Polymer monolith microextraction combined with electrothermal vaporization inductively coupled plasma mass spectrometry for the determination of trace Cd, Tl, and Pb in human serum and urine
Jun
Yin
,
Bin
Hu
*,
Man
He
,
Mingming
Zheng
and
Yu-Qi
Feng
*
Department of Chemistry, Wuhan University, Wuhan, 430072, P. R. China. E-mail: binhu@whu.edu.cn; yqfeng@public.wh.hb.cn; Fax: +86-27-68754067
First published on 24th October 2008
Abstract
A novel method of polymer monolith microextraction (PMME) using a poly(acrylamide-vinylpyridine-N,N′-methylene bisacrylamide) (AA-VP-Bis) monolithic column combined with electrothermal vaporization inductively coupled plasma mass spectrometry (ETV-ICP-MS) for the determination of trace Cd, Tl, and Pb with Pd as chemical modifier has been proposed. Several factors that influence the microextraction efficiency including pH value, sample flow rate, extraction time, sample volume, eluent volume, and coexisting ions, were investigated and the optimal microextraction conditions were established. The chemical modification of Pd in ETV-ICP-MS has been studied and the results indicated that the pyrolysis temperature for the target analytes could be increased up to 600 °C and the signal intensities were increased by 4.1, 2.1, and 13.8 times for Cd, Tl, and Pb, respectively. Under the optimized conditions, with a consumption of 0.4 mL sample, the limits of detection (LODs) were 1.1, 0.5, and 0.2 ng L−1 for Cd, Tl, and Pb, and the relative standard deviations (RSDs) were 10.3, 9.7, and 9.2% (C = 0.05 µg L−1, n = 5) for Cd, Tl, and Pb, respectively. The proposed method was successfully applied to the determination of trace Cd, Tl, and Pb in human serum and urine samples, and the recovery for the spiked samples were in the range of 90–110%. In order to validate the proposed method, certified reference material of GBW09103 human urine was analyzed, and the determined values were in good agreement with the certified values. The poly(AA-VP-Bis) monolith can be used more than 40 times without decrease in the extraction efficiency.
Introduction
The history of solid phase extraction (SPE)1,2 as an effective sample pretreatment technique can be traced back to the early 1950s.3 Compared with the traditional separation and preconcentration methods such as liquid–liquid extraction, SPE has the following advantages: (1) emulsification which happens frequently in liquid–liquid extraction is avoided, so that it gives a high recovery and enrichment factor; (2) the phase-separation step can be simplified, the analyte components are easily collected, and the operation is simple, convenient and rapid; (3) the volume of solvent can be obviously reduced, and the usage of highly toxic or flammable solvent is avoided; (4) the application range is extensive with various adsorbents for selection; (5) on-line automation can easily be realized. In view of the above advantages, SPE has now been widely applied for sample pretreatment in a variety of fields including environmental, biological, geological, and food analysis, etc.It should be addressed that the current study on SPE is mainly focused on two aspects: new functional adsorbents and miniaturization of extraction equipment. The properties of adsorbents decide the analytical performance and application range of the method. Therefore, the exploration of new functional adsorbents with good mass transferability, pH stability, good bioadaptability, and high adsorption capacity has become one of the hotspots in the study of SPE. Some new functional adsorbents, such as ion imprinted materials,4 nanometer-sized materials,5 monolithic materials,6 have already demonstrated their outstanding adsorption performance in SPE. In the past few years, more and more efforts have been directed towards the miniaturization of analytical instrument, in order to greatly reduce reagent consumption, increase the sample throughput, and achieve in-situ, on-line, in-vivo, and real-time analysis. Similarly, the miniaturization of extraction equipment, the conveniency of operation as well as the ability of easily combining with other analytical instrument have become the foreland of SPE researches. Currently, some miroextraction methods, such as solid phase microextraction (SPME),7capillary microextraction (CME),8 stirring bar sorptive extraction (SBSE),9 and microextraction in packed syringe (MEPS),10 have attracted the attention of analysts.
Monolithic material11,12 is a new type of separation medium with high speed, high efficiency and high throughput, and has been widely applied as the stationary phase in various chromatographic methods such as gas chromatography (GC), high performance liquid chromatography (HPLC) and capillary electrochromatography (CEC). The applications of monolithic materials in non-chromatographic methods,13,14 especially in the field of sample pretreatment,14 have also attracted the attention of analysts in recent years. The history of monolithic material in SPE is not very long. The first paper describing this entirely new approach was published in 1998. To achieve the desired increase in surface area, Xie et al.15 used commercial 80% divinylbenzene (remaining 20% being ethylstyrene) as the only monomer and dodecanol with toluene as porogens to obtain monolith with a surface area of 400 m2/g resulting from the presence of mesopores and micropores by thermally polymerizing them in a PEEK tube. Despite this very high surface area, such monoliths had excellent hydrodynamic properties because of the 6 µm large through pores. Since then, monolithic material was rapidly accepted as the excellent adsorption materials for solid phase extraction of different analytes from various samples. Schaller et al.16 successfully separated three antidepressants by capillary electrophoresis (CE) with in-line SPE using a monolithic adsorbent, and the enrichment factors were over 500. In 2006, Feng's group proposed a new type of miniaturized syringe extraction method named polymer monolith microextraction (PMME),17 which offers several attractive features. These include the advantages of the polymer monolith, such as easy preparation with good control of porosity, convective mass transfer procedure, bioadaptability, and pH stability; and the advantages of the extraction device, such as convenience, flexibility, and easy operation. PMME combined with CE, HPLC and LC-MS, respectively, was successfully applied in the analysis of opiates in human urine as well as other fields.18–21 However, no correlative research work has been found in inorganic elemental analysis up to date.
Atomic spectrometry techniques, such as flame atomic absorption spectrometry (FAAS), electrothermal atomic absorption spectrometry (ETAAS), inductively coupled optical emission spectrometry (ICP-OES) and inductively coupled plasma mass spectrometry (ICP-MS), are the most used analytical techniques for inorganic elemental analysis. Among them, ICP-MS has been accepted as a powerful technique for trace element analysis due to its outstanding advantages, such as low detection limits, wide linear dynamic range, multi-element detection capability, high sample throughput,22 and has a widespread application in an extensive field such as life science, food science, clinical chemistry, and geological chemistry. However, conventional pneumatic nebulization (PN)-ICP-MS has disadvantages of low intake efficiency (less than 2%), large sample consumption and difficult to analyze high salt-containing and viscous samples.
Electrothermal vaporization (ETV),23,24 as a sample introduction technique for ICP-MS, has the following outstanding advantages: (1) very high transport efficiency (∼80%); (2) vaporization and excitation/ionization processes are separated; (3) low sample consumption in microgram or microliter level; (4) the sensitivity and selectivity of the method could be further improved by using chemical modifier. Therefore, it has consistently received considerable attention in atomic/mass spectrometry.23,24 In spite of these notable advantages, in some environmental and biological samples with complex matrix, the level of the heavy metal ions is fairly low and the major constituents can cause matrix effects and spectral interference. In order to circumvent the above problems, the utilization of chemical modifier or the use of efficient separation/preconcentration has been proven to be very effective for ETV-ICP-MS.
As mentioned above, PMME is a novel miniaturized sample pretreatment technique and ETV-ICP-MS is a micro-sampling and high sensitivity method. If PMME is combined with ETV-ICP-MS, it will be extremely suitable for the determination of ultra-trace elements in samples with a complex matrix, especially a sample with a limited available volume, for example, biological fluids.
Cadmium is a non-essential toxic element and acute cadmium poisoning may produce degenerative changes in renal tubular cells. Cadmium has also been demonstrated to inhibit many enzymes and competes with calcium metabolism and alter phosphorylation patterns.25Thallium occurs in the environment naturally in small amounts. Excess of thallium would cause gastrointestinal irritation and disorders of the nervous system after exposure for relatively short times. In the long term, this has the potential to cause effects such as changes in blood chemistry, damage to liver, kidney, intestinal, and testicular tissue, and hair loss.26 Also, lead fulfils no essential function in the human body, and its toxic effect is primarily the inactivation of enzymes and proteinsvia the binding to sulfydryl groups.27 They are all considered to be high toxicity for human health and can be considered as biomonitors whose concentration in human body would well indicate human exposure to the background of harmful chemicals in the environment. Serum and urine analysis are often used for monitoring environmental and occupational exposure to trace elements because serum and urine are the most readily available biological fluid and most trace elements are mainly excreted via urine.
The aim of this work is to develop a new method of PMME-ETV-ICP-MS for the determination of trace Cd, Tl, and Pb in human serum and urine with Pd as chemical modifier. The factors that influence the extraction efficiency and the vaporization behaviors of analytes were systematically investigated, and the optimal operation conditions were established. The proposed method was applied to the determination of trace Cd, Tl and Pb in human serum and urine with satisfactory results.
Experimental
Apparatus
An Agilent 7500a ICP-MS (Agilent, Japan), equipped with a modified commercially available WF-4C graphite furnace (Beijing Second Optics, China) was used as electrothermal vaporizer. The transfer line consisted of a laboratory-built connecting interface28 and a polyethylene tube (6 mm id) with total length of 70 cm. The optimized operating conditions for ICP-MS and ETV are given in Table 1. A Mettler Toledo 320-S pH meter (Mettler Toledo Instruments Co. Ltd., Shanghai, China) was used for pH value measurements. A WX-3000 microwave accelerated digestion system (EU Chemical Instruments Co. Ltd., Shanghai, China) was used for sample digestion. A syringe infusion pump (WZB-50, Weikete High-Technology, Changsha, China) was employed for the delivery of solutions.| ICP-MS plasma | |
|---|---|
| Rf power | 1250 W |
| Outer gas flow rate | 15 L min−1 |
| Intermediate gas flow rate | 0.9 L min−1 |
| Nebulizer gas flow rate | 0.7 L min−1 |
| Sampling depth | 7.0 mm |
| Sampler/skimmer diameter orifice | Nickel 1.0 mm/0.4 mm |
| Time-resolved data acquisition | |
| Scanning mode | Peak-hopping |
| Dwell time | 20 ms |
| Integration mode | Peak area |
| Points per spectral peak | 1 |
| Elements and istopes | 111Cd, 205Tl, 208Pb |
| Electrothermal vaporizer | |
| Sample volume | 10 µL |
| Carrier gas flow rate | 0.4 L min−1 |
| Drying step | 100 °C ramp 10 s \ hold 10 s |
| Pyrolysis step | 600 °C ramp 10 s \ hold 10 s |
| Vaporization step | 2200 °C hold 4 s |
| Cleaning step | 2500 °C hold 3 s |
Reagents
All laboratory wares were made of polyethylene or polypropylene material and were thoroughly cleaned by soaking in nitric acid (1 + 1) for at least 24 h. Immediately prior to use, all acid-washed wares were rinsed with high purity deionized water (Millipore Corporation, Bedford, MA, USA).Acrylamide (AA) was purchased from Tianjin Chemical Plant (Tianjin, China). 4-Vinylpyridine (4-VP) was obtained from Acros (Sweden). N,N′-Methylene bisacrylamide (Bis), 2,2′-azobis(2-methylpropionitrile) (AIBN), dodecanol and DMSO were obtained from Shanghai Chemical Reagent Co. Ltd. (Shanghai, China) and were of analytical reagent grade.
The stock solutions of Cd, Tl, and Pb (1 g L−1) were prepared from their nitrate of analytical reagent grade (Shanghai Chemical, Shanghai, China) and diluting to a certain volume with high purity deionized water. Highly pure grade HNO3 was distilled before used. Certified reference material of human urine GBW09103 was obtained from Beijing Medical University (Beijing, China). Pd(NO3)3 (Shanghai Chemical, Shanghai, China) and other chemical reagents used in this work were of analytical grade, and high purity deionized water was used throughout.
Sample digestion
Pooled human serum collected from healthy adults was supplied by the Hospital of Wuhan University (Wuhan, China) and urine sample of the laboratory workers were collected. Microwave heating digestion with nitric acid was used to oxidize organic material in serum and urine samples prior to analysis.4 mL urine sample was transferred into PTFE insert, and 4 mL HNO3 was added. The insert was placed into the bottom of the digestion vessel and the digestion vessel was then closed and positioned inside the microwave oven. After that, the heating program was performed as 5 min at 15 atm and 200 °C in 600W. After cooling down, the vessel was opened and clear solution was observed. The digest was then heated to near dryness by a hot plate, and the residue was dissolved with high purity deionized water, and then diluted to 4 mL. Before used, the solution was adjusted to the required pH value with NH3·H2O–HNO3.
4 mL human serum sample was also prepared using above microwave digestion procedure. The vessel was opened after cooling down, and then H2O2 (30%, 0.5 mL) was added dropwise into the PTFE insert. The clear solution was heated to near dryness by a plate heater, and the residue was dissolved with high purity deionized water, and then diluted to 4 mL. Before used, the solution was adjusted to the required pH value with NH3·H2O–HNO3.
Preparation of the poly(AA-VP-Bis) monolithic capillary
The poly(AA-VP-Bis) monolithic capillary was synthesized inside a fused silica capillary (20 cm × 530 µm i.d., Yongnian, Hebei, China) by a heat-initiated polymerization method.29 Firstly, the capillary was derivatized with 3-(triethoxysilyl) propyl methacrylate. Then, the pre-polymerization mixture consisting of monomer AA, 4-VP, crosslinker Bis, porogenic solvent DMSO and dodecanol, initiator AIBN was filled into the capillary. The capillary was immediately sealed with silicon rubber and the reaction was initiated at 60 °C for 18 h, followed by washing with methanol to remove the unreacted component and porogenic solvent.The resulting polymer monolith was taken for FT-IR, elemental analysis, SEM and N2 adsorption characterization.29 The spectra of FT-IR indicated the presence of acrylamide and pyridyl groups. The composition of the polymer monolith includes 56.4% (C), 13.2% (N) and 6.0% (H). In SEM, the interconnected skeletons and interconnected textural pores of the monolith are easily observed, and the monolith was attached tightly to the inner-wall of the capillary. The average skeleton pore size of the monolith determined by N2 adsorption method was 3.3 nm.
PMME procedure
The extraction device is composed of an extraction pinhead and the syringe barrel, and the schematic setup is referred to in Ref. 17 and 19. Briefly, the original metallic needle of the pinhead was replaced by a 3 cm-long monolithic capillary. The extraction device could be used after the capillary was fixed in the pinhead by the adhesive.A WZB-50 syringe infusion pump was employed for the delivery of solution, and the whole PMME procedure included four steps: (1) Precondition. The syringe was filled with 0.15 mL of 0.1 mol L−1 phosphate buffer solution (pH = 7.0), and then ejected through the monolithic capillary at 0.2 mL min−1 by the infusion pump; (2) Preconcentration. 0.4 mL sample solution was ejected through the monolithic capillary at 0.2 mL min−1; (3) Washing. 0.05 mL high purity deionized water was ejected through the monolithic capillary at 0.1 mL min−1; (4) Elution. 0.05 mL HNO3 (1.0 mol L−1) was ejected through the monolithic capillary at 0.2 mL min−1. A whole PMME procedure could be finished in 4 min. The above analytical process could result in an 8-fold preconcentration factor.
ETV analysis
After the ETV unit was connected to the ICP-MS and the system was stabilized, 0.05 mL Pd(NO3)3 (1 g L−1) was added to the eluting solution of PMME, and 10 µL of the mixed solution obtained by ultrasonic vibration was injected into the graphite furnace. During the drying step of the temperature program, the dosing hole of the graphite furnace was kept open to remove the water and other vapors. When the dosing hole was sealed with a graphite probe 5–10 s prior to the high-temperature vaporization step, the vaporized analytes were swept into the plasma ionization source by a carrier gas of argon and the peak-hop transient mode of data acquisition was used to detect the ions selected.Results and discussion
Optimization of PMME parameters
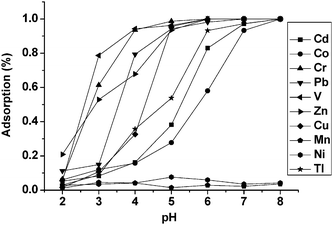 | ||
| Fig. 1 Effect of pH value on the adsorption of several metal ions on polymer monolith. Concentration of the target analyte: 0.5 µg L−1; sample volume: 0.4 mL; sample flow rate: 0.2 mL min−1. | ||
According to the theory of hard and soft acids and bases, the interaction force between soft acid and soft base, or hard acid and hard base, are stronger than the interaction force between soft acid and hard base, or hard acid and soft base. Since the amidocyanogen and pyridine in poly(AA-VP-Bis) monolith are nearly soft bases, they have only a slight interaction force with the hard acid metal ions such as Mn, which is coincident with the experimental results.
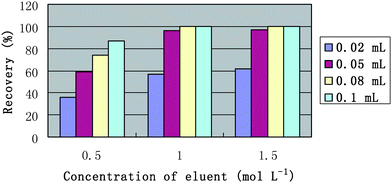 | ||
| Fig. 2 Effects of eluent concentration and volume on the recoveries of Cd. Cd: 0.5 µg L−1; sample volume: 0.4 mL; sample flow rate: 0.2 mL min−1; elution flow rate: 0.2 mL min−1. | ||
The elution flow rate was also studied, and no obvious difference was observed with the elution flow rate varying in the range of 0.05–0.25 mL min−1. In this work, 0.05 mL of 1.0 mol L−1 HNO3 with the elution flow rate of 0.20 mL min−1 was employed to simultaneously elute the analytes retained on the polymer monolith.
The optimized PMME operation parameters are summarized in Table 2.
Adsorption capacity and regeneration of polymer monolith
To study the adsorption capacity of poly(AA-VP-Bis) monolith, 10 mL sample solution containing 50 µg L−1 of each analyte was passed through the polymer monolith and the effluent was collected in batches for ICP-MS determination. The breakthrough point was defined as the 5% leakage of analytes, and the adsorption capacity evaluated from the breakthrough curve were 3.42, 2.73 and 3.16 µg m−1 for Cd, Tl and Pb, respectively.The regeneration of polymer monolith is one of the key factors in evaluating the performance of PMME. For the regeneration of the polymer monolith, 1.0 mol L−1 HNO3 solution and high purity deionized water were passed through the column. By this treatment, the poly(AA-VP-Bis) monolith could be re-used for more than 40 times with good stability (recovery higher than 90% for the target elements) under the optimized conditions.
Optimization of ETV parameters
In order to improve the vaporization behavior of volatile elements, Pd has been widely used in ETAAS, ETV-ICP-OES, and ETV-ICP-MS as an efficient chemical modifier.30–32 It has been confirmed that the usage of Pd as chemical modifier could obviously improve the analytical performance of these methods. In this work, the vaporization behavior of Cd, Tl, and Pb in ETV-ICP-MS with and without Pd as chemical modifier had been studied for comparison, and the experimental results demonstrated that the pyrolysis temperature for target analytes could be increased to 600 °C without any loss in the presence of Pd, and the signal intensities of Cd, Tl, and Pb were increased by 4.1, 2.1, and 13.8 times, respectively. Therefore, a pyrolysis temperature of 600 °C with pyrolysis time of 10 s and a vaporization temperature of 2200 °C with a vaporization time of 4 s were chosen as the ETV heating program for simultaneous determination of Cd, Tl and Pb by ETV-ICP-MS. In this work, effect of Pd concentration on the signal intensities of the analytes was also investigated, and it was found that the maximum signal intensities were obtained for Cd, Tl, and Pb with Pd concentration in the range of 500–800 mg L−1. In the subsequent experiment, 500 mg L−1 of Pd was employed as chemical modifier.Effects of coexisting ions
The effects of coexisting ions, as commonly expected in biological fluids, on the preconcentration and determination of Cd, Tl and Pb were examined. Sample solutions of 2.0 µg L−1analytes containing the added interfering ions were treated according to the proposed procedure. The tolerance limits of the coexisting ions, which were defined as 10% decrease of signal intensity, are summarized in Table 3. Table 3 also lists the average level of these coexisting ions in human serum and urine.33 As can be seen, the tolerance limits for the studied co-existing ions are higher than the average level of these coexisting ions in human serum and urine although a relative low tolerance to Cu, Zn, Al, Co and Fe was found for poly(AA-VP-Bis) monolith column, indicating that the proposed method could be employed for the analysis of trace Cd, Tl and Pb in human serum and urine.| Coexisting ions | Tolerance limit (mg L−1) | Concentration range in human serum33 (mg L−1) | Concentration range in urine33 (mg L−1) |
|---|---|---|---|
| Note:a mL L−1.b µg L−1. | |||
| K+ | > 10,000 | 140–215 | ≈ 1.9 |
| Na+ | > 10,000 | 2500–3560a | ≈ 2200 |
| Ca2+ | 500 | 91–106 | ≈ 120 |
| Mg2+ | 500 | 17–22 | ≈ 90 |
| Mn2+ | > 5,000 | 0.1–2.9b | 0.1–20 b |
| Ni2+ | > 5,000 | 0.05–1.3 b | 0.06–8 b |
| Cu2+ | 2 | 0.6–1.4 | 42–50 b |
| Zn2+ | 2 | 0.6–1.2 | 0.27–0.85 |
| Al3+ | 2 | 0.5–8 b | 2.3–110 b |
| Co2+ | 4 | 0.08–0.45 b | 0.2–135 b |
| Fe3+ | 4 | ≈ 0.17 | |
| Cl − | > 10,000 | ||
| Br − | > 10,000 | 3.5–5.5 b | ≈ 5 |
| SO42−, H2PO4−, CH3COO− | > 10,000 |
For comparison, the detection limits of the proposed method and the other similar methods reported in the literatures for Cd, Tl and Pb are listed in Table 4. As can be seen, the detection limits obtained by the proposed method are better than that obtained by isotope dilution (ID)-ETV-ICP-MS,34,35 slurry sampling ETV-ICP-MS,36capillary microextraction (CME)-ICP-MS,37 flow injection knotted reactor (FI-KR)-ICP-MS,38 flow injection solid phase extraction (FI-SPE)-ICP-MS,5 large-bore direct injection high efficiency nebulizer (LB-DIHEN)-ICP-MS,39 cloud extraction (CPE)-ETAAS40 and SPE-ICP-OES,41 but poorer than that obtained by single drop microextraction (SDME)-ETV-ICP-MS.42
| Analytical method | Sample | Limits of detection (ng L−1) | |||
|---|---|---|---|---|---|
| Cd | Tl | Pb | |||
| a ID: isotope dilution; SS: slurry sampling; CME: capillary microextraction; KR: knotted reactor; SPE: solid phase extraction; LB-DIHEN: large-bore direct injection high efficiency nebulizer; SDME: single drop microextraction; CPE: cloud point extraction. | |||||
| This method | PMME-ETV-ICP-MS | Urine and serum | 1.1 | 0.5 | 0.2 |
| Ref. 34 | aETV-ID-ICP-MS | Urine | 20 | — | 5 |
| Ref. 35 | aID ETV-ICP-MS | Fuel alcohol | 80 | 1.0 | 50 |
| Ref. 36 | a SS-ETV-ICP-MS | Sediment | 10 | 60 | 400 |
| Ref. 37 | aCME-ICP-MS | Urine | 4.5 | — | 3.7 |
| Ref. 38 | aKR-FI-ICP-MS | Natural water | 1.1 | — | 4.5 |
| Ref. 5 | aSPE-FI-ICP-MS | Natural water | 79 | — | 27 |
| Ref. 39 | aLB-DIHEN-ICP-MS | Urine | 10; 3 | — | 90; 6 |
| Ref. 40 | aCPE-ETAAS | Urine | 8 | — | — |
| Ref. 41 | aSPE-ICP-OES | Urine, blood and serum | 300 | — | 1100 |
| Ref. 42 | aSDME-ETV-ICP-MS | Urine and water | 0.27; 0.16 | — | — |
The proposed method was also applied to the analysis of real human serum and urine samples. The analytical results, together with the recoveries for the spiked samples, are given in Table 6. As can be seen, the concentrations of Cd, Tl and Pb in non-spiked serum sample are 0.09, 0.08 and 0.11 µg L−1, respectively, and the concentrations for Cd, Tl and Pb in non-spiked urine are 38, 0.37 and 29 µg L−1. The Cd, Tl and Pb concentrations in non-spiked human serum and urine samples obtained by the proposed method are within the range reported by Caroli et al.33 for normal human serum and urine. The recoveries for the target analytes in spiked serum and urine ranged from 90% to 110%.
| Sample | Element | Added (µg L−1) | Founda (µg L−1) | Recovery (%) | Average level (µg L−1)33 |
|---|---|---|---|---|---|
| a Values expressed as mean ± s (n = 3). | |||||
| Human serum | Cd | 0 | 0.09 ± 0.01 | — | 0.04–0.4 |
| 0.1 | 0.20 ± 0.01 | 110 | |||
| 0.2 | 0.28 ± 0.02 | 95 | |||
| 0 | 0.08 ± 0.01 | — | 0.05–0.3 | ||
| Tl | 0.1 | 0.17 ± 0.02 | 90 | ||
| 0.2 | 0.27 ± 0.02 | 95 | |||
| 0 | 0.11 ± 0.01 | — | — | ||
| Pb | 0.1 | 0.22 ± 0.02 | 110 | ||
| 0.2 | 0.29 ± 0.02 | 90 | |||
| Human urine | Cd | 0 | 38 ± 2 | — | 0.4–70 |
| 50 | 89 ± 4 | 102 | |||
| 100 | 135 ± 6 | 97 | |||
| 0 | 0.37 ± 0.03 | — | 0.02–8.9 | ||
| Tl | 0.5 | 0.91 ± 0.05 | 108 | ||
| 1.0 | 1.41 ± 0.09 | 104 | |||
| 0 | 29 ± 2 | — | 12–30 | ||
| Pb | 50 | 76 ± 3 | 94 | ||
| 100 | 126 ± 5 | 97 |
Conclusion
In this research, a novel poly(AA-VP-Bis) monolith microextraction method has been proposed for the separation/preconcentration of trace metal ions for the first time. The results indicated that poly(AA-VP-Bis) monolith could be used in PMME with high extraction efficiency, high adsorption capacity, good chemical stability, and good regeneration. Although a low tolerance to Cu, Zn, Al, Co and Fe was found for poly(AA-VP-Bis) monolith column and several of these elements, such as Fe and Zn, are also present at relatively high concentrations in biotissues/fluids, the tolerance limits of the poly(AA-VP-Bis) monolith column for these coexisting elements are higher than the average levels of these coexisting ions in biological fluids [30], indicating that the proposed method could be used in the analysis of biological fluid such as human serum and urine. The combination of miniaturized PMME technique and micro-sampling ETV technique is extremely suitable for the determination of ultra-trace elements in biological fluids, especially when the sample volume is limited.Acknowledgements
Financial support from the Science Fund for Creative Research Groups of NSFC (No. 20621502), National Nature Science Foundation of China (No. 20575048, 20375030), National Science Fund for Distinguished Young Scholars (No. 20625516) and MOE of China (NCET-04-0658) is gratefully acknowledged.References
- I. Liska, J. Chromatogr. A, 2000, 885, 3–16 CrossRef CAS.
- C. F. Poole, A. D. Gunatilleka and R. Sethuraman, J. Chromatogr. A, 2000, 885, 17–39 CrossRef CAS.
- H. Braus, F. M. Middleton and G. Walton, Anal. Chem., 1951, 23, 1160–1164 CrossRef CAS.
- T. P. Rao, S. Daniel and J. M. Gladis, Trac-Trends Anal. Chem., 2004, 23, 28–35 CrossRef.
- J. Yin, Z. C. Jiang, G. Chang and B. Hu, Anal. Chim. Acta, 2005, 540, 333–339 CrossRef CAS.
- H. F. Zou, X. D. Huang, M. L. Ye and Q. Z. Luo, J. Chromatogr. A, 2002, 954, 5–32 CrossRef CAS.
- J. Pawliszyn, J. Chromatogr. Sci., 2000, 38, 270–278 CAS.
- J. C. Wu and J. Pawliszyn, Anal. Chem., 2001, 73, 55–63 CrossRef CAS.
- C. Blasco, M. Fernandez, Y. Pico and G. Font, J. Chromatogr. A, 2004, 1030, 77–85 CrossRef CAS.
- M. Abdel-Rehim, J. Chromatogr. B, 2004, 801, 317–321 CrossRef.
- F. Svec, J. Sep. Sci., 2005, 28, 729–745 CrossRef CAS.
- S. Eeltink and F. Svec, Electrophoresis, 2007, 28, 137–147 CrossRef CAS.
- F. Svec, Electrophoresis, 2006, 27, 947–961 CrossRef CAS.
- F. Svec, J. Chromatogr. B, 2006, 841, 52–64 CrossRef CAS.
- S. F. Xie, F. Svec and J. M. J. Frechet, Chem. Mater., 1998, 10, 4072–4078 CrossRef CAS.
- D. Schaller, E. F. Hilder and P. R. Haddad, Anal. Chim. Acta, 2006, 556, 104–111 CrossRef CAS.
- M. Zhang, F. Wei, Y. F. Zhang, J. Nie and Y. Q. Feng, J. Chromatogr. A, 2006, 1102, 294–301 CrossRef CAS.
- H. J. Zhang, J. F. Huang, H. Wang and Y. Q. Feng, Anal. Chim. Acta, 2006, 565, 129–135 CrossRef CAS.
- H. J. Zhang, J. S. Li, H. Wang and Y. Q. Feng, Anal. Bioanal. Chem., 2006, 386, 2035–2042 CrossRef CAS.
- J. F. Huang, H. J. Zhang and Y. Q. Feng, J. Agr. Food Chem., 2006, 54, 9279–9286 CrossRef CAS.
- F. Wei, M. Zhang and Y. Q. Feng, Electrophoresis, 2006, 27, 1939–1948 CrossRef CAS.
- D. Beauchemin, Anal. Chem., 2006, 78, 4111–4136 CrossRef CAS.
- B. Hu, S. Q. Li, G. Q. Xiang, M. He and Z. C. Jiang, Appl. Spectrosc. Rev., 2007, 42, 203–234 CrossRef CAS.
- A. Martin-Esteban and B. Slowikowski, Crit. Rev. Anal. Chem., 2003, 33, 43–55 CrossRef CAS.
- A. A. Moshtaghie, A. Raisi and H. Goodarzi, J. Islam. Academ. Sci., 1991, 4, 192–195 Search PubMed.
- M. M. Hassanien, K. H. S. Abou-El-Sherbini and G. A. E. Mostafa, Talanta, 2003, 59, 383–392 CrossRef CAS.
- U. Ewers, H. W. Schlipköter, Blei. In: Merian(Ed.), Metalle in der Umwelt. Verbreitung, Analytik und biologische Relevanz. Verlag Chemie, 1984, Weinheim, pp. 351–374 Search PubMed.
- S. Q. Li, B. Hu and Z. C. Jiang, J. Anal. At. Spectrom., 2004, 19, 387–391 RSC.
- Y. Fan, M. Zhang and Y. Q. Feng, J. Chromatogr. A, 2005, 1099, 84–91 CrossRef CAS.
- A. C. Davis, C. P. Calloway and B. T. Jones, Talanta, 2007, 71, 1144–1149 CrossRef CAS.
- Y. J. Tseng, Y. D. Tsai and S. J. Jiang, Anal. Bioanal. Chem., 2007, 387, 2849–2855 CrossRef CAS.
- A. Viitak and A. B. Volynsky, Talanta, 2006, 70, 890–895 CrossRef CAS.
- S. Caroli, A. Alimonti, E. Coni, F. Petrucci, O. Senofonte and N. Violante, Crit. Rev. Anal. Chem., 1994, 24, 363 CrossRef CAS.
- K. H. Lee and S. H. Liu, Analyst, 1998, 123, 1557–1560 RSC.
- T. D. SaintPierre, V. L. A. Frescura and A. J. Curtius, Talanta, 2006, 68, 957–962 CrossRef CAS.
- L. F. Dias, G. R. Miranda, T. D. SaintPierre, S. M. Maia, V. L. A. Frescura and A. J. Curtius, Spectrochim. Acta, 2005, 60B, 117–124 CAS.
- Y. W. Wu, B. Hu, Z. C. Jiang, Y. Q. Feng, P. Lu and B. Z. Li, Rapid Commun. Mass Spectrom., 2006, 20, 3527–3534 CrossRef CAS.
- B. Dimitrova-Koleva, K. Benkhedda, E. Ivanova and F. Adams, Talanta, 2007, 71, 44–50 CrossRef CAS.
- M. G. Minnich, D. C. Miller and P. J. Parsons, Spectrochim. Acta Part B, 2008, 63, 389–395 CrossRef.
- P. R. Aranda, R. A. Gil, S. Moyano, I. D. Vito, L. D. Martinez,Talanta DOI:10.1016/j.talanta. 2008.07.009.
- J. S. Suleiman, B. Hu, C. Z. Huang and N. Zhang, J. Hazard. Mater., 2008, 157, 410–417 CrossRef CAS.
- L. B. Xia, B. Hu, Z. C. Jiang, Y. L. Wu and Y. Liang, Anal. Chem., 2004, 76, 2910–2915 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |