An improved injectable polysaccharide hydrogel: modified gellan gum for long-term cartilage regenerationin vitro
Yihong
Gong
,
Chunming
Wang
,
Ruenn Chai
Lai
,
Kai
Su
,
Feng
Zhang
and
Dong-an
Wang
*
Division of Bioengineering, School of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive, N1.3-B2-13, 637457, Republic of Singapore. E-mail: DAWang@ntu.edu.sg; Fax: (+65) 67911761; Tel: (+65) 6316 8890
First published on 26th January 2009
Abstract
Polysaccharide-based hydrogels have been proven promising in tissue engineering applications due to their good biocompatibility, controllable properties and abundance, and are therefore in great demand as therapeutic cell vehicles. Gellan gum, a natural polysaccharide and FDA-approved food additive, has been well exploited in food and pharmaceutical industries. For tissue engineering purposes, however, gellan-based hydrogels need to be modified in order to meet the requirement of encapsulating living cells while maintaining their injectability, because the gelling point of this temperature-dependent gel is too high (above 42 °C). This study started from chemically scissoring (via oxidative cleavage) the gellan backbones to optimize the gelation temperature for injection as a result of down-regulating their molecular size. Chondrocytes were then seeded into the modified gellan gels, the cytocompatibility and the capability to promote in vitrotissue regeneration of which were evaluated. Notably, chondrocytic constructs based on modified gellan gel were kinetically monitored for 150 days in comparison with those based on agarose gel, showing superiority for long-term cartilaginous development in terms of many aspects such as cell proliferation and specific matrix formation. Biochemical analysis, histological staining, and immunofluorescent observation indicate that the modified gellan is able to retain the chondrocytes viability, enhance the extracellular matrix (ECM) secretion and maintain normal phenotype, which demonstrates that gellan is a potential and promising injectable vehicle for therapeutic chondrocyte delivery.
Introduction
Hydrogels, originating from either synthetic polymers or natural biomolecules, have proven competent in mediating cell transplantation and accommodating tissue regeneration.1–5 Their most remarkable feature is their injectability—that the cell-suspended macromonomers can be injected to a target site and gelated via various curing means to form cell-laden constructsin situ.6–12 Moreover, the liquid form of gel precursors provides a spatial fit between the formed implant and any randomly shaped venues, while the rapid process of gelation leads to a uniform distribution of transplanted cells in the implant bulk. All these excellent properties bring more expectations and investigations on the development of novel hydrogel materials to meet the ever greater demands by biomaterialists and tissue engineers.Gellan gum, an FDA-approved food additive, is a linear, anionic extracellular polysaccharide secreted by the bacterium Sphingomonas elodea with repeating tetrasaccharide units of D-glucose, D-glucuronic acid, D-glucose, and L-rhamnose as shown in Fig. 1A.13–15 Gellan gum-based hydrogels can be conveniently prepared, which have shown favorable physical properties as well as biocompatibility, and thus have been well exploited in food and pharmaceutical industries. Recent investigations by Smith et al.16 and Wang et al.17 have demonstrated that various cell types immobilized into or onto gellan gel-based matrices could maintain high viability and appropriate functionalities, implying the potential use of gellan gum for tissue engineering purposes.
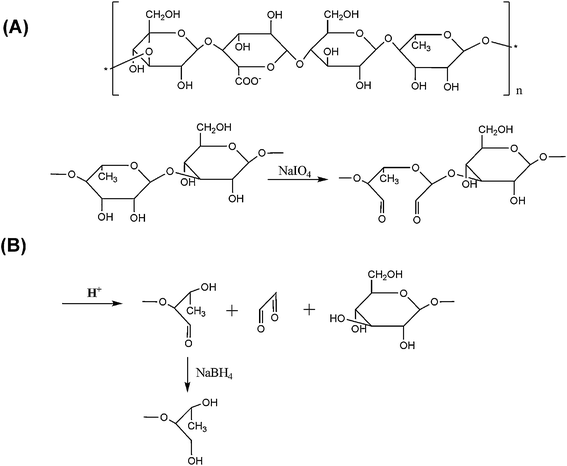 | ||
| Fig. 1 Schematic diagram of (A) the structure of gellan repeating units, and (B) NaIO4oxidation. | ||
However, a substantial problem remains for the gellan gel to qualify as genuinely injectable cell vehicle for practical use—its gelation temperature is too high. Gellan is a thermo-sensitive gel like agarose; its melting point (Tmelting ∼70 °C) is higher than its gelling point (gelation temperature, Tgelation). In the presence of cations (e.g., Na+, K+ and Ca2+), aqueous solutions of gellan undergo thermo-reversible gelation when cooled to below Tgelation, forming a gel that is stable below Tmelting. Therefore, the gellan solution is theoretically available to be injected into a defect site and form a gel in situ by cooling treatment. But in physiological cationic conditions, the Tgelation of commercially available and unmodified gellan is too high (>42 °C), and must be decreased to suspend the mammalian cells at physiological temperature (∼37.5 °C) for injectable purposes. To achieve a similar goal, Oliveira et al. adopted temperature- and pH-based technologies to fabricate gellan gum-based hydrogels into various structures, with encapsulated chondrocytes in these 3D matrices.18
In this study, we aimed to optimize the gelling point by adjusting the molecular weight of gellan. The molecular weight decrease may render the assembling and aggregation of gellan molecular chains more difficult in the gelation process, so as to lower the Tgelation in turn.19 A chemical scissoring process is designed via a NaIO4-based oxidative cleavage reaction on the adjacent dihydroxyl substrates throughout the gellan polysaccharide backbone as demonstrated in Fig. 1B. After modification, the injectable chondrocytic gellan gels are constructed and a long-term (150 d) performance in vitro is designed for practice to evaluate cartilaginous regeneration performance.
Materials and methods
Materials
All reagents unless otherwise specified were purchased from Sigma-Aldrich Inc. and used as received.Oxidation of gellan gum
Gellan (Phytagel™) was mixed with deionized water (5 g gum powder in 500 mL) and heated to 70 °C for 3–5 hrs to obtain a homogeneous aqueous solution. After the gellan solution was cooled to 40 °C, NaIO4 (0.25 M) was then added at different dosages (3 mL, 6 mL and 7.2 mL) with gentle stirring and reacted for 1–120 hrs to achieve various oxidization degrees. An equimolar amount of ethylene glycol was added at predefined reaction time points to stop the oxidation reaction. After 30 min reaction, 5ml NaBH4 (0.124 M) was added and reacted for 8 hrs to reduce the aldehyde groups. The resulting solution was loaded onto Sephadex® G25 SEC (mobile phase: deionized water) to remove all the small molecular impurities. Finally the product was lyophilized and stored in a dark desiccator. The details of the reaction conditions and product serial numbers are listed in Table 1. The sample serial numbers indicate the NaIO4 dosage and reaction time. Nuclear magnetic resonance spectroscopy (Bruker advance 300 NMR) was employed to investigate the molecular structure of the product before and after NaBH4reduction.| NaIO4/Gellan (×10−2mmol·g−1) | Oxidation time (h) | [η](×103mL·g−1) | M w (Da) | |
|---|---|---|---|---|
| Raw Gellan | 0 | 0 | 9.06 | 4.84 × 106 |
| G(15)-1h | 15 | 1 | 3.62 | 1.77 × 106 |
| G(30)-1h | 30 | 1 | 1.76 | 7.94 × 105 |
| G(15)-36h | 15 | 36 | 3.08 | 1.48 × 106 |
| G(30)-36h | 30 | 36 | 0.52 | 2.09 × 105 |
| G(36)-36h | 36 | 36 | 0.23 | 8.55 × 104 |
| G(30)-72h | 30 | 72 | 0.41 | 1.61 × 105 |
| G(30)-120h | 30 | 120 | 0.18 | 6.53 × 104 |
Intrinsic viscosities test and molecular weight
The intrinsic viscosities of raw gellan and oxidized gellan were determined at various concentrations (0.05%, 0.04%, 0.025% and 0.005%, w/v) in deionized water with a viscometer (AMVn Automated Microviscometer, Anton Paar) at 23 °C. Molecular weight (Mw) is calculated using the Kuhn–Mark–Houwink equation:| [η] = KMwα |
Gelation point test
The gellan was dissolved in PBS at various concentrations at 70 °C and the solutions were added into eppendorf® tubes (1 mL per tube). The tubes were then incubated in an accurate water bath with a temperature error of less than 0.1 °C. The initial temperature of the water bath was kept high to prevent gelation of the samples. Every 20 min, the water bath temperature was decreased by 1 °C. The tubes were brought for observation after the temperature has become stable for at least 10 min. Once the gellan solution was observed to lose its flow ability, the current temperature was recorded as the gelation temperature. In order to minimize the error of human observation, the Tgelation determinations of each concentration were repeated at least 5 times.Cytotoxicity test
According to the results of gelation point tests, G(15)-36h (Table 1) was chosen for the determination of the material biocompatibility. To determine the cytotoxicity of the oxidized gellan, human epidermis fibroblasts (hEFBs, passage 8–15, purchased from Cambrex, North Brunswick, NJ, USA) were incubated with the extractant of the hydrogel. The gellan gels were fabricated from 5% (w/v) G(15)-36h in PBS or 5% (w/v) raw gellan in deionized watervia injecting the hydrogel solution directly into a silica rubber mould (diameter = 16mm, height = 3–4mm). The hydrogels were then immersed in Dulbecco's minimum essential medium (DMEM) with the supplements of 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 10% (v/v) fetal bovine serum (FBS), 100 units/ml penicillin, and 100 µg/ml streptomycin at a ratio of 100 mg hydrogel/mL DMEM for 24 hrs at 37 °C. The extracted medium was collected for cytotoxicity determination. All cell culture reagents were purchased from Gibco (Invitrogen, Singapore).The hEFBs were seeded onto a 96-well culture plate (5 × 103cells per well) and cultivated in DMEM supplemented with 10% (v/v) FBS at 37 °C in 5% CO2 atmosphere. After 12 hrs, DMEM was removed and 200 µL extractant of G(15)-36h was added. Every 24 hrs, the medium was discarded and new extractant was added. The cell viability was tested using WST-1 assay {4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate assay, Roche Diagnostics, Germany}. Briefly, 10 µL of WST solution was added into each well (n = 3, for each time point). After 4 hrs incubation the absorbance at 450 nm of the medium was determined by a microplate reader (Multiskan® spectrum, Thermo). As a control, parallel experiments were carried out by culturing the hEFBs in raw gellan extractant and DMEM with 10% (v/v) FBS simultaneously. The morphology of the cells at day 7 was observed by fluorescence microscope using “live/dead” dye staining (Molecular probes, Invitrogen Singapore).
Chondrocyte isolation and culture
Chondrocytes were isolated from cartilage tissue of edible porcine articular cartilage. Briefly, cartilage tissue obtained from the joint of a 5-month-old pig was cut into small chips. Chondrocytes were isolated by incubating the cartilage pieces in DMEM culture medium with 10% (v/v) FBS containing 1 mg/mL collagenase type II at 37 °C for 12 hours under gentle stirring. The chondrocytes were centrifuged, and resuspended in DMEM supplemented with 20% (v/v) FBS, 0.01 M 4-(2-hydroxyethyl)-piperazine-1-ethanesulfonic acid (HEPES), 0.1 mM nonessential amino acids (NEAA), 0.4 mM proline, 0.05 mg/mL vitamin C, 100 units/mL penicillin and 100 µg/mL streptomycin. The cell suspension was then seeded in 75 mL tissue culture flask (Falcon, seeding density ∼2 × 104cells/cm2) and incubated in humidified air with 5% CO2 at 37 °C for routine culture.Chondrocyte encapsulation
After a confluent cell layer was formed (7–10 d), the chondrocytes were detached using 0.25% (w/v) trypsin in PBS. The gellan solution (37 °C, G(15)-36h, 1.8% dissolved in PBS, w/v) was added to re-suspend the chondrocytes. The hydrogel solution mixed with chondrocytes was injected onto the surface of silica rubber (40 µL each droplet). The seeding density was 1.2 × 107cells/mL. After gelation at room temperature for 15 min, the gellan beads were transferred to a 24-well cell culture plate and were then incubated under the conditions described previously. As a control, the chondrocytes were also encapsulated in agarose beads (Type VII, 1.8% w/v) following the same approach and with the same cell seeding density and initial volume (40µL each) and cultivated under the same conditions.Biochemical analysis
After 3–150 d culture, hydrogel/chondrocyte constructs were taken out, washed 3 times with deionized water to remove the salt in the medium followed by freeze drying (36 hrs), and digested with 1 mL of papain per sample for biochemical analysis.21,22DNA content was assessed by applying the Hoechst 33258 dye assay (7.7 pg DNA/cell).23,24Dimethylmethylene blue dye was added into the digested solution, and absorption at 525 nm was measured using a UV-VIS spectrophotometer (Multiskan® spectrum, Thermo) to quantify sulfated glycosaminoglycan (GAG) content.25,26 The total collagen was quantified from the hydroxyproline content after hydrolysis (6N HCl at 115 °C for 18 hrs) and reaction with p-dimethylaminobenzaldehyde and chloramine-T. Absorption at 550 nm was measured using the spectrophotometer.27,28 The percentages of components in the hydrogel were calculated according to the biochemical analysis results and the hydrogel dry weight.Histology
Samples from each time point (n = 2 for each group) were fixed in 4% (w/v) neutral buffered paraformaldehyde for 2 d. The fixed specimens were then embedded in paraffin and cross sectioned using a microtome. Sections from all groups were subsequently stained with hematoxylin-eosin (H&E), Safranin-O, or Masson's Trichome staining.For immunofluorescent staining, the specimens were incubated with 10% goat blocking serum (w/v, in PBS) for 20 minutes to suppress non-specific binding of IgG. The specimens were then incubated with the collagen II (2 ng/mL in PBS, MAB8887, Chemicon®) primary antibody at 4 °C overnight and washed with PBS. The specimens were incubated with Anti-IgG (5 ng/mL in PBS, Invitrogen Alexa Fluor®, 488) at room temperature for 1 hr in the dark followed by PBS washing 3 times. The specimens were incubated with the collagen I primary antibody (2 ng/mL, mouse monoclona IgG, Santa Cruz Biotechnology) at room temperature for 1 hr. The specimens were washed with PBS and Anti-IgG (5 ng/mL, Invitrogen Alexa Fluor®, 543) was added. After 1 hr incubation, the specimens were washed with PBS and then DAPI (1 ng/mL, Invitrogen) was utilized to locate the cell nucleus.
Statistical analysis
Where appropriate, ANOVA and Student’s t-test were performed to analyze biochemical results with 3 samples in each group and a P < 0.05 was considered to indicate a statistically significant difference. Data are presented as mean ± SD.Results
Gellan oxidation
NaIO4 was utilized to cleave the adjacent dihydroxyl groups in the polysaccharide chain, which led to the hydrolyzation of glycosidic linkages and degradation of gellan. NMR was used to trace the reaction process. As shown in Fig. 2 (I) and (II), a peak appears at 9.077 ppm, which represents the proton in the aldehyde group, after NaIO4 cleaving. The peak disappears after the reduction by NaBH4. | ||
| Fig. 2 1H NMR spectra (300 MHz, in D2O) of (I) G(15)-36h before reduction, (II) magnified spectrum of G(15)-36h before reduction at 9.0 ppm and (III) magnified spectrum of G(15)-36h after reduction at 9.0 ppm. | ||
Intrinsic viscosities [η] and Mw of raw and oxidized gellan are listed in Table 1. The [η] and Mw of oxidized gellan were reduced significantly, compared with those of raw gellan. With the increase of dosage of NaIO4 or oxidation time, the [η] and Mw were decreased.
The Tgelation of raw gellan as well as gellan oxidized to different extents were measured, and are shown in Fig. 3. The Tgelation was increased with the increase of polysaccharide concentration. However, raw gellan has a very high gelation temperature with the lowest of 44 °C at a concentration of 0.1% (gellan can hardly form a gel when the concentration is below 0.1%). After oxidation, the gelation temperature of gellan was reduced by the increases of oxidant dosage. For example, the gelation temperature of G(15)-1h is 49 °C at 3% concentration, while the gelation temperature of G(30)-1h at the same concentration is decreased to 22 °C. The oxidation time also has a great influence on the oxidized gellan gelation temperature. For instance, the gelation temperature of gellan at a concentration of 5% with the same oxidant dosage (NaIO4/gellan = 0.30, mmol/g) shifts from 37 °C [after 1h oxidation, G(30)-1h] to 27 °C [after 36 hrs oxidation, G(30)-36h] and finally to 22 °C [after 72 hrs oxidation, G(30)-72h].
 | ||
| Fig. 3 Gelation temperatures of oxidized gellan with different reaction conditions. | ||
Cytotoxicity test
Thy cytotoxicities of raw and oxidized gellan were determined by WST assay and living cell observation. As shown in Fig. 4 (A), the viability of the cells that were incubated with the extractant of raw or oxidized gellan increased as a function of culture time. No significant difference was observed in the cell viability profiles of these three groups. Fibroblasts stained with “live/dead” assay are shown in Fig. 4(B–D). The living and dead cells were, respectively, stained in green and red under fluorescence microscope. The majority of the fibroblasts from all three groups were found to be viable and dead cells were seldom found. Moreover, cells from all three groups maintained a normal spindle-like cell shape. As a summarization of above data, both raw and oxidized gellan showed no toxicity. | ||
| Fig. 4 Cytotoxicity evaluation of gellan via (A) cytoviability determination of human epidermis fibroblasts as a function of culture time, and cell morphology observation after incubation in the extractant of (B) raw gellan, (C) G(15)-36h and (D) DMEM at day 7 via “live/dead” staining. | ||
Cartilage tissue regeneration
Biocompatibility of the gellan gel was evaluated by culturing chondrocytes encapsulated in the gel with agarose gel as a control. Long term in vitro culture (150 d) was performed in order to reveal the neo tissue regeneration capability of this novel hydrogel. Fig. 5 (A1–A5) are images of oxidized gellan (G) and agarose (A) gross views, encapsulating porcine articular chondrocytes with initial seeding density 1.2 × 107cells/mL. Fig. 5 (B) shows the evolution of the average wet weight of oxidized gellan and agarose gels, with the initial hydrogel volume being 40 µL in both cases. It was shown that weight of both the gellan/cell and agarose/cell constructs increased moderately from day 3 to day 12. From day 12 to day 25, the weight increase in gellan was more radical compared with that in agarose. From day 25 to day 150, the weight of both hydrogels appeared to be constant with slight fluctuations. Fig. 5 (C) shows the trend of average cell number/construct: chondrocytes in both gellan and agarose proliferated very rapidly during the first 25 d, and then the cell number remained constant after undergoing some decrease. It should be noted that the chondrocytes cultured in gellan had much higher proliferation rate than those in agarose during the rapid cell proliferating period in the first 25 d and the average cell number per construct was always significantly higher than that in agarose at each time point. Fig. 6 shows fluorescent images of oxidized gellan/cell and agarose/cell constructs after “live/dead” staining. The images demonstrate that the chondrocytes remained viable in both gels. | ||
| Fig. 5 Gross view of gellan/chondrocytes constructs (left, labeled with “G”) and agarose/chondrocytes constructs (right, labeled with “A”) after (A1) 3, (A2) 25, (A3)40, (A4) 70, (A5) 150 d culture; (B) average gel weight and (C) average cell number in one construct of gellan/chondrocytes and agarose/chondrocytes constructs as a function of culture time (n = 3). *Differences between different constructs at the same culture time are not significant (p > 0.05) and **Differences between different constructs at the same culture time are significant (p < 0.05). | ||
 | ||
| Fig. 6 Fluorescent images to show the viability of chondrocytes encapsulated in agarose after (A) 3, (B) 12, (C) 25, and (D) 40 d culture, and in gellan after (E) 3, (F) 12, (G) 25, and (H) 40 d culture. | ||
Cross-section histochemistry staining of hydrogel/cell was utilized to further investigate the ECM secretion and neo tissue regeneration in Fig. 7. H&E (Fig. 7 A1 and C1) staining shows that the chondrocytes were distributed in the constructs uniformly at the beginning of culture. The matrix surrounding the chondrocytes was stained blue because of the basophilic mucus and proteoglycan secretion (Fig. 7 A2–A6 and C2–C6). It is noted that cartilage-like structure with mature chondrocytes and lacuna are more obvious in gellan/cell constructs rather than that in agarose/cell constructs after 70 d culture. Safranin-O staining (Fig. 7 B1–B6 and D1–D6) discloses the process of sulfated-GAG secretion. At the beginning of culture, the amount of sulfated-GAG (red or orange) was small and mainly centralized around the cells. As the culture time extended, the sulfated-GAG began to distribute all over the constructs and became more and more uniform. Compared with agarose/cell constructs at each time point, gellan/cell constructs had more sulfated-GAG secretion and more uniform matrix distribution. Collagen secretion was found to be similar to sulfated-GAG secretion (Fig. 8 A1–A6 and D1–D6). Large amounts of collagen secretion (blue) were found in the late stages of cell culture. Moreover, the collagen distribution in gellan/cells was more uniform than that in agarose/cells, especially after 70 d.
 | ||
| Fig. 7 Hematoxylin and eosin staining of agarose/cell constructs after (A1) 12, (A2) 25 (A3) 40, (A4) 70, (A5) 100, (A6) 150 d culture and gellan/cell constructs after (C1) 12, (C2) 25 (C3) 40, (C4) 70, (C5) 100, (C6) 150 d culture. Safranin-O staining of agarose/cell constructs after (B1) 12, (B2) 25 (B3) 40, (B4) 70, (B5) 100, (B6) 150 d culture and gellan/cell constructs after (D1) 12, (D2) 25 (D3) 40, (D4) 70, (D5) 100, (D6) 150 d culture. | ||
 | ||
| Fig. 8 Masson's Trichome staining (blue for total collagen) of agarose/cell constructs after (A1) 12, (A2) 25 (A3) 40, (A4) 70, (A5) 100, (A6) 150 d culture and gellan/cell constructs after (D1) 12, (D2) 25 (D3) 40, (D4) 70, (D5) 100, (D6) 150 d culture. Collagen II immunofluorescent staining (green for collagen II) of agarose/cell constructs after (B1) 12, (B2) 25 (B3) 40, (B4) 70, (B5) 100, (B6) 150 d culture and gellan/cell constructs after (E1) 12, (E2) 25 (E3) 40, (E4) 70, (E5) 100, (E6) 150 d culture. Collagen I immunofluorescent staining (red for collagen I) of agarose/cell constructs after (C1) 12, (C2) 25 (C3) 40, (C4) 70, (C5) 100, (C6) 150 d culture and gellan/cell constructs after (F1) 12, (F2) 25 (F3) 40, (F4) 70, (F5) 100, (F6) 150 d culture. | ||
In order to distinguish collagen type I and type II, immunofluorescent staining was performed. As shown in Fig. 8 B1–B6 and E1–E2, obvious collagen type II secretion (green) was detected in both gellan/cells and agarose/cells, and the distribution was similar with the collagen detected by Masson's Trichome staining. Compared with collagen type II, the secretion of collagen type I was rare and unremarkable (Fig. 8 C1–C6 and F1–F6).
The amount of GAG and collagen was quantified and normalized by cell number in the hydrogel/cells constructs. As shown in Fig. 9 (A), the amount of GAG secretion increased with the increase of culture time. The GAG secretion per cells had no significant difference between the gellan and agarose. Due the more rapid cell proliferation rate, the normalized collagen secretion per cell in gellan constructs in early culture stage (days 3–25) was lower than that in agarose. However, the average collagen secretion per cell in gellan increased faster than that in agarose and finally exceeded it after 100 d culture.
 | ||
| Fig. 9 The secretion of (A) GAG and (B) collagen in the agarose/cell and gellan/cell constructs as a function of culture time. The values have been normalized by the cell number in the constructs. * Differences between different constructs at the same culture time are not significant (p > 0.05) and ** differences between different constructs at the same culture time are significant (p < 0.05). | ||
The components of hydrogel/cells constructs can be roughly divided into three parts: collagen, GAG (the two main parts of the ECM) and other components (such as hydrogel, cells and some bio-molecules). The percentages of the three parts were calculated according to biochemical quantification and are shown in Fig. 10. At the beginning of the culture the collagen and GAG percentages were very low, while the other components (possibly mainly consisting of hydrogel) contributed the most part of the constructs’ dry weight. As the culture time extended, the percentages of collagen and GAG increased due to continuous ECM secretion. The component ratios in gellan/cells and agarose/cells were similar during days 25 to 70. However, the percentage of other components in gellan/cells decreased much faster than that in agarose/cells from days 70 to 150. Finally over 96% dry weight of gellan/cells constructs was contributed by collagen and GAG at day 150, while the percentage of other components still remained about 19% in agarose/cells constructs.
 | ||
| Fig. 10 Schematic diagram of major constituent percentages (dry weight) in agarose/cell constructs (upper) and gellan/cell constructs (lower) at different culture times (n = 3). | ||
Discussion
In order to endow gellan with the capability of injection with therapeutic cells, it was modified via NaIO4oxidation and Smith degradation in this study. The oxidation cleaved polymer chains into smaller segments, leading to the decrease in polymer molecular weight and partial destruction of cross-linking points. As a result, gelation becomes more difficult and the gelation temperature decreases. By increasing the oxidant dosage or oxidation period, the gelation temperature can be decreased to even lower, within the range of 25 °C to 37 °C. However, higher NaIO4 dosage would possibly exacerbate Smith degradation and generate more side products that are hardly controllable or predictable, and the reaction time applied in this study should be sufficiently long to achieve a uniform hydrolyzation of gellan to the desirable extent. Hence, we preferred to fabricate the gel in relatively “gentle” conditions (lower oxidant dosage with longer reaction time), and G(15)-36h was eventually chosen for the subsequent biological evaluations. The gelation temperature of 1.8% G(15)-36h solution is around 26 °C, which makes it possible to mix the cells in the hydrogel solution without any latent harm to the cells before injection. The hydrogel/cells suspension can be maintained in fluid state for a long time before being injected into the subcutaneous tissue and solidified by some cooling process such as blowing of cold air or ice compression. After solidification, the gel construction would be stable under Tmelting (∼70 °C) and act as a 3-D supporting matrix for tissue regeneration. The viscous gel solution allows the chondrocytes to be suspended uniformly with gentle vortex and no significant cell precipitation was observed in the gelation process. The fluorescence image (Fig. 6E) and cross section images (Fig. 7 C1, D1) confirmed that the distribution of cells was uniform. Moreover, this modification method introduces aldehyde groups with high reactivity to the polysaccharide chains along with decreasing the gelation temperature. This gives the oxidized gellan high potential to be further modified by conjugating biomolecules using aldehyde group based reactions such as Schiff-base reaction,17 although it has not been demonstrated in the current work. In this case, the oxidized gellan was reduced by NaBH4 treatment, which avoided the potentially toxic risk caused by aldehyde groups.Chondrocytes were encapsulated in modified gellan for evaluation of neo tissue regeneration; and agarose gel constructs were used as controls. Agarose has been widely used in cartilage regeneration as a popular hydrogel model and it has been shown that agarose can provide a good growth environment for chondrocytes and help the cells keep a differentiated phenotype as assessed by expression of type II collagen and synthesis of proteoglycans.5,29–32 Moreover, the gelation behaviors of gellan and agarose are similar, and therefore agarose should be a good reference system to evaluate modified gellan. The constructs were subjected to long term in vitro culture to get a bigger picture of the biocompatibility/performance of the hydrogels. The cell “live/dead” observation showed that most of the chondrocytes could remain viable in a normally round shape within the gellan gel during the culture (Fig. 6). ECM components, including GAG and collagen, were continuously secreted and deposited in gellan/chondrocytes constructs over the culturing period (Fig. 7). Collagen type I was rarely found in the section of gellan construct sections, which implies that the chondrocytes could maintain their original phenotype during long-term regenerationin vitro (Fig. 8). It is noted that neo tissue regeneration in gellan gel was much faster than that in agarose, which has been demonstrated by the comparison of gel weight and cell numbers (Fig. 5). In summary, the modified gellan is capable of encapsulating chondrocytes, enhancing cell proliferation, and promoting ECM secretion with much faster rate in gellan than that in agarose.
From histology section observation, the terminal ECM distribution in agarose (Fig. 7 A5–A6, B5–B6 and Fig. 8 A5–A6, B5–B6) was not uniform. Both collagen and GAG aggregated around the chondrocytes, while less ECM was found at other sites. The most probable reason for both the components and distribution of ECM in agarose could be nondegradability of agarosein vitro, which acts as a barrier preventing tissue regeneration in the later stages of culture. On the other hand, in gellan constructs, the chondrocytes and ECM tended to merge and cartilage-like tissue was formed after 100 d culture (Fig. 7 C5–C6 and D5–D6). The biochemical quantification results also show that the total weight percentage of collagen and GAG in the hydrogel increased with culture time. Collagen and GAG in the gellan construct were spread over more than 95% of the gel after 100 d of culture, while they occupied less than 80% of the gel space in agarose constructs. The superior performance of modified gellan might be partially attributed to NaIO4oxidation and molecular weight decrease. The modified gellan undergoes the process of Smith degradation during the preparation process, so that the cross-linking of sites by the cleaved molecular chain aggregation becomes weaker, possibly leading the gel to lapse into forms of small chips or dissolvable molecules during long-term incubation. It has been proved that the degradability of alginate, a polysaccharide with similar structure, can be controlled by NaIO4oxidation.33 In subsequent work, we plan to investigate the degradation properties of modified gellan, which may be potentially an important and useful feature of the gels.
Conclusion
In summary, a novel injectable hydrogel has been successfully developed basing on a FDA-approved polysaccharide, gellan gum, the molecular size of which has been chemically reduced and optimized to alter the working gelling point to the cell-favorable range and simultaneously maintain the injectable processability for transplantation. The yielded chondrocytic cell-laden gellan gels have been kinetically tested for nearly half a year. The results indicate that such modified gellan gels have superior performance in long-term culture in vitro in comparison with other popularly employed gels such as agarose. To summarise, this newly developed hydrogel-based scaffolding system is capable of providing a promising platform for cartilaginous regeneration.Acknowledgements
This research was financially supported by Grant ARC 10/06, Ministry of Education (MoE), Singapore.References
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920 CrossRef CAS.
- J. A. Hubbell, Bio-technology, 1995, 13, 565 CrossRef CAS.
- E. Lavik and R. Langer, Appl. Microbiol. Biot., 2004, 65, 1 Search PubMed.
- D. A. Wang, C. G. Williams, Q. A. Li, B. Sharma and J. H. Elisseeff, Biomaterials, 2003, 24, 3969 CrossRef CAS.
- Y. H. Gong, L. J. He, J. Li, Q. L. Zhou, Z. W. Ma, C. Y. Gao and J. C. Shen, J. Biomed. Mater. Res. B, 2007, 82B, 192 CrossRef CAS.
- G. M. Cruise, D. S. Scharp and J. A. Hubbell, Biomaterials, 1998, 19, 1287 CrossRef CAS.
- D. A. Wang, C. G. Williams, F. Yang, N. Cher, H. Lee and J. H. Elisseeff, Tissue Eng., 2005, 11, 201 CrossRef CAS.
- A. D. Augst, H. J. Kong and D. J. Mooney, Macromol. Biosci., 2006, 6, 623 CrossRef CAS.
- K. H. Bouhadir, K. Y. Lee, E. Alsberg, K. L. Damm, K. W. Anderson and D. J. Mooney, Biotechnol. Progr., 2001, 17, 945 CrossRef CAS.
- Y. Hong, H. Q. Song, Y. H. Gong, Z. W. Mao, C. Y. Gao and J. C. Shen, Acta Biomater., 2007, 3, 23 CrossRef CAS.
- D. A. Wang, S. Varghese, B. Sharma, I. Strehin, S. Fermanian, J. Gorham, D. H. Fairbrother, B. Cascio and J. H. Elisseeff, Nat. Mater., 2007, 6, 385 CrossRef CAS.
- X. Z. Shu, Y. C. Liu, F. Palumbo and G. D. Prestwich, Biomaterials, 2003, 24, 3825 CrossRef CAS.
- H. Grasdalen and O. Smidsrod, Carbohyd. Polym., 1987, 7, 371 CrossRef CAS.
- P. E. Jansson, B. Lindberg and P. A. Sandford, Carbohyd. Res., 1983, 124, 135 CrossRef CAS.
- I. B. Bajaj, P. S. Saudagar, R. S. Singhal and A. Pandey, J. Biosci. Bioeng., 2006, 102, 150 CrossRef CAS.
- M. A. Smith, R. M. Shelton, Y. Perrie and J. J. Harris, J. Biomater. Appl., 2007, 22, 241 CrossRef.
- C. M. Wang, Y. H. Gong, Y. M. Lin, J. B. Shen and D. A. Wang, Acta. Biomater., 2008, 4, 1226 CrossRef CAS.
- J. T. Oliveira, R. Picciochi, T. C. Santos, L. Martins, L. G. Pinto, P. B. Malafaya, R. A. Sousa, A. P. Marques, A. G. Castro, J. F. Mano, N. M. Neves and R. L. Reis, Tissue. Eng. A, 2008, 14, 748 Search PubMed (Meeting Abstract: OP165).
- S. Mashimo, N. Shinyashiki and Y. Matsumura, Carbohyd. Polym., 1996, 30, 141 CrossRef CAS.
- E. Dreveton, F. Monot, J. Lecourtier, D. Ballerini and L. Choplin, J. Ferment. Bioeng., 1996, 82, 27.
- G. A. Ameer, T. A. Mahmood and R. Langer, J. Orthop. Res., 2002, 20, 16 CrossRef CAS.
- R. L. Y. Sah, Y. J. Kim, J. Y. H. Doong, A. J. Grodzinsky, A. H. K. Plaas and J. D. Sandy, J. Orthop. Res., 1989, 7, 619 CrossRef CAS.
- Y. J. Kim, R. L. Y. Sah, J. Y. H. Doong and A. J. Grodzinsky, Anal. Biochem., 1988, 174, 168 CAS.
- L. E. Freed, A. P. Hollander, I. Martin, J. R. Barry, R. Langer and G. Vunjak-Novakovic, Exp. Cell Res., 1998, 240, 58 CrossRef CAS.
- R. W. Farndale, D. J. Buttle and A. J. Barrett, Biochim. Biophys. Acta., 1986, 883, 173 CrossRef CAS.
- L. E. Freed, J. C. Marquis, A. Nohria, J. Emmanual, A. G. Mikos and R. Langer, J. Biomed. Mater. Res., 1993, 27, 11 CrossRef CAS.
- A. P. Hollander, T. F. Heathfield, C. Webber, Y. Iwata, R. Bourne, C. Rorabeck and A. R. Poole, J. Clin. Invest., 1994, 93, 1722 CrossRef CAS.
- J. F. Woessner, Arch. Biochem. Biophys., 1996, 93, 440.
- H. A. Awad, M. Q. Wickham, H. A. Leddy, J. M. Gimble and F. Guilak, Biomaterials, 2004, 25, 3211 CrossRef CAS.
- R. G. LeBaron and K. A. Athanasiou, Biomaterials, 2000, 21, 2575 CrossRef CAS.
- P. D. Benya and J. D. Shafer, Cell, 1982, 30, 215 CrossRef CAS.
- B. Rahfoth, J. Weisser, F. Sternkopf, T. Aigner, K. von der Mark and R. Brauer, Osteoarthr. Cartilage., 1998, 6, 50 Search PubMed.
- H. J. Kong, E. Alsberg, D. Kaigler, K. Y. Lee and D. J. Mooney, Adv. Mater., 2004, 16, 1917 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |