Synthesis and mesomorphic properties of new V-shaped shape-persistent nematogens containing a thiazole or a thiadiazole bending unit
Matthias
Lehmann
*a,
Jens
Seltmann
a,
Alexander A.
Auer
b,
Eric
Prochnow
b and
Udo
Benedikt
b
aNon-Classical Synthetic Methods, Institute of Chemistry, Chemnitz University of Technology, Straße der Nationen 62, 09111, Chemnitz, Germany. E-mail: Matthias.Lehmann@chemie.tu-chemnitz.de; Fax: (+) 49 371 531 21229; Tel: +49 371 531 31205
bTheoretical Chemistry, Institute of Chemistry, Chemnitz University of Technology, Straße der Nationen 62, 09111, Chemnitz, Germany
First published on 27th February 2009
Abstract
Two series of new V-shaped molecules, containing a central thiazole or thiadiazole bending unit have been synthesised. The design is based on shape-persistent phenylene ethynylene scaffolds that were attached stepwise with high regioselectivity to a desymmetrised iodo and bromo functionalised bent core with an apex angle of about 160°. Molecular engineering results in materials that exhibit exclusively nematic liquid crystal phases (monotropic or enantiotropic). The phase behaviour was investigated by means of polarised optical microscopy (POM), differential scanning calorimetry (DSC) and X-ray diffraction.
Introduction
Bent shaped molecules came in the focus of liquid crystal research when Niori et al.1 discovered the exceptional properties of the so-called banana phases. Since then enormous efforts have been put into the research of this mesogen family,2 but also theoretical work on shape-persistent V-shaped molecules has been performed and the formation of the elusive thermotropic biaxial nematic phase was proposed to be feasible.3 Among this type of mesogens, molecules containing five-membered heterocycles such as 1,3,4-oxadiazoles4,5 and 1,3,4-thiadiazoles6–9 are frequently targeted structures, owing to their large dipole moment, promoting self-assembly in liquid crystalline (LC) phases with ferroelectric properties.10,11 Moreover, diaryl substituted five-membered heterocycles were also of interest as materials for light emitting diodes.12Indeed, the first widely accepted observation of a biaxial thermotropic nematic phase of rather small molecules was reported in the series of oxadiazole derivatives.4 Dipole moments along the apex angle were discussed to be responsible for the formation of thermotropic biaxial nematic phases.13 The latter was unexpected since earlier theoretical studies proposed molecules with an apex angle of about 109° to be favourable for the formation of biaxial nematic phases.3 The relationship between the structure of the bent core and mesomorphism was studied in the series of banana molecules14 and some hockey stick oligophenylenes.15 The latter mesogens without lateral chains indicated the importance of molecular shape and orientation of dipoles for the temperature range of nematic phases. In contrast, the banana molecules assembled in smectic phases; the variation of cores resulted in no facile interpretable structure–property relationship. Banana mesogens and the oxadiazole diester derivatives are not shape-persistent. Rotation about the ester single bonds results in a not well-defined molecular shape forming the LC. A wide range of shape persistent diaryl oxadiazole,16thiazole6,17,18 and thiadiazole6,16,18,19 derivatives as well as diarylethynyl thiophene20 and thiazole17 derivatives have been investigated. Although often nematic phases were observed at high temperatures, no indication for phase biaxiality was reported except for a thiazole derivative.2
Following general trends can be stressed: (i) less bended thiadiazole or thiazole derivatives show mesophases at a higher temperature than mesogens with a stronger bended oxadiazole or oxazole cores; (ii) derivatives with non-C2-symmetric cores (oxazole, thiazole) show lower transition temperatures than the bent mesogens with C2-symmetric cores (oxadiazole, thiadiazole) and (iii) bis(arylethynyl) derivatives possess transitions at lower temperature when compared with diaryl substituted compounds.
In our attempt to study the LC behaviour of bent core nematogens as a function of the bending angle, we focused on molecules with a bent core substituted by a phenylene ethynylene arm scaffold with 2,5-dialkoxy chains at the middle benzene unit of the arm. The latter promotes the formation of a nematic phase, known from stilbenoid star compounds and polymers21,22 and decreased the crystallisation tendency. Such a molecular design provides shape-persistent mesogens with clearing temperatures below 200 °C. In the series of fluorenone derivatives with a bent angle of 90° and oxadiazole mesogens with a bent angle of 136°, nematic phases were observed. These phases did not crystallize upon cooling, which allowed the investigation of their biaxial character. For both cores evidence for the biaxial character of the thermotropic nematic phase has been presented.23,24
Herein, we report the synthesis and mesomorphic behaviour of a library of new shape-persistent thiazole and thiadiazole derivatives with a bending angle of approximately 160° and lateral hexyloxy chains. The molecular design is shown in Fig. 1. The terminal aromatic groups have been varied on each individual arm, in order to study the effect of dipoles, which have previously been found to be essential for the formation of mesophases in this mesogen family. For this reason, cyanophenyl or pyridyl groups have been attached. The latter is known to form hydrogen bonds with hydrogen-bond acceptors, which can induce LC behaviour of bent molecules.25
 | ||
| Fig. 1 Prepared V-shaped molecules. | ||
Synthesis
In order to vary independently the individual oligophenylene ethynylene arms by a convergent synthesis, i.e. the coupling of arms with a terminal ethynyl group in the final step, the cores 5 and 6 were synthesized (Scheme 1). These building blocks allow the stepwise Sonogashira–Hagihara cross coupling, due to the different reactivity of iodoaryl and bromoaryl bonds. Heterocycles 5 and 6 were prepared by treatment of 3 and 4 with P2S5. Arms 7–9 were synthesised by a previously published route26 using 4-iodopyridine or cyano substituted iodobenzenes. Coupling of a first arm to the cores 5 and 6 at room temperature (rt) resulted in hockey stick intermediates 10–15 (Scheme 2), which could be isolated in excellent yields (82–95%). A second coupling step of the intermediate products 10–15 with one of the arms 7–9 at 60 °C led to the desired V-shaped thiadiazoles 1a–f (36–74%) and thiazoles 2a–i (40–69%) (Fig. 2). All compounds were carefully purified and characterised by 1H, 13C NMR, mass spectrometry and elemental analysis.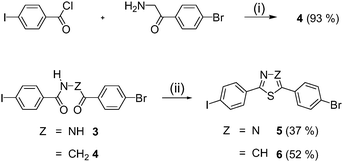
 | ||
| Fig. 2 Prepared V-shaped thiadiazole derivatives 1a–1f and thiazole derivatives 2a–2i. | ||
Core structure and dipoles
The different bent cores, thiazole and thiadiazole, have been chosen in order to study the influence of dipoles in the centre of V-shaped molecules with the same bending angle. Fig. 3 shows the two bending cores with the experimental and calculated dipoles of the parent heterocycles compared to the oxadiazole building block, that was previously shown to promote biaxiality in nematic LC.24 Interestingly, the dipoles of the sulfur heterocycles point to the sulfur atoms. Consequently, in all V-shaped mesogens with these heterocyclic cores, the dipoles of the core direct from the apex backwards along the bisect of the molecule. Thereby, the dipole of the thiadiazole (3.11 D) is larger than that of the oxadiazole (3.06 D). In contrast, the thiazole core possesses only a smaller dipole of 1.47 D, with an antiparallel (1.25 D) and a transversal contribution (0.78 D) relative to the apex of the molecule. In addition to various dipole–dipole and van der Waals interactions, π–π interactions33 may also influence the mesomorphic behaviour. Especially, electron poor and electron rich building blocks are known to interact favourable.34 An electron rich unit, the 2,5-dialkoxy-substituted benzene ring, is located in the middle of the arm scaffold. In the series of the electron-deficient five-membered heterocycles, the HOMO and LUMO energies were recently calculated for the diphenyl substituted molecules and indicated that the thiadiazole derivatives possessed the highest electron deficiency (LUMO is lowest in energy).35 This might result in favourable π-stacking donor–acceptor interactions and consequently in a stabilisation of the mesophases. | ||
| Fig. 3 Experimental27–29 and calculated30 (B3LYP/DZP)31,32 dipole moments of five-membered heterocyclic central cores. | ||
 | ||
| Scheme 2 Synthesis of the intermediate products. | ||
Thermotropic properties
The mesomorphic properties of the materials were studied by differential scanning calorimetry (DSC) and polarised optical microscopy (POM) and the results are collected in Table 1 and visualised in Scheme 3. | ||
| Scheme 3 Thermotropic behaviour of V-shaped mesogens. First column represents the second heating, the second column the first cooling cycle. | ||
| Compound | Rate 10 °C/min (onset [°C]/ΔH [kJ mol−1])a | ΔSN [J mol−1K−1] |
|---|---|---|
| a Data are given for the second heating and for monotropic samples for the first cooling. b Data are given for the first heating. c Data are given for the first cooling. d Determined by POM. | ||
| 11 | Cr 156.6/53.9bN 197.6/0.5I | 1.1 |
| 14 | Cr 163.4/57.5N 169.4/0.3I | 0.7 |
| 1a | Cr 185.9/60.8bN 235.0/1.5bI | 3.0 |
| 1b | Cr 163 Id | — |
| 1c | Cr 143.5/61.8N 162.6/0.3I | 0.7 |
| 1d | Cr 141.8/45.1 (N 123.3/0.1) I | 0.3 |
| 1e | Cr 183.4/69.3 (N 181.8/0.6) I | 1.3 |
| 1f | Cr 147.2/70.6 (N 107.2/0.2) I | 0.5 |
| 2a | Cr 153.4/81.0bN 189.8/0.3bI | 0.6 |
| 2b | Cr 136 Id | — |
| 2c | Cr 123.2/73.0N 135.2/0.1I | 0.2 |
| 2d | Cr 123.5/51.2bN 130.3/0.2cI | 0.5 |
| 2e | Cr 108.4/34.5 (N 103.1/0.1) I | 0.3 |
| 2f | Cr 124.9/58.5bN 143.6/0.5I | 1.2 |
| 2g | Cr 133.4/74.0bN 154.8/0.4I | 0.9 |
| 2h | Cr 139 Id | — |
| 2i | Cr 128.1/72.6b (N 73.3/0.1) I | 0.3 |
From the six intermediate products 10–15 only the molecules 11 and 14 with a CN group at the para position (p-CN) of the terminal phenyl ring exhibit a liquid crystal phase. The clearing temperature is higher and the temperature range of the mesophase is much larger for the thiadiazole derivative 11.
Almost all V-shaped molecules exhibit mesophases and crystallisation is often suppressed (Scheme 3). Thus, even for enantiotropic mesophases the second heating cycle does not reveal a Cr–N transition. Crystalline compounds 1b, 2b, 2h and monotropic and enantiotropic LC phases of 1e and 2f, which crystallise relatively fast, are exceptions. Thus as reported earlier,23,24 the substitution pattern with internal hexyloxy chains prevents rapid crystallisation in most materials. As discussed in detail below, the thermotropic behaviour appears to depend on a subtle equilibrium between dipoles in the shape-persistent scaffold.
In the series of thiadiazoles derivatives, the p-CN group leads to the highest clearing temperatures, enthalpies and entropies (e.g.1a). The introduction of a CN group at the meta-position (m-CN) considerably decreases the transition temperatures (1c). In addition the enthalpy and entropy of the N–I transition has been significantly reduced. The mesophase stability was found to be decreased, too. Whereas 1a has a LC interval of 49 °C, 1c only exists over a temperature range of 19 °C. Consequently, further exchange of the p-CN by a m-CN group results in the loss of LC properties (1b). Analogous thermotropic behaviour was observed for the pyridyl derivatives 1d–f, i.e. the m-CN group reduces transition temperatures, enthalpies and entropies. The pyridyl group introduces a smaller dipole moment along the shape-persistent arm when compared with the p-CN function. This leads only to minor changes in the melting temperatures, but a strong decrease of the clearing temperatures when derivatives are considered in which one p-CN group is exchanged by a pyridyl group (c.p. 1a and 1e; 1c and 1f). All the nematic mesophases of pyridyl derivatives are therefore only monotropic, a result that emphasises the importance of large dipoles at the terminal aromatic unit of the mesogens for the stability of nematic mesophases.
Similar behaviour was monitored for the thiazole derivatives: (i) p-CN groups stabilise the nematic mesophase (2a); (ii) m-CN substituents decrease clearing temperatures to a larger extent than melting temperatures (2c, 2d) and eventually a non-mesomorphic compound with two m-CN units is obtained (2b); (iii) pyridyl groups again destabilise the mesophases and depending on the substitution pattern enantiotropic (2f, 2g), monotropic (2e, 2i) or non-LC materials (2h) are found. Two dissimilar V-shaped mesogens exist for thiazole derivatives where the lateral contributions of the core dipoles point to either the one or the other of the two different terminal aryl units. In this way, the thermotropic behaviour is influenced by a delicate interplay of the various dipole moments in the molecular scaffold. For example, if the terminal p-CN group is aligned antiparallel to the transversal dipole of the bent core, as in 2f, the same mesophase range but by 10 °C lower transition temperatures are found compared to the material 2g, where these dipoles are parallel. Similarly, compound 2h is non-mesomorphic, whereas mesogen 2i with the reverse substitution pattern forms a monotropic LC phase.
The transition enthalpies and entropies are also changing according to the substitution pattern and are smallest for compound 2c (Table 1). Theory predicts a vanishing transition entropy, i.e. a second order transition, when the material transforms from the isotropic phase directly to a biaxial nematic phase. Thus, a very small transition entropy may indicate that the elusive biaxial nematic phase might be observed for materials such as 1d, 2c, 2e or 2i.
POM investigations exhibit for all V-shaped LC-materials Schlieren textures pointing clearly to the nematic character of the mesophases (Fig. 4). The observation of almost exclusive two-brushed defects for the V-shaped mesogens (Fig. 4B–D) may be indicative for the biaxial nature of their phase.36 For further information on the uniaxial or biaxial character of the nematic phases well aligned samples are needed, which could only be obtained for compounds 1c, 1d and 2e. Fig. 5 presents temperature-dependent conoscopic observations for compound 1c, that could be aligned between two glass plates by cooling slowly from the isotropic to the nematic phase. At high temperatures just after the I–N transition, the nematic mesophase is uniaxial and optically positive, which is evidenced by the conoscopic cross at all rotation angles and the color sequence when introducing the lambda compensation plate (Fig. 5A and B). Consequently, the large refractive index is aligned parallel with the propagation direction of light. This is in contrast to the oxadiazole or fluorenone derivatives, for which optical negative phases have been monitored.23,24
 | ||
| Fig. 4 Schlieren textures of (A) 14 at 166 °C, (B) 2d at 129 °C (C) 2c at 138 °C and (D) 1c at 100 °C. | ||
 | ||
| Fig. 5 (A) Conoscopic picture of homeotropically aligned Nu phase of 1c at 137 °C; (B) conoscopic cross with the lambda compensation plate introduced at 153 °C; (C) splitted isogyres at 85 °C; (D): conoscopy with circular polarisers inserted, the two dark spots indicate the optical axes of the nematic phase. | ||
Decreasing the temperature to 85 °C results in a separation of two isogyres (Fig. 5C) and the circular polariser visualises the two optical axes (Fig. 5D). These observations point to a biaxial nematic phase, however, the results were obtained for all materials only at temperatures below the melting transition where the nematic phase was metastable and therefore the onset of crystallisation did not allow a more detailed study.
X-Ray diffraction (XRD)
XRD patterns from oriented nematic mesophases were obtained by cooling the sample from the isotropic liquid to the liquid crystal state in the presence of a magnetic field (2 T). The sample was then transferred to the XRD sample holder at the temperature chosen for the measurements. For almost all compounds the substitution pattern with alkoxy chains prevents the materials from rapid crystallisation and consequently a study at low temperatures was possible. Some of the materials crystallise relatively fast, and could therefore not be analysed (1e, 2f).Fig. 6 shows the XRD pattern of compound 2g that was typically observed in the thiazole and thiadiazole family. The small angle reflection (i) at the meridian with d = 14 Å appears to be broad with a correlation length of about three molecules and is related to distances along the bisect of the molecules as indicated in Fig. 7A. The second reflection at 3.8 Å originates from the π–π-distance of the nematogens (Fig. 7B). A third weak reflection (d = 9.7 Å) is present at the equator with a correlation length of about nine repeating units. This signal must be associated with a direction orthogonal to the bisect and the aromatic planes, consequently it is attributed to a periodicity along the molecular long axis. A periodic electron density related to the molecular length is expected to be in the range of 39.9 Å; such a XRD feature at small angles was not observed. However, if the molecules above and below the reference mesogen shift by 9.7 Å along the molecular long axis, then electron poor thiadiazole rings interact with the electron rich alkoxy substituted aromatics. Such a translation would give rise to a periodicity of about 9–10 Å, which explains the small angle reflection on the equator (Fig. 6A and 7A). Two further diffuse and weak XRD intensities can be detected on the equator at wide angles (Fig. 6B). One corresponds to the halo and can be attributed to the liquid-like aliphatic chains (d = 4.2 Å). The other at d = 5.2 Å cannot be rationalised by distances within or between the phenylene ethynylene scaffolds, but may originate from average separations of aliphatic chains in the direction of the mesogen long axis. The results for all other mesogens are similar and are summarised in Table 2.
| Compound | T/°Ca | Reflection | d/Å | ξ/db |
|---|---|---|---|---|
| a Temperatures in parenthesis are temperatures of alignment in the magnetic field. b ξ is the correlation length calculated from the half width of the reflections by the Scherrer formula.37 The value ξ/d describes the correlation of mesogens in units of molecular distances. | ||||
| 1a | 25 (225) | (i) | 12.2 | 6 |
| (ii) | 9.3 | 7 | ||
| (iii) | 3.6 | 15 | ||
| (iv) | 5.6 | 6 | ||
| (v) | 4.0 | 4 | ||
| 1c | 25 (145) | (i) | 12.4 | 7 |
| (ii) | 9.8 | 9 | ||
| (iii) | 3.6 | 14 | ||
| (v) | 4.3 | 4 | ||
| 1d | 25 | (i) | 15.0 | 4 |
| (ii) | 10.0 | 14 | ||
| (iii) | 4.0 | 5 | ||
| (iv) | 5.2 | 5 | ||
| (v) | 4.1 | 5 | ||
| 1e | 25 (180) | (i) | 15.2 | 3 |
| (iii) | 3.7 | 5 | ||
| (v) | 4.1 | 4 | ||
| 2a | 25 (175) | (i) | 14.0 | 4 |
| (ii) | 10.1 | 17 | ||
| (iii) | 3.8 | 7 | ||
| (v) | 4.1 | 5 | ||
| 2c | 25 (125) | (i) | 14.2 | 4 |
| (ii) | 10.0 | 4 | ||
| (iii) | 3.8 | 8 | ||
| (iv) | 5.6 | 4 | ||
| (v) | 4.3 | 6 | ||
| 2d | 25 (125) | (i) | 14.5 | 3 |
| (ii) | 9.9 | 7 | ||
| (iii) | 3.9 | 8 | ||
| (iv) | 5.6 | 4 | ||
| (v) | 4.3 | 7 | ||
| 2f | 25 (140) | (i) | 14.6 | 4 |
| (ii) | 9.7 | 10 | ||
| (iii) | 3.9 | 6 | ||
| (v) | 4.1 | 5 | ||
| 2g | 25 (80) | (i) | 14.0 | 3 |
| (ii) | 10.0 | 9 | ||
| (iii) | 3.9 | 6 | ||
| (iv) | 5.2 | 7 | ||
| (v) | 4.0 | 6 |
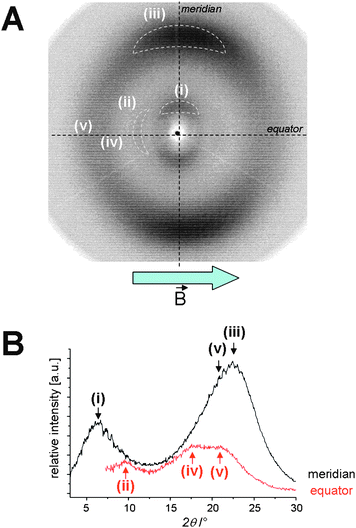 | ||
| Fig. 6 (A) X-Ray diffraction pattern of 2g aligned in the magnetic field by cooling from the isotropic phase to 80 °C and subsequently rapidly cooled to 25 °C. (B) Integration of the averaged XRD intensity along the meridian (black) and the equator (red). | ||
 | ||
| Fig. 7 Model of intermolecular orientation within the nematic phase and interpretation of the reflections (i)–(iii). (A) Top view. (B) Side view. | ||
The interpretation of the X-ray pattern is based on the assumption that two different domains are formed during the alignment procedure, which are schematically drawn in Fig. 7. Otherwise it would not be possible to explain the simultaneous appearance of reflections at the meridian of the X-ray photograph corresponding to the distances along the apex and the π–π-stacking. Such findings are in contrast to earlier studies on fluorenones and oxadiazoles, where the latter reflections revealed either on the meridian or at the equator of the X-ray pattern depending on the alignment procedure. The presence of two domains in this study is yet not fully understood, but may originate from the different alignment behaviour of this mesogen family (optical positive versus optical negative materials for fluorenone and oxadiazole derivatives) and from the interplay of surface interactions and magnetic field alignment. However, the results indicate that in the individual domains of the nematic phases the three molecular axes are aligned along different directors.
Conclusions
We described the synthesis of two series of new, shape-persistent, V-shaped molecules with a phenylene ethynylene scaffold and thiazole or thiadiazole bending units. The convergent route allows for the preparation of symmetric and non-symmetric derivatives in high yields. Almost all compounds exhibit nematic phases. The mesophase range and the clearing temperatures are controlled by the type of terminal aromatic groups. In general m-CN substituted phenyl units and pyridyl building blocks lower the clearing temperatures but also decrease substantially the mesophase stability compared with p-CN substituted compounds. Mesophases can be supercooled and are even metastable at room temperature. POM, DSC and XRD studies reveal features which are indicative for nematic phases with phase biaxiality, thus this family of materials seems to provide a promising leading structure for the engineering of nematogens with room temperature stable, thermotropic biaxial nematic mesophases in the future.Experimental
Chemicals were obtained from Acros and Sigma-Aldrich and used as received. The synthesis of compound 338 was described previously. Column chromatography was carried out on silica 60 (Merck, mesh 70–230). PFT 1H and 13C NMR spectra were recorded in CDCl3 with a Varian Oxford 400 MHz spectrometer with the residual solvent signal at 7.26 ppm as a reference. Mass spectra were obtained on a Finnigan MAT95 (FD MS). Elemental analysis was carried out in the microanalytical laboratory at the University of Mainz. POM observations were made with a Zeiss Axioscop 40 equipped with a Linkam THMS600 hot stage. DSC was performed using a Perkin Elmer Pyris 1.X-Ray diffraction measurements were carried out on powder samples in glass capillaries of 1.5 mm diameter. The WAXS measurements were performed by using a standard copper anode (2.2 kW) source with pinhole collimation equipped with an X-ray mirror (Osmic typ CMF15-sCu6) and a Bruker detector (High-star) with 1024 × 1024 pixels. The diffraction data were calibrated by using silver behenate as a calibration standard.39 The X-ray patterns were evaluated using the datasqueeze software (http://www.datasqueezesoftware.com/).
N-[2-(4-Bromophenyl)-2-oxoethyl]-4-iodobenzoicamid (4)
To a solution of 1.0 g (4.0 mmol) 2-amino-4′-bromoaceto-phenon hydrochloride and 1.1 g (8.0 mmol) K2CO3 in 30 ml of a 1 : 1-mixture of THF and water 1.1 g (4.1 mmol) 4-iodo-benzoylchloride, dissolved in 20 ml THF, was added dropwise. After stirring over night and removing the THF under vacuum, the yellow precipitate was collected by filtration and washed with water; yield 1.6 g (93%). 1H NMR (400 MHz, DMSO-d6): δ = 4.75 (d, 2H, CH2, 3J = 5.6); 7.67 (2H, AA′BB′, CH); 7.78 (2H, AA′BB′, CH); 7.89 (2H, AA′BB′, CH); 7.97 (2H, AA′BB′, CH); 8.97 (t, 1H, N–H, 3J = 5.6); 13C NMR (100 MHz, DMSO-d6): δ 46.5 (CH2); 99.2 (Cq, C–I); 127.7 (Cq, C–Br); 129.3, 129.9, 131.9 (aromat. CH); 133.2, 134.0 (Cq); 137.3 (aromat. CH); 165.9 (Cq, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 194.6 (HN–CO).
O); 194.6 (HN–CO).
5-(4-Bromophenyl)-2-(iodophenyl)-1,3,4-thiadiazole (5)
The mixture of 1.0 g (2.3 mmol) 4-bromo-N′-(4-iodobenzoyl) benzoic acid hydrazide and 2.0 g (9.0 mmol) P2S5 was dissolved in 100 ml pyridine and heated for 96 h at 100 °C. After evaporation of the solvent the residue was treated with CH2Cl2 and active coal. Filtration with cellite and removing the solvent gave the crude product which was crystallized from EtOH to obtain a white solid; yield 0.44 g (43%). 1H NMR (400 MHz, CDCl3): δ 7.64 (2H, AA′BB′, CH), 7.74 (2H, AA′BB′, CH), 7.87 (m, 4H, CH); 13C NMR (100 MHz, CDCl3): δ 97.8 (Cq, C–I); 125.8 (Cq, C–Br); 128.9, 129.0 (Cq); 129.3, 129.4, 132.5, 138.4 (aromat. CH); 167.4, 167.6 (Cq, NC–S); MS (FD): m/z (%) 441.6 (98, M+˙), 443.6 (100, [M + 2]+˙); EA: Calc. for C14H8BrIN2S: C 37.95, H 1.82, N 6.32, S 7.24. Found: C 37.95, H 1.87, N 6.35, S 7.36.
General method for preparation of intermediate products (10–15)
The mixture of 1.0 eq of thiadiazole 5 or thiazole 6, 1.0 eq. of the corresponding terminal alkyne 7–9, 0.1 eq of Pd(PPh3)4 and 0.05 eq of CuI in piperidine is stirred for 12 h at room temperature. Afterwards the solvent is removed under vacuum and the products are isolated by column chromatography using mixtures of EtOAc/hexane or CH2Cl2/hexane, respectively.2-(4-Bromophenyl)-5-{4-[4-(3-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (10)
Hexane/CH2Cl2 = 1/2 (Rf = 0.15), yellow solid; yield 0.14 g (82%). 1H NMR (400 MHz, CDCl3): δ 0.90 (m, 6H, CH3), 1.37 (m, 8H, CH2), 1.55 (m, 4H, CH2), 1.87 (m, 4H, CH2); 4.05 (t, 4H, OCH2, 3J = 6.4), 7.02, 7.04 (2s, 2H, CH), 7.47 (ddd, 1H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH), 7.61 (ddd, 1H, 3J = 7.8, 4J = 1.55, 4J = 1.4, CH), 7.64 (4H, AA′BB′, CH), 7.73 (ddd, 1H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH), 7.80 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH), 7.89 (2H, AA′BB′, CH), 8.00 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.2 (CH3), 22.8, 25.9, 29.4, 31.7 (CH2), 69.7 (OCH2); 88.5, 88.9, 92.6, 94.6 (Cq, C≡C); 113.0, 113.4, 114.3 (Cq); 116.8, 116.9 (aromat. CH); 118.2 (Cq, CN); 125.2, 125.8, 126.5 (Cq); 127.9 (aromat. CH); 129.1 (Cq); 129.4 (aromat. CH); 129.6 (Cq); 131.5, 132.4, 132.6, 134.9, 135.7 (aromat. CH); 153.8, 153.9 (Cq, C–O); 167.3, 167.8 (Cq, NC–S); MS (FD): m/z (%) 740.8 (92.6, M+˙), 742.8 (100 [M + 2]+˙); EA: Calc. for C43H40BrN3O2S: C 69.53, H 5.43, N 5.66, S 4.32. Found: C 69.39, H 5.39, N 5.65, S 4.46.
2-(4-Bromophenyl)-5-{4-[4-(4-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (11)
Hexane/CH2Cl2 = 1/2 (Rf = 0.1), yellow solid; yield 0.12 g (95%). 1H NMR (400 MHz, CDCl3): δ 0.90 (m, 6H, CH3), 1.38 (m, 8H, CH2), 1.55 (m, 4H, CH2), 1.87 (m, 4H, CH2), 4.05 (t, 4H, OCH2, 3J = 6.4), 7.02 (s, 1H, CH), 7.05 (s, 1H, CH), 7.59–7.67 (m, 8H, CH); 7.90 (2H, AA′BB′, CH); 8.01 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.2 (CH3); 22.8, 25.9, 29.4, 31.7 (CH2); 69.6, 69.7 (OCH2); 88.8, 90.6, 93.4, 94.7 (Cq, C≡C); 111.5, 113.3, 114.5 (Cq); 116.8, 116.9 (aromat. CH); 118.7 (Cq, CN); 125.8, 126.5 (Cq); 128.0 (aromat. CH); 128.5, 129.1 (Cq); 129.4 (aromat. CH); 129.6 (Cq); 132.1, 132.2, 132.4, 132.6 (aromat. CH); 153.8, 154.0 (Cq, C–O); 167.3, 167.8 (Cq, NC–S); MS (FD): m/z (%) 741.2 (93, M+˙), 743.2 (100, [M + 2]+˙); EA: Calc. for C43H40BrN3O2S: C 69.53, H 5.43, N 5.66, S 4.32. Found: C 69.48, H 5.44, N 5.49, S 4.28.
2-(4-Bromophenyl)-5-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (12)
Hexane/EtOAc = 4/1 (Rf = 0.1), orange solid; yield 0.22 g (90%). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 6H, CH3), 1.37 (m, 8H, CH2), 1.55 (m, 4H, CH2), 1.86 (m, 4H, CH2), 4.05 (t, 4H, OCH2, 3J = 6.4), 7.03 (s, 1H, CH), 7.04 (s, 1H, CH), 7.37 (2H, AA′BB′, CH), 7.64 (4H, AA′BB′, CH), 7.89 (2H, AA′BB′, CH), 8.00 (2H, AA′BB′, CH); 8.60 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.2 (CH3); 22.8, 25.9, 29.4, 31.7 (CH2); 69.7 (OCH2); 88.8, 90.7, 92.3, 94.7 (Cq, C≡C); 113.2, 114.6 (Cq); 116.8, 117.0, 125.5 (aromat. CH); 125.8, 126.4 (Cq); 127.9 (aromat. CH); 129.1 (Cq); 129.4 (aromat. CH); 129.6, 131.6 (Cq); 132.4, 132.6, 149.9 (aromat. CH); 153.8, 154.1 (Cq, C–O); 167.3, 167.8 (Cq, NC–S); MS (FD): m/z (%) 716.9 (90, M+˙), 718.9 (100, [M + 2]+˙); EA: Calc. for C41H40BrN3O2S: C 68.51, H 5.61, N 5.85, S 4.46. Found: C 68.50, H 5.81, N 5.67, S 4.39.
2-(4-Bromophenyl)-5-{4-[4-(3-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (13)
Hexane/CH2Cl2 = 1/2 (Rf = 0.2), yellow solid; yield 0.15 g (86%). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 6H, CH3), 1.37 (m, 8H, CH2), 1.56 (m, 4H, CH2), 1.87 (m, 4H, CH2); 4.04 (t, 2H, OCH2, 3J = 6.4); 4.05 (t, 2H, OCH2, 3J = 6.4), 7.01, 7.04 (2s, 2H, CH), 7.47 (m, 3H, CH), 7.55 (2H, AA′BB′, CH), 7.60 (m, 3H, CH), 7.73 (ddd, 1H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH), 7.80 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH), 7.95 (2H, AA′BB′, cH), 8.02 (s, 1H,N–CH); 13C NMR (100 MHz, CDCl3): δ 14.17, 14.21 (CH3); 22.80, 22.81, 25.9, 29.38, 29.42, 31.70, 31.74 (CH2); 69.6, 69.7 (OCH2); 88.3, 88.6, 92.5, 95.0 (Cq, C≡C); 113.0, 113.2, 114.6 (Cq); 116.8, 116.9 (aromat. CH); 118.2 (Cq, CN); 122.5, 125.22, 125.24 (Cq); 126.3, 128.2, 129.4 (aromat. CH); 130.3 (Cq); 131.5, 132.3, 132.4 (aromat. CH); 133.2 (Cq); 134.9, 135.6 (aromat. CH); 138.6 (Cq); 139.9 (aromat. CH); 153.8, 154.0 (Cq, C–O); 166.7 (Cq, NC–S); MS (FD): m/z (%) 740.1 (87, M+˙), 742.2 (100, [M + 2]+˙); EA: Calc. for C43H40BrN3O2S: C 71.24, H 5.57, N 3.78, S 4.32. Found: C 70.81, H 5.44, N 3.78, S 4.21.
2-(4-Bromophenyl)-5-{4-[4-(4-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (14)
Hexane/CH2Cl2 = 1/2 (Rf = 0.2), yellow solid; yield 0.14 g (86%). 1H NMR (400 MHz, CDCl3): δ 0.90 (m, 6H, CH3), 1.36 (m, 8H, CH2), 1.55 (m, 4H, CH2), 1.86 (m, 4H, CH2), 4.05 (t, 4H, OCH2, 3J = 6.4), 7.02 (s, 1H, CH), 7.04 (s, 1H, CH), 7.47 (2H, AA′BB′, CH); 7.56 (2H, AA′BB′, CH); 7.61 (m, 6H, CH); 7.96 (2H, AA′BB′, CH); 8.03 (s, 1H,N–CH); 13C NMR (100 MHz, CDCl3): δ 14.19, 14.22 (CH3); 22.8, 25.87, 25.90, 29.38, 29.42, 31.71, 31.74 (CH2); 69.6, 69.8 (OCH2); 88.2, 90.7, 93.3, 95.1 (Cq, C≡C); 111.5, 113.1, 114.8 (Cq); 116.8, 116.9 (aromat. CH); 118.7 (Cq, CN); 122.5, 125.2 (Cq); 126.4, 128.2 (aromat. CH); 128.5, 130.3 (Cq); 132.1, 132.2, 132.3, 132.5 (aromat. CH); 133.2, 138.6 (Cq); 139.9 (aromat. CH); 153.8, 154.0 (Cq, C–O); 167.7 (Cq, NC–S); MS (FD): m/z (%) 739.9 (87, M+˙), 741.9 (100, [M + 2]+˙); EA: Calc. for C44H41BrN2O2S: C 71.24, H 5.57, N 3.78. Found: C 70.89, H 5.56, N 3.70.
2-(4-Bromophenyl)-5-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (15)
Hexane/EtOAc = 3/1 (Rf = 0.25), orange solid; yield 0.12 g (83%). 1H NMR (400 MHz, CDCl3): δ 0.90 (t, 3H, CH3, 3J = 6.4), 0.91 (t, 3H, CH3, 3J = 6.4), 1.36 (m, 8H, CH2), 1.55 (m, 4H, CH2), 1.86 (m, 4H, CH2), 4.04 (t, 4H, OCH2, 3J = 6.4), 7.03 (s, 1H, CH), 7.04 (s, 1H, CH), 7.38 (2H, AA′BB′, CH), 7.47 (2H, AA′BB′, CH), 7.56 (2H, AA′BB′, CH), 7.61 (2H, AA′BB′, CH), 7.95 (2H, AA′BB′, CH), 8.02 (s, 1H,N–CH); 8.61 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.21 (CH3); 22.80, 25.87, 25.89, 29.37, 29.41, 31.70, 31.73 (CH2); 69.66, 69.76 (OCH2); 88.2, 90.8, 92.2, 95.1 (Cq, C≡C); 113.0, 114.8 (Cq); 116.8, 117.0 (aromat. CH); 122.5, 125.2 (Cq); 125.6, 126.4, 128.2 (aromat. CH); 130.3, 131.7 (Cq); 132.3, 132.4 (aromat. CH); 133.2, 138.6 (Cq); 139.9 (aromat. CH); 149.9 (aromat. CH); 153.8, 154.1 (Cq, C–O); 166.7 (Cq, NC–S); MS (FD): m/z (%) 716.1 (87, M+˙), 718.1 (100, [M + 2]+˙); EA: Calc. for C42H41BrN2O2S: C 70.28, H 5.76, N 3.90, S 4.47. Found: C 69.94, H 5.61, N 3.86, S 4.41.
General method for preparation of V-shaped molecules 1a–f and 2a–i
The mixture of 1.0 eq of thiadiazole derivatives 10–12 or thiazole derivatives 13–15, 1.0 eq of the corresponding terminal alkine 7–9, 0.2 eq of Pd(PPh3)4 and 0.1 eq of CuI in piperidine is stirred for 12 h at 60 °C. Afterwards the solvent is removed under vacuum and the products are isolated by means of column chromatography using mixtures of EtOAc/hexane or CH2Cl2/hexane, respectively.2,5-Bis-{4-[4-(4-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (1a)
Hexane/CH2Cl2 = 1/3 (Rf = 0.1), bright yellow solid; yield 58 mg (5). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.87 (m, 8H, CH2); 4.04 (t, 8H, OCH2, 3J = 6.4); 7.02, 7.04 (2s, 4H, CH); 7.65 (m, 12H, CH); 8.01 (4H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.2 (CH3); 22.8, 25.9, 29.4, 31.7 (CH2); 69.7 (OCH2); 88.8, 90.6, 93.4, 94.7 (Cq, C≡C); 111.5, 113.3, 114.5 (Cq); 116.8, 116.9 (aromat. CH); 118.3 (Cq, CN); 126.4 (Cq); 127.9 (aromat. CH); 128.5, 129.7 (Cq); 132.1, 132.2, 132.4 (aromat. CH); 153.8, 154.0 (Cq, C–O); 167.7 (Cq, NC–S); MS (FD): m/z (%) 1087.6 (100, M+˙); EA: Calc. for C72H72N4O4S: C 79.38, H 6.66, N 5.14, S 2.94. Found: C 78.96, H 6.83, N 5.19, S 3.01.
2,5-Bis-{4-[4-(3-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (1b)
Hexane/CH2Cl2 = 1/3 (Rf = 0.1), yellow solid; yield 36 mg (36 %). 1H NMR (400 MHz, CDCl3): δ 0.90 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.87 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02, 7.05 (2s, 4H, CH); 7.47 (ddd, 2H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH); 7.61 (ddd, 2H, 3J = 7.8, 4J = 1.55, 4J = 1.4, CH); 7.65 (4H, AA′BB, CH); 7.74 (ddd, 2H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH); 7.80 (ddd, 2H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 8.01 (4H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.2 (CH3); 22.6, 25.9, 29.4, 31.7 (CH2); 69.7 (OCH2); 88.4, 88.8, 92.6, 94.6 (Cq, C≡C); 113.0, 113.4, 114.3 (Cq); 116.8, 116.9 (aromat. CH); 118.3 (Cq, CN); 125.2, 126.5 (Cq); 128.0, 129.4 (aromat. CH); 129.7 (Cq); 132.4, 135.0, 135.7 (aromat. CH); 153.9, 154.0 (Cq, C–O); 167.7 (Cq, NC–S); MS (FD): m/z (%) 1087.6 (8, M+˙), 544.4 (100, [M + 1]2+); EA: Calc. for C72H72N4O4S: C 79.38, H 6.66, N 5.14, S 2.94. Found: C 78.96, H 6.84, N 5.09, S 3.01.
2-{4-[4-(3-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]-ethynylphenyl}-5-{4-[4-(4-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (1c)
Hexane/CH2Cl2 = 2/1 (Rf = 0.25), bright yellow solid; yield 51 mg (50 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.87 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.03, 7.04 (2s, 4H, CH); 7.47 (ddd, 1H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH); 7.63 (m, 9H, CH); 7.73 (ddd, 1H, 3J = 7.8, 4J = 1.65, 4J = 1.4, CH); 7.80 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 8.02 (m, 4H, CH); 13C NMR (100 MHz, CDCl3): δ 14.0, 14.1 (CH3); 22.8, 25.9, 29.38, 29.41, 31.70, 31.73 (CH2); 69.64, 69.67, 69.72 (OCH2); 88.5, 88.79, 88.83, 90.6, 92.6, 93.4, 94.6, 94.7 (Cq, C≡C); 111.5, 113.0, 113.3, 113.4, 114.3, 114.5 (Cq); 116.8, 116.9 (aromat. CH); 118.2, 118.7 (Cq, CN); 125.2, 126.4, 126.5 (Cq); 127.9 (aromat. CH); 128.5 (Cq); 129.4 (aromat. CH); 129.70, 129.74 (Cq); 131.5. 132.1, 132.2, 132.4, 134.9, 135.7 (aromat. CH); 153.8, 153.9, 154.0 (Cq, C–O); 167.72, 167.74 (Cq, NC–S); MS (FD): m/z (%) 1088.7 (100, M+˙); EA: Calc. for C72H72N4O4S: C 79.38, H 6.66, N 5.14, S 2.94. Found: C 78.76; H 6.53; N 5.12; S 2.86.
2,5-Bis-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (1d)
Hexane/EtOAc = 1/1 (Rf = 0.3), orange solid; yield 60 mg (74 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.55 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.04 (t, 8H, OCH2); 7.03 (s, 2H, CH), 7.04 (s, 2H, CH); 7.37 (4H, AA′BB′, CH); 7.64 (4H, AA′BB′, CH); 8.00 (4H, AA′BB′, CH); 8.60 (4H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.3 (CH3); 22.8 (CH2); 25.9 (CH2); 29.4 (CH2); 31.7 (CH2); 69.7 (OCH2); 88.8, 90.7, 92.3, 94.7 (Cq, C≡C); 113.2, 114.6 (Cq); 116.8, 117.0, 125.5 (aromat. CH); 126.4 (Cq); 128.0 (aromat. CH); 129.7, 131.6 (Cq); 132.4, 149.9 (aromat. CH); 153.8, 154.1 (Cq, C–O); 167.7 (Cq, NC–S); MS (FD): m/z 1039.7 (100, M+˙).
2-{4-[4-(4-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl)-ethynylphenyl]-5-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (1e)
Hexane/EtOAc = 3/1 (Rf = 0.3), yellow solid; yield 49 mg (57 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.03 (4H, CH); 7.37 (2H, AA′BB′, CH); 7.64 (m, 8H, CH); 8.00 (4H, AA′BB′, CH); 8.60 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.21 (CH3); 22.78, 22.80, 25.87, 25.89, 29.37, 29.41, 31.70, 31.73 (CH2); 69.67, 69.73 (OCH2); 88.8, 90.58, 90.69, 92.28, 93.42, 94.72 (Cq, C≡C); 111.5, 113.2, 113.4, 114.3, 114.6 (Cq); 116.81, 116.86, 116.89, 116.99 (aromat. CH); 118.7 (Cq, CN); 125.5 (aromat. CH); 126.4 (Cq); 128.0 (aromat. CH); 128.5, 129.7, 131.7 (Cq); 132.1, 132.2, 132.4, 149.9 (aromat. CH); 153.8, 154.0, 154.1 (Cq, C–O); 167.7 (Cq, NC–S); MS (FD): m/z (%) 1063.6 (100, M+˙). EA: Calc. for C70H72N4O4S: C 78.91, H 6.81, N 5.26, S 3.01. Found: C 78.48, H 7.03, N 5.20, S 3.03.
2-{4-[4-(3-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]-ethynylphenyl}-5-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3,4-thiadiazole (1f)
Hexane/EtOAc = 3/1 (Rf = 0.22), orange solid; yield 50 mg (48 %). 1H NMR (400 MHz, CDCl3): δ 0.90 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.03 (s, 4H, CH); 7.37 (2H, AA′BB′, CH); 7.46 (ddd, 1H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH) 7.59 (ddd, 1H, 3J = 7.8, 4J = 1.55, 4J = 1.4, CH); 7.64 (4H, AA′BB′, CH); 7.72 (ddd, 1H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH); 7.79 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 8.00 (4H, AA′BB′, CH); 8.60 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.17, 14.21 (CH3); 22.78, 22.80, 25.86, 25.89, 29.37, 29.38, 31.70, 31.73 (CH2); 69.67, 69.72 (OCH2); 88.54, 88.76, 88.82, 90.69, 92.26, 92.55, 94.59, 94.71 (Cq, C≡C); 113.0, 113.2, 113.4, 114.3, 114.6 (Cq); 116.82, 116.84, 116.88, 116.99 (aromat. CH); 118.2 (Cq, CN); 125.2 (Cq); 125.5 (aromat. CH); 126.4, 126.5 (Cq); 127.9, 129.4 (aromat. CH); 129.69, 129.74, 131.5, 131.7 (Cq); 132.37, 132.38, 134.9, 135.6, 149.9 (aromat. CH); 153.82, 153.84, 153.94, 154.08 (Cq, C–O); 167.71, 167.73 (Cq, NC–S); MS (FD): m/z (%) 1063.5 (100, M+˙). EA: Calc. for C70H72N4O4S: C 78.91, H 6.81, N 5.26, S 3.01. Found: C 78.67, H 6.99, N 5.03, S 3.17.
2,5-Bis-{4-[4-(4-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2a)
Hexane/CH2Cl2 = 3/1 (Rf = 0.1), orange solid; yield 38 mg (54 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02 (s, 2H, CH); 7.04 (s, 2H, CH); 7.61 (m, 14H, CH); 7.97 (2H, AA′BB′, CH); 8.08 (s, 1H,N–CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.21 (CH3); 22.80, 25.89, 25.92, 29.41, 29.45, 31.72, 31.75 (CH2); 69.7, 69.8 (OCH2); 87.6, 88.3, 90.7, 93.32, 93.35, 95.10, 95.13 (Cq, C≡C); 111.6, 113.1, 113.2, 114.9, 115.0 (Cq); 116.9, 117.0 (aromat. CH); 118.7 (Cq, CN); 123.5, 125.3 (Cq); 126.4, 126.6 (aromat. CH); 128.6, 131.3 (Cq); 132.1, 132.2, 132.3, 132.5 (aromat. CH); 133.3, 139.3 (Cq); 140.1 (aromat. CH); 153.8, 154.1 (Cq, C–O); 166.8 (Cq, NC–S); MS (FD): m/z (%) 1087.5 (100, M+˙); EA: Calc. for C73H73N3O4S: C 80.55, H 6.76, N 3.86, S 2.95. Found: C 80.53, H 6.87, N 3.77, S 2.86.
2,5-Bis-{4-[4-(3-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2b)
Hexane/EtOAc = 10/1 (Rf = 0.1), yellow solid; yield 37 mg (46 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.38 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02 (s, 2H, CH); 7.04 (s, 2H, CH); 7.47 (ddd, 2H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH); 7.61 (m, 7H, CH); 7.73 (ddd, 2H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH); 7.80 (ddd, 2H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 7.97 (2H, AA′BB′, CH); 8.09 (s, 1H,N–CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.22 (CH3); 22.80, 25.9, 29.42, 29.45, 31.72, 31.75 (CH2); 69.7, 69.8 (OCH2); 87.6, 88.3, 88.64, 88.65, 92.45, 92.48, 94.97, 95.02 (Cq, C≡C); 113.07, 113.15, 113.24, 114.68, 114.75 (Cq); 116.9, 117.0 (aromat. CH); 118.3 (Cq, CN); 123.5, 125.3 (Cq); 126.4, 126.6, 129.4 (aromat. CH); 131.3 (Cq); 132.5, 132.3, 132.5 (aromat. CH); 133.3 (Cq); 135.0, 135.7 (aromat. CH); 139.3 (Cq); 140.1 (aromat. CH); 153.81, 153.86, 154.0 (Cq, C–O); 166.8 (Cq, NC–S); MS (FD): m/z (%) 1087.6 (54, M+˙), 543.8 (100, M2+).
2-{4-[4-(4-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-5-{4-[4-(3-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2c)
Hexane/CH2Cl2 = 1/2 (Rf = 0.1), yellow solid; yield 35 mg (40 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02 (s, 2H, CH); 7.04 (s, 2H, CH); 7.47 (ddd, 1H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH); 7.61 (m, 11H, CH); 7.73 (ddd, 1H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH); 7.80 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 7.97 (2H, AA′BB′, CH); 8.09 (s, 1H,N–CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.22 (CH3); 22.8, 23.0, 25.86, 25.89, 29.38, 29.41, 31.70, 31.73 (CH2); 69.63, 69.66, 69.73, 66.75 (OCH2); 87.6, 88.2, 88.6, 90.6, 92.4, 93.3, 94.9, 95.1 (Cq, C≡C); 111.5, 113.0, 113.1, 114.6, 114.8 (Cq); 116.8, 116.9 (aromat. CH); 118.2, 118.7 (Cq, CN); 123.5, 125.18, 125.21 (Cq); 126.4, 126.6 (aromat. CH); 128.5 (Cq); 129.4 (aromat. CH); 131.2 (Cq); 131.5, 132.1, 132.2, 132.3, 132.4 (aromat. CH); 133.3 (Cq); 134.9, 135.6 (aromat. CH); 139.2 (Cq); 140.0 (aromat. CH); 153.74, 153.76, 153.96, 154.01 (Cq, C–O); 166.7 (Cq, NC–S); MS (FD): m/z (%) 1087.6 (100, M+˙).
2-{4-[4-(3-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-5-{4-[4-(4-cyanophenylethinyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2d)
Hexane/CH2Cl2 = 1/2 (Rf = 0.1), yellow solid; yield 50 mg (57 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02 (s, 2H, CH); 7.04 (s, 2H, CH); 7.47 (ddd, 1H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH); 7.61 (m, 11H, CH); 7.73 (ddd, 1H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH); 7.80 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 7.97 (2H, AA′BB′, CH); 8.09 (s, 1H,N–CH); 13C NMR (100 MHz, CDCl3): δ 14.21 (CH3); 22.8, 25.88, 25.90, 29.40, 29.42, 31.71, 31.74 (CH2); 69.7, 66.8 (OCH2); 87.6, 88.3, 88.6, 90.7, 92.5, 93.3, 95.0, 95.1 (Cq, C≡C); 111.5, 113.0, 113.03, 113.2, 114.6, 114.9 (Cq); 116.80, 116.84, 116.96 (aromat. CH); 118.3, 118.7 (Cq, CN); 123.5, 125.24, 125.26 (Cq); 126.4, 126.6 (aromat. CH); 128.5 (Cq); 129.4 (aromat. CH); 131.3 (Cq); 131.5, 132.1, 132.2, 132.3, 132.4 (aromat. CH); 133.3 (Cq); 134.9, 135.0, 135.7 (aromat. CH); 139.2 (Cq); 140.0 (aromat. CH); 153.77, 153.80, 153.99, 154.06 (Cq, C–O); 166.8 (Cq, NC–S); MS (FD): m/z (%) 1087.5 (100, M+˙); EA: Calc. for C73H73N3O4S: C 80.55, H 6.76, N 3.86, S 2.95. Found: C 80.66, H 6.60, N 3.87, S 3.05.
2,5-Bis-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2e)
Hexane/EtOAc = 2/1 (Rf = 0.2), orange solid; yield 45 mg (49 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.04 (t, 8H, OCH2, 3J = 6.4); 7.03 (s, 2H, CH); 7.04 (s, 2H, CH); 7.38 (4H, AA′BB′, CH); 7.60 (m, 6H, CH); 7.96 (2H, AA′BB′, CH); 8.08 (s, 1H,N–CH); 8.60 (4H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.21 (CH3); 22.78, 22.80, 25.86, 25.89, 29.37, 29.41, 31.70, 31.73 (CH2); 69.66, 69.75 (OCH2); 87.6, 88.2, 90.8, 92.15, 92.18, 95.08, 95.11 (Cq, C≡C); 112.85, 112.95, 114.9, 115.0 (Cq); 116.8, 117.0 (aromat. CH); 123.5, 125.2 (Cq); 125.5, 126.4, 126.6 (aromat. CH); 131.3, 131.7 (Cq); 132.3, 132.4 (aromat. CH); 133.3 (Cq); 139.2 (Cq); 140.0, 149.9 (aromat. CH); 153.73, 153.76, 154.11, 154.12 (Cq, C–O); 166.7 (Cq, NC–S); MS (FD): m/z (%) 1039.6 (100, M+˙).
2-{4-[4-(4-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-5-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2f)
Hexane/EtOAc = 3/1 (Rf = 0.2), yellow solid; yield 36 mg (51 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02, 7.03 (2s, 2H, CH); 7.04 (s, 2H, CH); 7.38 (2H, AA′BB′, CH); 7.57–7.65 (m, 10H, CH); 7.96 (2H, AA′BB′, CH); 8.08 (s, 1H,N–CH); 8.60 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.21 (CH3); 22.79, 22.80, 25.87, 25.90, 29.38, 29.42, 31.71, 31.74 (CH2); 69.65, 69.68, 69.77 (OCH2); 87.6, 88.2, 90.7, 90.8, 92.2, 93.3, 95.08, 95.13 (Cq, C≡C); 111.5, 112.9, 113.1, 114.8, 115.0 (Cq); 116.82, 116.83, 116.95, 117.1 (aromat. CH); 118.7 (Cq, CN); 123.5, 125.2 (Cq); 125.6, 126.4, 126.6 (aromat. CH); 128.5, 131.3, 131.7 (Cq); 132.1, 132.2, 132.3, 132.4 (aromat. CH); 133.3, 139.2 (Cq); 140.1, 149.9 (aromat. CH); 153.75, 153.80, 154.04, 154.13 (Cq, C–O); 166.8 (Cq, NC–S); MS (FD): m/z (%) 1063.6 (100, M+˙); EA: Calc. for C71H73N3O4S: C 80.11, H 6.91, N 3.95, S 3.01. Found: C 79.48, H 6.94, N 3.96, S 2.86.
5-{4-[4-(4-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl)ethynylphenyl]-2-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethinylphenyl}-1,3-thiazole (2g)
Hexane/EtOAc = 3/1 (Rf = 0.2), yellow solid; yield 43 mg (53 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.57 (m, 8H, CH2); 1.87 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02, 7.03, 7.04, 7.05 (4s, 4H, CH); 7.38 (2H, AA′BB′, CH); 7.57–7.65 (m, 10H, CH); 7.97 (2H, AA′BB′, CH); 8.09 (s, 1H,N–CH); 8.61 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.19, 14.22 (CH3); 22.8, 25.89, 25.91, 29.39, 29.42, 29.85, 31.72, 31.75 (CH2); 69.67, 69.69, 69.78 (OCH2); 87.6, 88.2, 90.7, 90.8, 92.2, 93.3, 95.10, 95.13 (Cq, C≡C); 111.5, 112.97, 113.02, 114.91, 114.94 (Cq); 116.8, 116.9, 117.0, 117.1 (aromat. CH); 118.7 (Cq, CN); 123.5, 125.2 (Cq); 125.6, 126.4, 126.6 (aromat. CH); 128.5, 131.3, 131.8 (Cq); 132.1, 132.2, 132.3, 132.4 (aromat. CH); 133.3, 139.3 (Cq); 140.1, 149.8 (aromat. CH); 153.77, 153.79, 154.07, 154.13 (Cq, C–O); 166.8 (Cq, NC–S); MS (FD): m/z (%) 1063.5 (100, M+˙); EA: Calc. for C71H73N3O4S: C 80.11, H 6.91, N 3.95, S 3.01. Found: C 79.60, H 6.99, N 3.83, S 2.98.
2-{4-[4-(3-Cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-5-{4-[4-(4-pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2h)
Hexane/EtOAc = 3/1 (Rf = 0.2), yellow solid; yield 60 mg (69 %). 1H NMR (400 MHz, CDCl3): δ 0.91 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02, 7.03 (2s, 2H, CH); 7.04 (s, 2H, CH); 7.38 (2H, AA′BB′, CH); 7.47 (ddd, 1H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH); 7.60 (m, 7H, CH); 7.73 (ddd, 1H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH); 7.80 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 7.97 (2H, AA′BB′, CH); 8.08 (s, 1H,N–CH); 8.60 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.17, 14.21 (CH3); 22.79, 22.80, 25.89, 25.91, 29.40, 29.44, 31.72, 31.74 (CH2); 69.72, 69.81 (OCH2); 87.6, 88.3, 88.6, 90.8, 92.2, 92.5, 95.0, 95.1(Cq, C≡C); 112.9, 113.1, 113.2, 114.7, 115.1 (Cq); 116.9, 117.0, 117.1 (aromat. CH); 118.2 (Cq, CN); 123.5, 125.3 (Cq); 125.6, 126.4, 126.6, 129.4 (aromat. CH); 131.3 (Cq); 131.5 (aromat. CH); 131.7 (Cq); 132.3, 132.4 (aromat. CH); 133.3 (Cq); 135.0, 135.7 (aromat. CH); 139.2 (Cq); 140.1, 149.9 (aromat. CH); 153.79, 153.84, 154.0, 154.2 (Cq, C–O); 166.8 (Cq, NC–S); MS (FD): m/z (%) 1063.8 (100, M+˙).
2-{4-[4-(4-Pyridylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-5-{4-[4-(3-cyanophenylethynyl)-2,5-bis-(hexyloxy)phenyl]ethynylphenyl}-1,3-thiazole (2i)
Hexane/EtOAc = 3/1 (Rf = 0.2), yellow solid; yield 45 mg (60 %). 1H NMR (400 MHz, CDCl3): δ 0.90 (m, 12H, CH3); 1.37 (m, 16H, CH2); 1.56 (m, 8H, CH2); 1.86 (m, 8H, CH2); 4.05 (t, 8H, OCH2, 3J = 6.4); 7.02, 7.03, 7.04, 7.05 (4s, 4H, CH); 7.38 (2H, AA′BB′, CH); 7.47 (ddd, 1H, 3J = 7.8, 3J = 7.8, 5J = 0.6, CH); 7.61 (m, 7H, CH); 7.73 (ddd, 1H, 3J = 7.8, 4J = 1.65, 5J = 1.4, CH); 7.80 (ddd, 1H, 4J = 1.65, 4J = 1.55, 5J = 0.6, CH); 7.97 (2H, AA′BB′, CH); 8.09 (s, 1H,N–CH); 8.60 (2H, AA′BB′, CH); 13C NMR (100 MHz, CDCl3): δ 14.18, 14.22 (CH3); 22.8, 25.87, 25.91, 29.40, 29.42, 31.72, 31.75 (CH2); 69.7, 69.8 (OCH2); 87.6, 88.2, 88.6, 90.8, 92.2, 92.5, 95.0, 95.1 (Cq, C≡C); 112.96, 113.03, 113.1, 114.7, 114.9 (Cq); 116.8, 116.9, 117.0, 117.1 (aromat. CH); 118.3 (Cq, CN); 123.5, 125.3 (Cq); 125.6, 126.4, 126.6, 129.4 (aromat. CH); 131.3 (Cq); 131.5 (aromat. CH); 131.7 (Cq); 132.3, 132.4 (aromat. CH); 133.3 (Cq); 135.0, 135.7 (aromat. CH); 139.3 (Cq); 140.1, 150.0 (aromat. CH); 153.8, 154.0, 154.1 (Cq, C–O); 166.8 (Cq, NC–S); MS (FD): m/z (%) 1063.7 (17, M+˙), 1064.5 (100, [M + H]+).
Acknowledgements
We are grateful to the Bundesministerium für Bildung und Forschung (BMBF) Deutsche Forschungsgemeinschaft (DFG) for their financial support. We thank Prof. Herbert Meier, Annette Oehlhof and Dr Norbert Hanold for the completion of FD mass spectrometry and elemental analysis at the University of Mainz.References
- T. Niori, T. Sekine, J. Watanabe, T. Furukawa and H. Takezoe, J. Mater. Chem., 1996, 6, 1231 RSC.
- H. Takezoe and Y. Takanishi, Jpn. J. Appl. Phys., 2006, 45, 597 CrossRef CAS.
- (a) P. I. C. Teixeira, A. J. Masters and B. M. Mulder, Mol. Cryst. Liq. Cryst., 1998, 323, 167 CrossRef CAS; (b) G. R. Luckhurst, Thin Solid Films, 2001, 393, 40 CrossRef CAS.
- (a) B. R. Acharya, A. Primak and S. Kumar, Phys. Rev. Lett., 2004, 92, 145506 CrossRef; (b) L. A. Madsen, T. J. Dingemans, M. Kakata and E. T. Samulski, Phys. Rev. Lett., 2004, 92, 145505 CrossRef CAS.
- D. Apreutesi and G. Mehl, J. Mater. Chem., 2007, 17, 4711 RSC and references therein.
- K. Dimitrova, J. Hauschild, H. Zaschke and H. Schubert, J. Prakt. Chem., 1980, 6, 933 CrossRef.
- (a) M. Parra, J. Alderete, C. Zuniga and S. Hernandez, Liq. Cryst., 2002, 5, 647 CrossRef; (b) M. Parra, J. Vergara, J. Alderete and C. Zuniga, Liq. Cryst., 2004, 11, 1531.
- M. Sato and S. Ujile, Adv. Mater., 1996, 8, 567 CrossRef CAS.
- Y. Xu, B. Li, H. Liu, Z. Guo, Z. Tai and Z. Xu, Liq. Cryst., 2002, 2, 199 CrossRef.
- R. Lunkwitz, C. Tschierske, A. Langhoff, F. Gießelmann and P. Zugenmaier, J. Mater. Chem., 1997, 9, 1713 Search PubMed.
- C. Tschierske, D. Joachimi, G. Y. Bak, H. Zaschke, B. Linstroem, H. Kresse and D. Demus, Ferroelectrics, 1991, 1–4, 289.
- S. Nakashima, M. Sato and I. Yamaguchi, Polym. Int., 2008, 57, 39 CrossRef CAS.
- M. A. Bates, Chem. Phys. Lett., 2007, 437, 189 CrossRef CAS.
- J. Thisayukta, Y. Nakayama and J. Watanabe, Liq. Cryst., 2000, 27, 1129 CrossRef CAS.
- T. J. Dingemans, N. S. Murthy and E. T. Samulski, J. Phys. Chem. B, 2001, 105, 8845 CrossRef CAS.
- S. Kang, Y. Saito, N. Watanabe, M. Tokita, Y. Takanishi, H. Takezoe and J. Watanabe, J. Phys. Chem. B, 2006, 110, 5205 CrossRef CAS.
- C. H. Lee and T. Yamamoto, Mol. Cryst. Liq. Cryst., 2001, 363, 77 CrossRef CAS.
- K. Dölling, H. Zaschke and H. Schubert, J. Prakt. Chem., 1979, 321, 643 CrossRef.
- A. Mori, A. Sekiguchi, K. Masui, T. Shimada, M. Horie, K. Osakada, M. Kawamoto and T. Ikeda, J. Am. Chem. Soc., 2003, 125, 1700 CrossRef CAS.
- A. J. Paraskos and T. M. Swager, Chem. Mater., 2002, 14, 4543 CrossRef CAS.
- M. Lehmann and J. Levin, Mol. Cryst. Liq. Cryst., 2004, 411, 273–281 CrossRef.
- (a) H. Meier, M. Lehmann, H. C. Holst and D. Schwöppe, Tetrahedron, 2004, 60, 6881–6888 CrossRef CAS; (b) Z. Bao, Y. Chen, R. Cai and L. Yu, Macromolecules, 1993, 26, 5281 CrossRef CAS; (c) S. H. Chen, A. C. Su, S. R. Han, S. A. Chen and Y. Z. Lee, Macromolecules, 2004, 37, 181 CrossRef CAS.
- M. Lehmann, S.-W. Kang, C. Köhn, S. Haseloh, U. Kolb, D. Schollmeyer, Q. Wang and S. Kumar, J. Mater. Chem., 2006, 16, 4326 RSC.
- M. Lehmann, C. Köhn, H. Kresse and Z. Vakhovskaya, Chem. Commun., 2008, 1768 RSC.
- (a) M. Parra, P. Hidalgo and J. Alderete, Liq. Cryst., 2005, 32, 449 CrossRef CAS; (b) M. Parra, J. Alderete, C. Zuñica, V. Jimenez and P. Hidalgo, Liq. Cryst., 2003, 30, 297 CrossRef CAS; (c) P. J. Martin and D. W. Bruce, Liq. Cryst., 2007, 34, 767 CrossRef CAS.
- (a) M. Lehmann and J. Levin, Mol. Cryst. Liq. Cryst., 2004, 411, 273–281 CrossRef; (b) S. Höger, Z. Naturforsch., 1998, 53b, 960–964 Search PubMed.
- J. Sandstrom, Adv. Heterocycl. Chem., 1968, 9, 615.
- J. Sheridan, Physical Methods in Heterocyclic Chemistry, ed. A. R. Katritzky, Academic Press, New York- London, vol. 6, 1974, p. 53 Search PubMed.
- Comprehensive Hetero-cyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon Press, Oxford, vol. 5, 1984, p. 10 Search PubMed.
- Geometry optimisations and calculations of molecular dipole moments have been carried out using the TURBOMOLE program package: R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 1631 Search PubMed ; TURBOMOLE V5-7-1 Current version: See http://www.turbomole.com/.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- A. Schäfer, H. Horn and R. J. Ahlrichs, Chem. Phys., 1992, 97, 2571.
- S. Grimme, Angew. Chem., 2008, 120, 3478 CrossRef; S. Grimme, Angew. Chem., Int. Ed., 2008, 47, 3430 CrossRef.
- R. J. Bushby et al., Angew. Chem., Int. Ed., 2000, 39, 2333 CrossRef.
- E. Jansson, P. C. Jha and H. Ågren, Chem. Phys., 2006, 330, 166 CrossRef CAS.
- C. Chiccoli, I. Feruli, O. D. Lavrentovich, P. Pasini, S. V. Shiyanovskii and C. Zannoni, Phys. Rev. E, 2002, 66 Search PubMed 030701.
- (a) R. Jenkens, R. L. Snyder in Chemical Analysis “Introduction to X-ray Powder Diffractometry”, Wiley, New York, vol. 138, 1996 Search PubMed; (b) P. Scherrer, Nachr. Ges. Wiss. Göttingen, Math.-Phys. Kl., 1918, 2, 96 Search PubMed.
- Y. J. Cheng and T. Y. Luh, Chem. Eur. J., 2004, 10, 5361 CrossRef CAS.
- T. C. Huang, H. Toraya, T. N. Blanton and J. Wu, J. Appl. Crystallogr., 1993, 26, 180 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |