Use of an ambient ionization flowing atmospheric-pressure afterglow source for elemental analysis through hydride generation
Gregory D.
Schilling
a,
Jacob T.
Shelley
a,
José A. C.
Broekaert
b,
Roger P.
Sperline
c,
M. Bonner
Denton
c,
Charles J.
Barinaga
d,
David W.
Koppenaal
d and
Gary M.
Hieftje
*a
aDepartment of Chemistry, Indiana University, Bloomington, IN 47405, USA. E-mail: hieftje@indiana.edu; Fax: +1 (812) 855-0958; Tel: +1 (812) 855-2189
bInstitut für Anorganische und Angewandte Chemie, Universität Hamburg, Hamburg, Germany
cDepartment of Chemistry, University of Arizona, Tucson, AZ 85721, USA
dPacific Northwest National Laboratory, Richland, WA 99352, USA
First published on 22nd October 2008
Abstract
An ambient mass spectrometry ionization source based on an atmospheric-pressure flowing afterglow has been coupled to a Mattauch-Herzog mass spectrograph capable of simultaneous acquisition of a range of mass-to-charge values by means of a Faraday-strip array detector. The flowing afterglow was used as the ionization pathway for species produced by hydride generation. This ionization strategy circumvents problems, such as discharge instabilities or memory effects, induced by introducing the gaseous sample into the discharge. The generated spectra show both atomic and molecular peaks; calibration curves were calculated for both peak types with limits of detection for arsenic below 10 ppb. This study demonstrates the ability to use an ambient mass spectrometry source, commonly used for molecular analyses, for the detection of gas phase elemental species with the possibilty of performing speciation by coupling with a separation technique.
Introduction
Ambient mass spectrometry (AMS) has become a very active area of research through the introduction of desorption electrospray ionization (DESI)1 and direct analysis in real time (DART).2 These techniques allow the direct desorption/ionization of a very broad range of samples, and with no sample preparation.1,3–11 By means of predominantly proton or charge transfer ionization mechanisms, these sources produce simple mass spectra in which the molecular or protonated molecular ion generally dominates.3 Overall, AMS sources are attractive because of high sample throughput, ease of use, low initial costs, and low operating expense.Recently, a new ambient ionization source has been developed that utilizes the flowing afterglow of a helium atmospheric-pressure glow discharge (GD) to achieve desorption and/or ionization.12,13 Named the Flowing Atmospheric-Pressure Afterglow (FAPA), this source uses a beam of highly energetic helium metastable (He*) species to create a high flux of reagent ions, such as protonated water clusters, nitrogen dimer ions, and oxygen ions, to efficiently ionize analyte species that are placed within the afterglow region. This ionization mechanism, comparable to chemical ionization, results in simple mass spectra with little or no fragmentation, just as with other ambient ionization sources. Generally, the protonated molecular ion is formed, but spectra are often difficult to interpret due to the formation of clusters and adducts. As a result, ambient ionization sources generally are not used for elemental analysis; however, with a suitable sample-introduction method atomic information can be acquired.
Hydride generation (HG) enables the production of gaseous hydride species and is a standard method14 for several elements, including arsenic, germanium, antimony, selenium, tellurium, and others.15 These volatile hydrides are produced by mixing an analyte-containing acid solution, usually hydrochloric acid, and a solution of sodium borohydride. Upon mixing these components, the reaction in equation 1 proceeds. Although hydrogen is produced, there is ongoing debate about its form.16 The hydrogen can then combine with an anaylte species, such as those listed above, to form a volitile hydride, as in equation 2, where A is the analyte species, m isthe valence of the analyte A in solution, and n is the valence of analyte A in the hydride.17
| BH4− + 3H2O + H+ → H3BO3 + 8H | (1) |
| Am+ + (m + n)H → AHn + mH+ | (2) |
Previously, HG has been coupled to mass spectrometry through the use of inductively coupled plasma (ICP)20 or GD21 ionization sources. With these sources, the hydride species must be introduced into the plasma, which can change the plasma characteristics.21–26 This problem can become even more serious when HG is used in conjunction with transient sample introduction techniques in which the gaseous sample composition continuously changes. An example is chromatographic separations, which are generally essential for speciation analysis.
In the present paper, the use of HG with the FAPA source is used to achieve the advantages of HG while circumventing problems caused by sending high concentrations of hydride species into a plasma. The aim is to evaluate the use of a simple, low cost ionization source as an alternative method for performing elemental analyses by HG. Furthermore, the use of HG with an ambient ionization source provides a step towards coupling such sources to gaseous sample introduction techniques such as gas chromatography.
