.3,8 However, it took 2 min for the scanner to scan all the cells. Because intracellular Ca2+ mobilizations reach their peak within 30 s and then gradually decrease to a basal level in about 5 min, a cell scanner is not ideal for monitoring the time course of Ca2+ mobilization in detail.
Moreover, intracellular Ca2+ mobilization in B-cells is induced not only by BCR-mediated signals but by other stimuli (e.g., stimuli viaryanodine receptors,19,20 or mechanical stress21) as well. We have noticed that [Ca2+]i in a few percent of the B-cell population increases in the absence of antigen stimulation. Such noises made it difficult to detect a rare antigen-specific B-cell population with frequencies of 0.01 to 0.1% in all the B-cells.22–31 To overcome the difficulties, we have developed a novel cell microwell-array chip system equipped with charge-coupled device (CCD) time-lapse scanner (MAC-CCD system). The system can monitor the alteration of [Ca2+]i in individual cells every 10 s. To distinguish the BCR-mediated Ca2+ mobilizations from the non-specific Ca2+ mobilizations, we applied a correlation method32 that is a robust and general technique for pattern recognition. Here we show that the MAC-CCD system with the correlation method is useful for comprehensive analysis of intracellular Ca2+ mobilizations of more than 15,000–20,000 individual cells and demonstrate that the system can detect a rare cell population. This system may contribute to high throughput and comprehensive analysis of a large number of cells on a single-cell basis.
Experimental
System description
MAC-CCD system consists of microwell-array chips and CCD-scanner (Fig. 1). A microwell-array chip consists of 50 to 270 clusters of 900 microwells.3 Each microwell is cylindrical in shape and can accommodate only a single cell. The CCD scanner detects the fluorescence intensity of the individual cells in the microwells every 10 s per scanning point. For the detection of antigen-specific B-cells, cells were loaded with Ca2+ indicator, fluo-4, and applied to a microwell-array chip. Then, cellular fluorescence before (scanning points 1 to 10) and post (scanning points 11 to 40) stimulation with antigen was detected and the fluorescence intensity of individual cells was quantified with software. Cells that responded to antigen were determined with the correlation method. Then, positively detected cells were retrieved with a micromanipulator, and the cDNAs of the antibodies were amplified from each retrieved cell by single-cell 5′-RACE.33Antibody cDNAs were transfected into 293T cells and the antigen specificity of the recombinant antibodies in culture supernatants is examined by ELISA.3 | ||
| Fig. 1 Overview of MAC-CCD system. Protocols for detecting antigen-specific B-cells and producing antigen-specific antibodies with MAC-CCD system is depicted schematically. | ||
Correlation method
Antigen-stimulated B-cells were selected by analyzing the time course of the alteration of fluo-4 fluorescence intensities of individual cells with the correlation method.32 The decision for selecting the B-cells that responded to antigen was performed according to a correlation value (C value). The C value for each cell is calculated by the following equation:
where σi is the normalized fluo-4 fluorescence intensity at the i-th scanning point of a cell of interest and Ti is the average of normalized fluo-4 fluorescence intensity at the i-th scanning point of the cells that responded to antigen. The C value is ≤1. If the C value of a cell of interest is above a preset criterion and close to 1, the observed fluorescence alteration of the cell was matched with that of the template and we selected the cell as a B-cell that responded to antigen.
Materials and methods in the electronic supplementary information
The detailed materials and methods are described in the ESI.†Results and discussion
To evaluate the utility of the MAC-CCD system for analyzing the intracellular Ca2+ mobilization of individual cells, we have employed MD4 transgenic mice whose transgene encodes the HEL-specific antibody (HyHEL10).34B-cells from MD4 mice express HEL-specific antibodies as B-cell receptors on the cell surface. Stimulation of MD4 B-cells with HEL induces a transient increase in intracellular Ca2+ mobilization, which can be monitored with a fluorescent calcium indicator, fluo-4.3 MD4 B-cells or normal B-cells were loaded with fluo-4, applied on the microwell-array chips, and their fluorescence intensities monitored with the MAC-CCD system. The fluorescence intensity of cells in 45,000 wells was obtained in 10 s for a scanning point at single-cell levels. During the initial 100 s (scanning points 1 to 10), cellular fluorescence was monitored without stimulation. The cells were then stimulated with 10 µg/ml HEL on the chip, and thereafter their fluorescence was monitored for an additional 300 s (scanning points 11 to 40). Fluorescence signals of 100 randomly selected MD4 B-cells and normal B-cells are shown in Fig. 2A and B, respectively. Fluorescence alteration of individual cells is demonstrated in Fig. 2C and D Stimulation of MD4 B-cells with HEL induced a steep ascent of fluo-4 fluorescence and followed by a slow descent (Fig. 2A and C). In contrast, we did not observe the corresponding alteration of fluorescence in normal B-cells, but did observe a sharp increase and the following sharp decrease in fluo-4 fluorescence in some cells (Fig. 2B and D). The results confirmed that the MAC-CCD system could distinguish the pattern of fluo-4 fluorescence alteration in antigen-stimulated B-cells and that in unstimulated B-cells. | ||
| Fig. 2 Time-lapse analysis of antigen-induced intracellular Ca2+ alteration with the MAC-CCD system. MD4 B-cells (A, C) or normal B-cells (B, D) on a microwell-array chip were stimulated with 10 µg/ml HEL. Fluo-4 fluorescence intensities of individual cells were monitored with the MAC-CCD system. Fluorescence alterations of 100 representative cells are shown in a graph (A, B) or that of each cell is separately shown in each graph (C, D). Arrowheads indicate the point of stimulation with HEL. The vertical axis indicates fluorescence intensity; the horizontal axis indicates scanning points 1 to 40. | ||
For the selection of antigen-responding B-cells with intracellular Ca2+ mobilization, we have employed a correlation method.32 For preparing the template that shows the typical intracellular Ca2+ mobilization of the B-cells activated with antigen, we used the data of fluo-4 fluorescence intensities of MD4 B-cells that were stimulated with HEL. As shown in Fig. 2A, some MD4 B-cells did not respond to HEL. In order to exclude the data of those non-responding cells, we evaluated the fluorescence data from various points of view. First, we defined the resting intensity (Srest) of each cell as the mean fluo-4 fluorescence intensity before stimulation (at 1 to 5 scanning points). We then defined the maximum intensity (Smax) for each cell as the maximum fluorescence intensity during scanning points 11 to 13 just after stimulation with HEL. Fig. 3A and B show the histogram of Srest and Smax for each cell, respectively. Srest and Smax for the major cell population were 1.79 ± 1.46 and 12.27 ± 8.08, respectively. Next, we calculated the ratio of Smax to Srest (r = Smax/Srest) for each cell, whose histogram is shown in Fig. 3C. The value of r for the major cell population was 5.66 ± 3.82. From these results, we arbitrarily defined the B-cells responding to antigen as those whose Smax is > 4 and r > 2. Furthermore, we excluded the data of the cells whose fluorescence intensity became smaller than that before stimulation, Si <Sresti ≥ 13. 84.6 ± 7.3% of MD4 B-cells stimulated with HEL fell in these criteria. We used the data of those cells for preparing the template. To that end, we normalized the fluorescence data of cells after stimulation as follows:

 | ||
| Fig. 3 Analysis of fluo-4 fluorescence intensities of MD4 B-cells stimulated with HEL and definition of the template for the correlation method. MD4 B-cells on a microwell-array chip were stimulated with HEL and the fluo-4 fluorescence was monitored with the MAC-CCD system. (A) Histogram analysis of fluo-4 fluorescence in MD4 B-cells before stimulation with HEL. The X-axis shows the mean fluorescence intensity of scanning points 1 to 5 (Srest) in logarithmic scale. (B) Histogram analysis of fluo-4 fluorescence in MD4 B-cells after stimulation with HEL. The X-axis shows the maximum fluorescence intensity during scanning points 11 to 13 (Smax) in logarithmic scale. The cutoff value for arbitrary definition of the B-cells responding to HEL is indicated with a dotted line. (C) Histogram analysis of the ratio of Smax to Srest for single MD4 B-cells. The cutoff value for arbitrary definition of the B-cells responding to HEL is indicated with a dotted line. (A-C) Representative data of four separate experiments are shown. (D) Template of intracellular Ca2+ response of HEL-activated MD4 B-cells. The arrow indicates the point of antigen stimulation. The X-axis shows the scanning points and the Y-axis shows the normalized mean fluorescence of HEL-activated MD4 B-cells. The template was prepared from four separate experiments. | ||
and we set the values before stimulation to zero as follows:
σ i = 0, i = 1 to 10.
Then, Ti was defined as the average of σi of all B-cells responding to HEL as follows:

where n is the number of HEL-responding B-cells. Ti was used as the template of the MD4 B-cells for analyzing intracellular Ca2+ response with the correlation method (Fig. 3D).
To evaluate the efficiency of the correlation method for selecting antigen-specific B-cells, we prepared B-cell preparations that contained 100%, 3.7%, 1.2%, 0.41%, 0.14%, and 0.046% MD4 B-cells by mixing them with normal B-cells, loaded them with fluo-4, and stimulated them with HEL. B-cells that responded to HEL were determined by the correlation method using the template prepared as described above (Fig. 3D). Supplementary Fig. 1† demonstrates the fluo-4 fluorescence alteration of 100 representative cells that showed high C values of more than 0.99 in 100% MD4 B-cell preparation. Those cells showed the typical alteration of fluo-4 fluorescence. We then analyzed 3.7%, 1.2%, 0.41%, 0.14%, and 0.046% MD4 B-cells using MAC-CCD system and correlation method. When we analyzed the alteration of fluo-4 fluorescence of 100%, 3.7%, and 1.2% MD4 B-cells, the number of positively detected cells with the MAC-CCD system and correlation method (C value > 0.875) decreased proportionally according to the percentage of MD4 B-cells (Fig. 4A and ESI Table 1).† When we analyzed MD4 B-cells of a frequency less than 0.41%, the number of positively detected cells did not decrease proportionally, indicating that the system might be detecting background noise (false-positive cells). When the C value was set to 0.975, the number of positively detected cells decreased proportionally to the 0.41% MD4 B-cells, although the number of detected cells was less than that in the analysis with the C value of 0.875 (Fig. 4A). To confirm whether the positively detected cells were MD4 B-cells, we retrieved the cells that showed top 16 highest C values with a micromanipulator and amplified the HyHEL10 antibody cDNA from the individual cells by single-cell RT-PCR. When the cells were selected from the 100%, 0.41%, 0.14%, and 0.046% MD4 B-cells, HyHEL10 antibody cDNA was amplified from 95%, 62%, 41%, and 24% of the retrieved cells, respectively (Fig. 4B). These results demonstrated that the MAC-CCD system with the correlation method could detect a very rare population, as few as 0.046%.
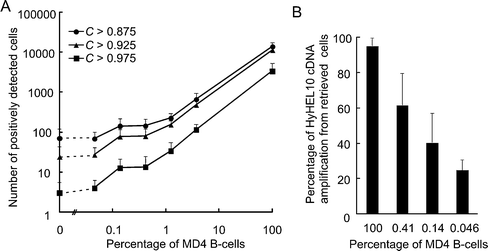 | ||
| Fig. 4 Analysis of fluo-4 fluorescence intensities of MD4 B-cells with correlation method. Mixtures of MD4 and normal B-cells that consisted of 100%, 3.7%, 1.2%, 0.41%, 0.14%, and 0.046% MD4 B-cells were prepared and stimulated with HEL on a microwell-array chip. The fluo-4 fluorescence of individual cells was monitored with the MAC-CCD system as in Fig. 3. The pattern of fluorescence alteration was then analyzed with the correlation method. (A) Number of positively detected cells in various percentages of MD4 B-cell preparations with the correlation method using different C values (> 0.875, closed circle; > 0.925, closed triangle; > 0.975, closed rectangle). The typical example shown is representative of four separate experiments. (B) Efficiency of HyHEL10 antibody cDNA amplification from selected and retrieved MD4 B-cells. MD4 B-cell preparations with various percentages of MD4 B-cells were stimulated and analyzed with the MAC-CCD system using the correlation method. Cells with a high C value were selected and transferred to each reaction tube. HyHEL10 antibody cDNA was amplified with RT-PCR from single cells and analyzed with agarose gel electrophoresis. The frequencies of HyHEL10 antibody cDNA amplification selected from various percentages of MD4 B-cell preparations are shown. The result represents three or four separate experiments. | ||
Next, we examined the feasibility of the system for detecting antigen-specific B-cells from human peripheral blood. We attempted to find type A influenza nucleoprotein (A-NP)-specific B-cells in peripheral blood lymphocytes from four healthy influenza-vaccinated volunteers and produce human monoclonal antibodies to A-NP. B-cells were prepared from the peripheral blood, loaded with fluo-4, applied on microwell-array chips, and stimulated with A-NP (10 µg/ml) on the chips. Fluorescence intensity alterations were monitored with the CCD scanner, and analyzed by the correlation method using the template shown in Fig. 3D. Because C values of MD4 B-cells from which we could amplify HyHEL10 antibody cDNA were ranged from 0.90 to 0.99 (data not shown), we selected B-cells whose C value was more than 0.90 as candidates of A-NP specific B-cells. Among them, we retrieved 95 cells with a high C value and amplified IgG antibody cDNA from individual cells with the single-cell 5′-RACE method.33 We obtained 16 recombinant IgG antibody proteins. Competitive ELISA showed that six of them (1–2A, 4–7A, 6–8A, 7–2A, 9–4A and 8–5A) specifically bind to A-NP (Fig. 5). These results demonstrate that the MAC-CCD system with the correlation method could be used to detect the B-cells that produce antigen-specific antibodies from human peripheral blood lymphocytes.
 | ||
| Fig. 5 Antigen specificity of human recombinant antibodies to influenza A-NP obtained with the MAC-CCD system. B-cells from peripheral blood of influenza-vaccinated volunteers were analyzed for reactivity of influenza A-NP using the MAC-CCD system and the correlation method. Cells with a high C value were recovered and antibody cDNAs were amplified with the single-cell 5′-RACE method. Amplified antibody cDNAs were transfected into 293T cells and antigen specificity of the recombinant antibodies were examined with ELISA. Thereafter, antigen specificities of the antibodies that positively reacted to A-NP (1–2A, 4–7A, 6–8A, 7–2A, 9–4A, and 8–5A) were confirmed with competitive ELISA. Binding of the antibodies to plate-coated influenza A-NP was examined in the presence of various doses (0, 1, or 5 µg/ml) of A-NP. | ||
Conclusions
In this study, we presented a MAC-CCD system that could monitor the Ca2+ mobilization of a large number of cells at the single-cell level every 10 s, which enabled detailed analysis of intracellular Ca2+ alteration. Through its use, we were able to obtain six human monoclonal antibodies against influenza A-NP. In recent years, the demand for monoclonal antibodies for diagnosis and therapy35 has been increasing, and various techniques for making monoclonal antibodies have been developed.3,4,6,36–44 The development of human monoclonal antibodies is especially important because of the few side effects accompanying antibody-based therapeutics. Our MAC-CCD system can detect antigen-specific B-cells directly in human peripheral blood lymphocytes and rapidly produce human monoclonal antibodies from the retrieved cells. Influenza is a zoonosis, and the recent occurrences of highly pathogenic avian influenza A virus (H5N1 subtype) infections have raised concerns for pandemic infection in the near future. Hence, effective vaccines and antibody-based therapeutics against the H5N1 virus should be urgently developed.45 From that perspective, it is worthwhile that we were able to obtain human monoclonal antibodies against the influenza virus directly from the peripheral blood. The MAC-CCD system provides an important tool for the development and generation of antibody therapeutics.Acknowledgements
We thank Hiroyoshi Nakazato and members of the Toyama New Industry Organization (TONIO) for helpful discussions, Nano System Solutions for design of CCD scanner, Tsutomu Obata for providing microwell-array chips, Kaoru Hata for her valuable secretarial work, and Kyowa Medex Co., Ltd, for providing type-A influenza nucleoprotein. This work was supported by a grant from the Toyama Medical-Bio Cluster (Promotion of the Cooperative Link of Unique Science and Technology for Economy Revitalization) project sponsored by the Ministry of Education, Culture, Sports and Science, Japan.Notes and References
- B. Angres, Expert Rev Mol Diagn, 2005, 5, 769–779 Search PubMed.
- K. Ino, M. Okochi, N. Konishi, M. Nakatochi, R. Imai, M. Shikida, A. Ito and H. Honda, Lab Chip, 2008, 8, 134–142 RSC.
- Y. Tokimitsu, H. Kishi, S. Kondo, R. Honda, K. Tajiri, K. Motoki, T. Ozawa, S. Kadowaki, T. Obata, S. Fujiki, C. Tateno, H. Takaishi, K. Chayama, K. Yoshizato, E. Tamiya, T. Sugiyama and A. Muraguchi, Cytometry A, 2007, 71, 1003–1010 CrossRef.
- K. Tajiri, H. Kishi, Y. Tokimitsu, S. Kondo, T. Ozawa, K. Kinoshita, A. Jin, S. Kadowaki, T. Sugiyama and A. Muraguchi, Cytometry A, 2007, 71, 961–967 CrossRef.
- M. C. Park, J. Y. Hur, K. W. Kwon, S. H. Park and K. Y. Suh, Lab Chip, 2006, 6, 988–994 RSC.
- J. C. Love, J. L. Ronan, G. M. Grotenbreg, A. G. van der Veen and H. L. Ploegh, Nat Biotechnol, 2006, 24, 703–707 CrossRef CAS.
- M. Deutsch, A. Deutsch, O. Shirihai, I. Hurevich, E. Afrimzon, Y. Shafran and N. Zurgil, Lab Chip, 2006, 6, 995–1000 RSC.
- S. Yamamura, H. Kishi, Y. Tokimitsu, S. Kondo, R. Honda, S. R. Rao, M. Omori, E. Tamiya and A. Muraguchi, Anal Chem, 2005, 77, 8050–8056 CrossRef CAS.
- A. Revzin, K. Sekine, A. Sin, R. G. Tompkins and M. Toner, Lab Chip, 2005, 5, 30–37 RSC.
- I. Biran and D. R. Walt, Anal Chem, 2002, 74, 3046–3054 CrossRef CAS.
- M. Engelke, N. Engels, K. Dittmann, B. Stork and J. Wienands, Immunol Rev, 2007, 218, 235–246 CrossRef CAS.
- M. Nishida, K. Sugimoto, Y. Hara, E. Mori, T. Morii, T. Kurosaki and Y. Mori, Embo J, 2003, 22, 4677–4688 CrossRef CAS.
- A. K. Abbas and A. H. Lichtman, Cellular and Molecular Immunology 5th edition, Updated edition, 2005, Elsevier, Amsterdam Search PubMed.
- P. Launay, H. Cheng, S. Srivatsan, R. Penner, A. Fleig and J. P. Kinet, Science, 2004, 306, 1374–1377 CrossRef CAS.
- C. M. Fanger, M. Hoth, G. R. Crabtree and R. S. Lewis, J Cell Biol, 1995, 131, 655–667 CrossRef CAS.
- M. R. Gold and A. L. DeFranco, Adv Immunol, 1994, 55, 221–295 Search PubMed.
- G. R. Crabtree and N. A. Clipstone, Annu Rev Biochem, 1994, 63, 1045–1083 CrossRef CAS.
- Hamamatsu Photonics (Hamamatsu, Japan, http://jp.hamamatsu.com/en/index.html) FDSS/IMACS-Lab can analyze up to 5,000 single cell levels using 4x objective lens. (http://jp.hamamatsu.com/products/life-science/pd264/5099/imacslab/index_ja.html).
- Y. Sei, K. L. Gallagher and J. W. Daly, Cell Calcium, 2001, 29, 149–160 CrossRef CAS.
- Y. Sei, K. L. Gallagher and A. S. Basile, J Biol Chem, 1999, 274, 5995–6002 CrossRef CAS.
- Q. H. Liu, X. Liu, Z. Wen, B. Hondowicz, L. King, J. Monroe and B. D. Freedman, J Immunol, 2005, 174, 68–79 CAS.
- S. Sasaki, M. C. Jaimes, T. H. Holmes, C. L. Dekker, K. Mahmood, G. W. Kemble, A. M. Arvin and H. B. Greenberg, J Virol, 2007, 81, 215–228 CAS.
- E. Tuaillon, Y. A. Tabaa, G. Petitjean, M. F. Huguet, G. Pajeaux, J. M. Fondere, B. Ponseille, J. Ducos, P. Blanc and J. P. Vendrell, J Immunol Methods, 2006, 315, 144–152 CrossRef CAS.
- M. A. Shokrgozar, M. R. Sam, A. Amirkhani and F. Shokri, Scand J Immunol, 2006, 64, 536–543 Search PubMed.
- I. J. Amanna and M. K. Slifka, J Immunol Methods, 2006, 317, 175–185 CrossRef CAS.
- E. J. Blink, A. Light, A. Kallies, S. L. Nutt, P. D. Hodgkin and D. M. Tarlinton, J Exp Med, 2005, 201, 545–554 CrossRef CAS.
- S. Crotty, R. D. Aubert, J. Glidewell and R. Ahmed, J Immunol Methods, 2004, 286, 111–122 CrossRef CAS.
- J. Bell and D. Gray, J Exp Med, 2003, 197, 1233–1244 CrossRef CAS.
- M. A. Shokrgozar and F. Shokri, Immunology, 2001, 104, 75–79 CrossRef CAS.
- R. Nanan, D. Heinrich, M. Frosch and H. W. Kreth, Vaccine, 2001, 20, 498–504 CrossRef CAS.
- H. Leyendeckers, M. Odendahl, A. Lohndorf, J. Irsch, M. Spangfort, S. Miltenyi, N. Hunzelmann, M. Assenmacher, A. Radbruch and J. Schmitz, Eur J Immunol, 1999, 29, 1406–1417 CrossRef CAS.
- B. V. K. V. Kumar A. Mahalanobis and R. D. Juday, Correlation Pattern Recognition, 2005, CAMBRIDGE University press, New York Search PubMed.
- T. Ozawa, H. Kishi and A. Muraguchi, Biotechniques, 2006, 40 Search PubMed , 469–470, 472, 474, passim.
- C. C. Goodnow, J. Crosbie, S. Adelstein, T. B. Lavoie, S. J. Smith-Gill, R. A. Brink, H. Pritchard-Briscoe, J. S. Wotherspoon, R. H. Loblay and K. Raphael, and et al. Nature, 1988, 334, 676–682 Search PubMed.
- J. M. Reichert, C. J. Rosensweig, L. B. Faden and M. C. Dewitz, Nat Biotechnol, 2005, 23, 1073–1078 CrossRef CAS.
- E. Traggiai, S. Becker, K. Subbarao, L. Kolesnikova, Y. Uematsu, M. R. Gismondo, B. R. Murphy, R. Rappuoli and A. Lanzavecchia, Nat Med, 2004, 10, 871–875 CrossRef CAS.
- A. Karpas, A. Dremucheva and B. H. Czepulkowski, Proc Natl Acad Sci U S A, 2001, 98, 1799–1804 CrossRef CAS.
- E. T. Boder and K. D. Wittrup, Nat Biotechnol, 1997, 15, 553–557 CAS.
- G. Winter, A. D. Griffiths, R. E. Hawkins and H. R. Hoogenboom, Annu Rev Immunol, 1994, 12, 433–455 CrossRef CAS.
- B. Gustafsson and J. Hinkula, Hum Antibodies Hybridomas, 1994, 5, 98–104 Search PubMed.
- G. Winter and C. Milstein, Nature, 1991, 349, 293–299 CrossRef CAS.
- J. McCafferty, A. D. Griffiths, G. Winter and D. J. Chiswell, Nature, 1990, 348, 552–554 CrossRef CAS.
- D. Kozbor, J. C. Roder, T. H. Chang, Z. Steplewski and H. Koprowski, Hybridoma, 1982, 1, 323–328 CAS.
- D. Kozbor and J. C. Roder, J Immunol, 1981, 127, 1275–1280 CAS.
- T. Horimoto and Y. Kawaoka, Trends Mol Med, 2006, 12, 506–514 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed materials and methods information. See DOI: 10.1039/b810438g |
| This journal is © The Royal Society of Chemistry 2009 |