Experimental
Sample introduction system
The hydride generation apparatus used in these studies has been describe previously,24 so only a brief description and relevant changes will be described here. The HG process was performed by mixing a solution of 5% sodium borohydride, stabilized in 5% sodium hydroxide, with a 1 M hydrochloric acid solution in a glass tee. The HCl solution contained the analyte of interest. The resulting mixture was pumped at a rate of 0.5 or 1.0 mL/min through an approximately 150 cm PTFE reaction coil using a 4-head peristaltic pump (Gilson, Middletown, WI). The coil was used to ensure complete reactant mixing and hydride species formation. The combined gas and solution mixture was then passed through a gas-liquid separator (GLS) filled with glass beads to provide ample surface area. A sweep gas inlet to the GLS was closed off during these experiments. After the GLS, a peristaltic pump (Gilson, Middletown, WI) was used to drive the liquid to waste, while the hydride vapor flowed through a condensing column, held at a temperature of ∼5 °C by means of a recirculating chiller (ThermoNeslab M75, Thermo-Electron Corporation, Waltham, MA), to remove residual water vapor from the gas mixture. The remaining hydride vapor was passed through a 915 µm i.d. glass capillary to the FAPA source. The glass capillary was held in an x,y,z fiber-optic positioner (Newport Corporation, Irvine, CA) in order to tune its position in the afterglow. A diagram of the FAPA and MHMS interface can be found in Fig. 1. | ||
| Fig. 1 Diagram of the FAPA system and the MHMS interface. | ||
Flowing atmospheric-pressure afterglow
The FAPA consists of a tungsten-pin anode and brass-plate cathode. The two electrodes are held in a Teflon®cell that has a side inlet for helium (1–3 L/min, 99.999% ultra high purity, Airgas, Radnor, PA), which served as the discharge gas. The discharge was sustained by a high voltage (EH Series, Glassman High Voltage, Inc., Whitehouse Station, NJ) power supply connected in series with the two electrodes. Three 1.25 kΩ, 50 W resistors were placed in series in the return path to the power supply. In all cases the discharge was sustained in a current-controlled mode. The discharge cell was held in a locally built x,y-translation stage to tune its position with respect to the mass spectrometer interface. The x,y-translation stage was placed on optical rails to allow optimization of the z position (distance from the discharge cell to the MHMS interface).Mattauch-Herzog mass spectrograph and focal plane camera
The MHMS and FPC have been described in detail previously,27–30 so only a brief description and pertinent changes for the current work will be given here.Slight modifications to the MHMS-ICP interface were made in order to efficiently sample ions from the FAPA source. These changes include the use of a traditional mass spectrometer sampling cone in which the conical portion was machined off, referred to here as the sampling plate. A flat front plate was used to replace the cone vacuum orifice, and the front plate was electrically isolated from the sampling plate by means of a Viton® o-ring. Additionally, an electrically isolated aluminum ion lens was placed between the sampling plate and the skimmer cone. The sampling plate and the skimmer cone are electrically connected and their potential was held at 1 kV by a high-voltage power supply (SL-10, Spellman, Plainville, NY). The potential of the front plate can be varied by using the combination of two power supplies (0–6 kV, SL-30, Spellman, Plainville, NY and 0–60 V dc, 6024A, Hewlett Packard (Agilent), Santa Clara, CA). The low-voltage power supply was used for fine tuning the potential of the front plate. Because of the large potential difference between the FAPA and the MHMS interface, no ions were detected unless the FAPA potential was referenced to the potential of the front plate instead of to ground. Therefore, both the FAPA and the fine tuning front-plate power supplies were referenced to the high voltage front-plate power supply. The ion lens between the sampling plate and the skimmer cone was maintained at the same potential as the front plate high-voltage power supply. No changes after the skimmer cone were made to the MHMS instrument.
The second-generation FPC detector was used in the current studies. This version consists of 128 gold Faraday strips, each of which is 45-µm wide; the strips are set on 50-µm centers. The gain of each Faraday strip can be independently selected between two levels. The high gain level was used for all experiments except where indicated. Furthermore, nondestructive readouts were used to minimize read noise associated with the detector electronics.
Time-of-flight mass spectrometer
Because the MHMS can measure only the low (atomic) mass-to-charge range, a LECO HT Unique® (LECO Corp. St. Joseph, MI) time-of-flight (TOF) mass spectrometer was used to obtain full mass spectra for the molecules created in the HG process. Only two differences were needed in the source configuration during coupling with the Unique®spectrometer. First, the sampling cone was held at a relatively low potential (∼60 V), so there was no need to reference source power supplies with the inlet potential. Second, the spectrometer was designed for conventional APCI and ESI sources, so the vacuum system could not cope with high helium flow rates (>0.9 L/min). As a result, the FAPA had to be operated at lower flow rates. The consequences of this change will be addressed in the Results and Discussion section.Sample preparation
Sodium borohydride solutions were prepared fresh daily and were stabilized with sodium hydroxide. Except where noted, solutions of sodium borohydride were prepared by dissolving 5 g of sodium borohydride (98.5% powder, Sigma-Aldrich, St. Louis, MO) and 5 g of sodium hydroxide (97.0% pellets, EM Science, Gibbstown, NL) in 100 mL of distilled, deionized water. Analyte solutions were prepared in 1 M hydrochloric acid obtained through dilution of concentrated hydrochloric acid (ACS Grade, Mallinckrodt Baker, Inc., Phillipsburg, NJ). Analyte stock solutions were prepared at the 10 ppm level and serial dilutions with deionized water were made to obtain lower concentrations.Results and discussion
FAPA background characterization
The mass spectral background of the FAPA source was determined in the absence of the HG system and is shown in Fig. 2. The background spectrum is shown in the form of a bar graph due to the nature of the current MHMS-FPC system. At present, the FPC is only wide enough to acquire between 2 and 30 m/z values simultaneously, depending on the mass range of observation. For this reason, it is necessary to vary the magnetic field to acquire an entire spectrum. Therefore, only a narrow range of background peaks was focused on the FPC at a time, and the peaks were individually integrated. The height of the bars in the graph represents the integrated peak area at each m/z value. The major background species from the FAPA are protonated water clusters. These cluster species are believed to ionize analytes in the afterglow region through proton transfer reactions,13 analogous to atmospheric-pressure chemical ionization (APCI) mechanisms.31,32 In addition to the water clusters, other ions from atmospheric components such as NO+ and O2+ are also present in the background. Furthermore, very minor peaks are observable at nearly all m/z values below m/z 200. These peaks are believed to be clusters or adducts of various atmospheric components or contaminants in the gas. No attempt was made to further characterize these peaks. Future experiments will be aimed at controlling the atmosphere in order to reduce the effects of such peaks. For all following experiments, data were blank subtracted using the HG system with blank solutions of 1M HCl and 5% NaBH4.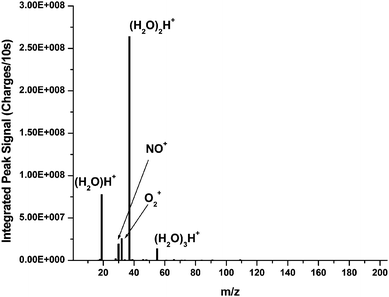 | ||
| Fig. 2 Background spectrum of the FAPA source without the HG system. The major peaks are protonated water clusters and ionized atmospheric components. | ||
Optimization of hydride generation
No attempt was made to fully optimize the HG introduction system for the present study. Instead, previous experience with the system was used as a guide. However, two parameters that were experimentally optimized were acid concentration and sample flow rate. The 75As+, 75AsH+, and 75AsH2+ signals were monitored for both 1 M and 2 M HCl acid concentrations; a plot of these data is shown in Fig. 3. The signal for these three species drops by approximately a factor of two when the 2 M HCl solution is used. It was hypothesized that the higher acid concentration produced too much H2 for the FAPA to accept; however, this hypothesis was not verified. In addition, the sample flow rate was varied from 0.5 to 1 mL/min; no variation in the signal levels for 75As+, 75AsH+, and 75AsH2+ was observed. Therefore, the higher flow rate was used for all subsequent experiments in order to minimize the washout time of the HG system. | ||
| Fig. 3 Spectrum containing the As+, AsH+, amd AsH2+ peaks obtained using two different acid concentrations for HG. The higher acid concentration yields a reduction in signal level. | ||
Hydride generation of arsenic
 | ||
| Fig. 4 Spectra of the clusters formed during the HG of arsenic by using the FAPA-MHMS-FPC system. a) Full spectrum accumulated from many mass windows by varying the magnetic field of the MHMS, and b) zoomed-in spectrum acquired using only two mass windows. Each point in b corresponds to a different Faraday strip of the FPC array detector. | ||
 | ||
| Fig. 5 Washout curve and precision for the HG-FAPA System. Complete washout occurs in about 20 minutes. The precision of the first 20 points is 7.4% RSD. | ||
 | ||
| Fig. 6 Noise power spectra for the As+ peak (m/z 75), a background peak (m/z 55), and the dark current from the detector. The major features are from power line noise and the largest noise level occurs for the analyte peak. Some flicker noise is present and is also greatest for the analyte peak. | ||
 | ||
| Fig. 7 Calibration curves for three analyte peaks of arsenic. The As+ and AsO+ curves roll off at higher concentrations, whereas the As2O3+ shows upward curvature. This behavior is due to increased formation of clusters at higher concentrations. The plots have been normalized, so slopes should not be compared quantitatively. | ||
The linear portion of the calibration plots, at lower concentrations, were used to calculate detection limits (3σ), which were found to be 7 ppb, 2 ppb, and 30 ppb for As+, AsO+, and As2O3+, respectively. These values are slightly worse than or on par with a gas sampling GD that was used in our laboratory with the HG system;21 yet, they are about 100 times worse than values obtained with an atmospheric-pressure glow discharge that was directly coupled to the interface of a TOFMS.24 However, in that study the hydride species were sent through the discharge rather than into the afterglow of the discharge. When the analytes are introduced into the discharge, only atomic As was detected, so only one peak had to be analyzed. In the present study, the As species are spread across several cluster peaks, making quantification more difficult. Certainly, work in the future should focus on reducing these cluster species if elemental analysis continues to be a goal. When the detection limits here are compared to those from a more commonly used method, such as HG-ICPMS, the values are between 10–10000 times worse.21,35 However, considering the added complexity and cost of operating an ICP, this technique might be desirable if sensitivity is not the dominant concern.

Conclusions
A hydride generation system has been coupled to an atmospheric-pressure GD ambient ionization source. This union demonstrates not only the ability to couple a gaseous sample introduction system to ambient mass spectrometry, but to obtain elemental information as well. The HG system provides high selectivity and could be coupled with liquid chromatography to perform speciation analysis for analytes such as arsenic. In addition, the system can provide quantitative information; however, care must be taken when considering the linear working range due to the formation of clusters at higher concentrations. This successful coupling would suggest that other gaseous sample introduction techniques, such as gas chromatography, could be effectively coupled with such an ionization source. Furthermore, with the soft ionization of the FAPA, future studies could be aimed at obtaining chemical species information by regulating the atmosphere in which the source is operating, and thereby controlling the degree of adduct or cluster formation. Controlling the atmosphere and the cluster formation would also be desirable in optimizing the sensitivity of the described system.Acknowledgements
Support for this work was provided by the U.S. Department of Energy, Office of Nonproliferation Research and Engineering. Pacific Northwest National Laboratory is operated by Battelle Memorial Institute for the Department of Energy under Contract DE-AC06-76RLO-1830. José A. C. Broekaert would like to thank the DFG for travel funds. The authors would also like to thank LECO Corporation for the loan of the Unique(R) TOFMS instrument.References
- Z. Takats, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef CAS.
- R. B. Cody, J. A. Laramee and H. D. Durst, Anal. Chem., 2005, 77, 2297–2302 CrossRef CAS.
- R. G. Cooks, Z. Ouyang, Z. Takats and J. M. Wiseman, Science, 2006, 311, 1566–1570 CrossRef CAS.
- F. M. Fernandez, R. B. Cody, M. D. Green, C. Y. Hampton, R. McGready, S. Sengaloundeth, N. J. White and P. N. Newton, ChemMedChem, 2006, 1, 702–705 CrossRef.
- K. Kpegba, T. Spadaro, R. B. Cody, N. Nesnas and J. A. Olson, Anal. Chem., 2007, 79, 5479–5483 CrossRef CAS.
- C. Petucci, J. Diffendal, D. Kaufman, B. Mekonnen, G. Terefenko and B. Musselman, Anal. Chem., 2007, 79, 5064–5070 CrossRef CAS.
- C. Y. Pierce, J. R. Barr, R. B. Cody, R. F. Massung, A. R. Woolfitt, H. Moura, H. A. Thompson and F. M. Fernandez, Chem. Commun., 2007, 807–809 RSC.
- M. S. Bereman, T. I. Williams and D. C. Muddiman, Anal. Chem., 2007, 79, 8812–8815 CrossRef CAS.
- I. Cotte-Rodriguez, C. C. Mulligan and G. Cooks, Anal. Chem., 2007, 79, 7069–7077 CrossRef CAS.
- G. Huang, H. Chen, X. Zhang, R. G. Cooks and Z. Ouyang, Anal. Chem., 2007, 79, 8327–8332 CrossRef CAS.
- C. C. Mulligan, D. K. MacMillan, R. J. Noll and R. G. Cooks, Rapid Commun. Mass Spectrom., 2007, 21, 3729–3736 CrossRef CAS.
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Anal. Chem., 2008, 80, 2654–2663 CrossRef CAS.
- F. J. Andrade, J. T. Shelley, W. C. Wetzel, M. R. Webb, G. Gamez, S. J. Ray and G. M. Hieftje, Anal. Chem., 2008, 80, 2646–2653 CrossRef CAS.
- A. E. Greenberg, R. R. Trussell, L. S. Clesceriand M. A. H. Franson, eds., Standard Methods for the Examination of Water and Wastewater, American Public Health Association, Washington, D.C., 1985 Search PubMed.
- P. Pohl, Trends Anal. Chem., 2004, 23, 87–101 CrossRef CAS.
- A. R. Kumar and P. Riyazuddin, Anal. Sci., 2005, 21, 1401–1410 CrossRef CAS.
- Hydride Generation Atomic Absorption Spectrometry, ed. J. T. Dedina and L. Dimiter, John Wiley & Sons, Ltd, West Sussex. 1995 Search PubMed.
- R. Cornelis, Handbook of Elemental Speciation: Techniques and Methodology, John Wiley and Sons, Ltd., West Sussex, England, 2003 Search PubMed.
- A. R. Kumar and P. Riyazuddin, Int. J. Environ. Anal. Chem., 2007, 87, 469–500 CrossRef CAS.
- M. J. Powell, D. W. Boomer and R. J. McVicars, Anal. Chem., 1986, 58, 2864–2867 CrossRef CAS.
- W. C. Wetzel, J. A. C. Broekaert and G. M. Hieftje, Spectrochim. Acta, Part B, 2002, 57B, 1009–1023 CrossRef.
- M. Grotti, C. Lagomarsino and J. M. Mermet, J. Anal. At. Spectrom., 2006, 21, 963–969 RSC.
- A. Menendez, J. Pisonero, R. Pereiro, N. Bordel and A. Sanz-Medel, J. Anal. At. Spectrom., 2003, 18, 557–563 RSC.
- W. C. Wetzel, F. J. Andrade, J. A. C. Broekaert and G. M. Hieftje, J. Anal. At. Spectrom., 2006, 21, 750–756 RSC.
- V.-D. Hodoroaba, V. Hoffmann, E. B. M. Steers and K. Wetzig, J. Anal. At. Spectrom., 2000, 15, 1075–1080 RSC.
- V.-D. Hodoroaba, E. B. M. Steers, V. Hoffmann and K. Wetzig, J. Anal. At. Spectrom., 2001, 16, 43–49 RSC.
- J. H. Barnes, G. D. Schilling, R. Sperline, M. B. Denton, E. T. Young, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Anal. Chem., 2004, 76, 2531–2536 CrossRef.
- J. H. I. V. Barnes, G. D. Schilling, M. B. Denton, D. W. Koppenaal and G. M. Hieftje, J. Anal. At. Spectrom., 2003, 18, 1015–1018 RSC.
- T. W. Burgoyne, G. M. Hieftje and R. A. Hites, J. Am. Soc. Mass Spectrom., 1997, 8, 307–318 CrossRef CAS.
- A. K. Knight, R. P. Sperline, G. M. Hieftje, E. Young, C. J. Barinaga, D. W. Koppenaal and M. B. Denton, Int. J. Mass Spectrom., 2002, 215, 131–139 Search PubMed.
- J. Sunner, G. Nicol and P. Kebarle, Anal. Chem., 1988, 60, 1300–1307 CrossRef CAS.
- M. M. Shahin, J. Chem. Phys., 1966, 45, 2600 CrossRef CAS -&.
- C. C. Mulligan, D. R. Justes, R. J. Noll, N. L. Sanders, B. C. Laughlin and R. G. Cooks, Analyst, 2006, 131, 556–567 RSC.
- G. D. Schilling, F. J. Andrade, J. H. Barnes, R. P. Sperline, M. B. Denton, C. J. Barinaga, D. W. Koppenaal and G. M. Hieftje, Anal. Chem., 2006, 78, 4319–4325 CrossRef CAS.
- B. Klaue and J. D. Blum, Anal. Chem., 1999, 71, 1408–1414 CrossRef CAS.
- J. Shelley, F. Andrade, S. Ray, J. Wiley and G. Hieftje, 34th Annual Conference of the Federation of Analytical Chemistry and Spectroscopy Societies, Memphis, TN, 2007 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |