Genome-wide transcriptome analysis of 150 cell samples†
Daniel
Irimia‡
*a,
Michael
Mindrinos‡
*b,
Aman
Russom
a,
Wenzhong
Xiao
b,
Julie
Wilhelmy
b,
Shenglong
Wang
c,
Joe Don
Heath
c,
Nurith
Kurn
c,
Ronald G.
Tompkins
a,
Ronald W.
Davis
b and
Mehmet
Toner
a
aBioMEMS Resource Center, Center for Engineering in Medicine and Surgical Services, Massachusetts General Hospital, Shriners Hospital for Children, and Harvard Medical School, Boston, MA 02114, USA. E-mail: dirimia@hms.harvard.edu
bStanford Genome Technology Center and Department of Biochemistry, Stanford University, Stanford, CA 94305, USA. E-mail: mindrinos@stanford.edu
cNuGEN Technologies Inc., San Carlos, CA 94070, USA
First published on 12th November 2008
Abstract
A major challenge in molecular biology is interrogating the human transcriptome on a genome wide scale when only a limited amount of biological sample is available for analysis. Current methodologies using microarray technologies for simultaneously monitoring mRNA transcription levels require nanogram amounts of total RNA. To overcome the sample size limitation of current technologies, we have developed a method to probe the global gene expression in biological samples as small as 150 cells, or the equivalent of approximately 300 pg total RNA. The new method employs microfluidic devices for the purification of total RNA from mammalian cells and ultra-sensitive whole transcriptome amplification techniques. We verified that the RNA integrity is preserved through the isolation process, accomplished highly reproducible whole transcriptome analysis, and established high correlation between repeated isolations of 150 cells and the same cell culture sample. We validated the technology by demonstrating that the combined microfluidic and amplification protocol is capable of identifying biological pathways perturbed by stimulation, which are consistent with the information recognized in bulk-isolated samples.
Insight, innovation, integrationWe have designed a microfluidic device for serial processing of cells that overcomes degradation problems during RNA isolation and developed a new protocol for whole genome amplification starting from 300 pg of total RNA. By combining the microfluidic device for the purification of total cellular RNA with the powerful linear whole transcriptome amplification protocol we have accomplished robust genome -wide expression analysis from small biological samples. In this way we have identified biological pathways perturbed by stimulation in 150 cell samples, and verified the accuracy of the information by direct comparison with whole genome expression in samples with 1 ng RNA. |
Introduction
As a major technology development in molecular biology, DNA microarrays can simultaneously monitor mRNAtranscript levels on a genome -wide scale,1,2 enabling the discovery of complex relationships of gene expression in biological and clinical samples.3,4 Intricate expression patterns in cancer, inflammation and stem cells, which could ultimately be exploited for clinical applications, can be uncovered using microarray technologies.4–7 For example, microarray analysis has been demonstrated to help reveal patterns of gene expression that correlate with aggressiveness and invasion of breast cancer,8 lung cancer9 melanoma,10 or lymphoma.11 However, an important limitation of the microarray technology that constrains its use in clinical medicine is the large amount of initial sample required for RNA preparation and processing for hybridization to the arrays. These requirements are in competition with the need for comprehensive analysis of smaller, more homogeneous samples arising from the physiologically more relevant roles that small groups of cells can play e.g. in early metastasis,12 circulating tumor cells,13stem cell regeneration and differentiation,14etc.Currently, more than 5000 cells are required as starting material to extract enough RNA and process it for high quality microarray analysis.15–17 For sample sizes lower than few thousand cells, the analysis of gene expression is challenging and requires either a priori knowledge of the genes of interest or the use of amplification protocols to enhance the signals.18,19 Methods based on exponential amplification (polymerase chain reaction, PCR) can usually probe at most tens of pre-selected genes at the same time, and can only be employed as verification techniques, when the existence of target genes has already been identified through other methods.18,20 A number of protocols have been reported, that attempt to interrogate gene expression of a few hundred genes at a time and rely on multiple rounds of linear amplification.21,22 Poor reproducibility of the results, especially for genes differentially expressed with less than 4–5 fold difference, represents a major barrier in the practical impact of these protocols.23 Parallel efforts using microfluidic techniques are still limited to few tens of genes that can be probed simultaneously.24–26
To close the gap between the sensitivity of verification tools such as PCR and fluorescent in situ hybridization (FISH) and the comprehensive analysis of genome wide interrogation of the transcriptome with DNA microarrays, new approaches that are more efficient in sample preparation and novel technologies in sample processing with high sensitivity and low noise are needed. One critical issue for global gene expression profiling of samples with fewer than one thousand cells is the loss of nucleic acid material during the nucleic acid purification process. In this context, microfluidic technologies hold great promise for processing small amounts of nucleic acids.25,27–29 One limitation, however, is that when the protocols for genome wide interrogation are pushed towards smaller and smaller samples, down to single cell analysis, the lack of reference samples could be a source of systematic error in analysis.30–33 The accuracy of the information uncovered using these technologies could not be estimated independently because it is not possible to distinguish between the biological noise in gene expression and potential noise introduced by the isolation and amplification protocols.
Here we report the development of an accurate technique for genome wide analysis of samples as small as 150 cells, which combines a new high quality RNA purification protocol implemented in microfluidic format and a new ultra-sensitive amplification protocol. We have designed a microfluidic device in which the precise temporal sequence, timing, and stoichiometry of the chemical reactions for RNA isolation are controlled from one outflow rate and the hard-wired channel sizes of the chip. We have optimized a very sensitive amplification protocol, capable of accurate whole genome amplification of samples down to 300 pg total RNA. Finally, we demonstrated that the combined microfluidic and amplification protocol is capable of identifying biological pathways perturbed by stimulation in 150 cell samples, and validated the technology by comparing these pathways with the information obtained from bulk-isolated samples.
Methods
Microfluidic chip design
A microfluidic device was designed to implement a sequential protocol for total RNA isolation. The protocol involves five successive steps. To implement these, a continuous fluid flow is established inside the device in one main channel to which five different solutions are then added at precise timing defined by the location of the accessory channels and the flow rate in the main channel (Fig. 1A). In the first three steps cells were lysed in the presence of high concentrations of chaotropic agent guanidinium thiocyanate (GTC), the cell lysate was enzymatically degraded in the presence of proteinase K, and the RNA was captured on a silica column. The ratio of mixing for the reagents was defined by the proportionality between the hydraulic resistance of the main channel and the accessory channels (Fig. 1B and C). GTC not only lyses cells, releasing the RNA, but at the same time it inhibits the endogenous nucleases, preserving the integrity of the RNA. The short diffusion distance between the two adjacent streams also assured that the exposure to high chaotropic agent concentrations is uniform and lysing conditions for all cells are equal, independent of the number of cells in the original sample (Fig. 1D). RNases were removed by the addition of proteinase K to the stream of cell lysate and enzymatic activity took place under conditions of reduced concentration of the chaotropic salt. Further downstream, the concentration of salt was increased again and the nucleic acids released from the cells were captured on a silica particle column (Fig. 1B). In a fourth step, enzymatic DNase treatment was performed on the silica column to degrade the contaminating DNA. In the last step, RNA was recaptured on a second silica column, cleaned, and eluted in distilled water. This RNA is of a quality suitable for further amplification steps. | ||
| Fig. 1 Microfluidic device for the isolation of RNA from small numbers of cells. A: Overall protocol for high quality RNA isolation. Mammalian cells are captured and lysed on the chip, in the presence of a high concentration of chaotropic agent. Cellular proteins are degraded enzymatically and nucleic acids are trapped on a silica column. After enzymatic degradation of the DNA, the RNA is eluted in distilled water and further amplified off the chip using a pre-commercial whole transcriptome amplification system. B: Hydraulic diagram of the flow inside the chip. All solutions necessary for the processing of cells and degradation of proteins are introduced on the chip at the same pressure. The target mixing ratio and timing of the different fluids is achieved by the differences in hydraulic resistance of the microfluidic channels. Additional controls are implemented off chip for the RNA washing and elution from the silica column. C: Overall image of the RNA isolation chip. All solutions are introduced to the chip from pipette tips inserted into the elastomeric device. Solutions of different colors are flown through the chip to illustrate the sequence of mixing reactions. The region of the channels where protein degradation takes place is heated to 54 °C while the rest of the chip is maintained at 4 °C. D. Cell lysis inside the microfluidic channels. Mammalian cells are lysed in less than 3 s after the contact with the high concentration chaotropic agent, which also inhibits the enzymes that may degrade the RNA. | ||
Microfabrication
The microfluidic device was fabricated in polydimethylsiloxane (PDMS, Corning) on glass using standard microfabrication technology (Fig. S1A–F† ). A microfabricated mold was first manufactured by patterning a 30 μm thick layer of SU8 photoresist (Microchem, Newton, MA) on top of a silicon wafer by exposure to 350 nm ultraviolet light through a Mylar mask (Fineline Imaging, Colorado Springs, CO). After developing the first layer using standard photolithography techniques, a second layer of 300 μm SU8 was patterned using a second mask. The two-layer SU8 structure that was photo-patterned on the silicon was used to cast a PDMS piece, replicating the microfluidic network. To finalize the device, the PDMS piece was bonded on a glass slide (1 × 3 inches, Fisher Scientific) after exposure to oxygen plasma (100 mTorr, 2% oxygen, 98% nitrogen, 20 s) and heating on a hotplate (70 °C, 5 min). Immediately after bonding, a 0.1% solution of (heptadecafluoro-1,1,2,2-tetrahydrodecyl)dimethylchlorosilane (fluorosilane, Gelest, Morrisville, PA) in ethanol (Sigma-Aldrich, St. Louis, MO) was flowed into the device for 3 min, using a regular 1 mL syringe. After the surface modification, the fluorosilane solution was removed by an ethanol wash and the ethanol subsequently removed by drying on a hotplate, at 100 °C, for 8 h. Silica particles of 50 to 100 μm size (S0507, Sigma-Aldrich, St. Louis, MI) were suspended into diethyl pyrocarbonate (DEPC) treated distilled water supplemented with 1 M MgCl2 (Ambion, Austin, TX). The particles were loaded in 100 μL plastic pipette tips (Fisher) and introduced into the column chambers through the access holes. The silica particles were trapped inside the microfluidic channels during device fabrication using a weir structure (Fig. S1H† ). After filling each chamber the pipette tip is removed and replaced by Tygon tubing (Small Parts) pinched with forceps.Microfluidic setup
The microfluidic devices were first primed with ethanol, with special care to remove all air bubbles. Reservoirs of 40 μL of each of ethylenediamine tetraacetic acid buffered solution (Tris EDTA) (Sigma Aldrich, St. Louis, MI), 4 M guanidinium thiocyanate (GTC, Ambion), proteinase K (Ambion), binding buffer 1 (GTC, Ambion), and binding buffer 2 (ethanol, Sigma), were implanted at the inlets of the device by forcing 100 μL pipette tips (Ambion) into the inlet holes of the device (Fig. 1C). Flow was established by suction from the outlet in a syringe driven by a syringe pump (Harvard Apparatus, Holliston, MA) which was set at 2.5 μL min−1. After establishing the flow, the device was placed on two Peltier devices side by side and temperatures set for 54 °C and 10 °C, using two separate temperature controllers. The device was positioned such that the “proteinase K treatment loop” was placed over the hot surface and the silica columns are placed over the cooler surface.After the stabilization of the flow and temperature, cells were introduced into the device by replacing the Tris buffer pipette tip with a second tip holding 50 μL of cell suspension. A liquid interface formed by a small droplet of phosphate buffered saline (PBS) buffer solution placed at the inlet before replacing the pipette prevented the introduction of bubbles inside the device during the pipette tips swap. Gentle tapping of the cell reservoir from time to time was required to avoid cell sticking to surfaces and clogging of the tip. Two parallel streams, one with cells and one with 4 M concentration chaotropic agent (GTC) were mixed continuously in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio. The high GTC concentration is also critical for the efficient inhibition of the intracellularRNA degrading enzymes. The length and flow rate in the lysing channel were calculated such that after 8 s the proteinase K was added to the cell lysate for a 3 min total reaction time. The proteolytic action of proteinase K was enhanced by the higher temperature of the digestion channel, and by the dilution of the GTC to a 0.8 M concentration optimal for the activity of the enzyme. After the digestion of associated proteins, salt concentration of the cell lysate was increased again to 2.4 M by the addition of GTC, and binding conditions of the nucleic acids to silica optimized by the addition of ethanol into the main channel. At the end of the first processing series the high salt cell lysate was passed through a silica column and nucleic acids captured on the column.
:5 ratio. The high GTC concentration is also critical for the efficient inhibition of the intracellularRNA degrading enzymes. The length and flow rate in the lysing channel were calculated such that after 8 s the proteinase K was added to the cell lysate for a 3 min total reaction time. The proteolytic action of proteinase K was enhanced by the higher temperature of the digestion channel, and by the dilution of the GTC to a 0.8 M concentration optimal for the activity of the enzyme. After the digestion of associated proteins, salt concentration of the cell lysate was increased again to 2.4 M by the addition of GTC, and binding conditions of the nucleic acids to silica optimized by the addition of ethanol into the main channel. At the end of the first processing series the high salt cell lysate was passed through a silica column and nucleic acids captured on the column.
Cells entering the cell channel were counted manually under the microscope. When the target number of cells was reached, the cell reservoir was removed. The height difference between the lysing solution reservoir and the cell inlet assured the backward flow of lysing buffer and prevented any uncounted cells or any amount of cell lysate in the cell reservoir from entering the device. The flow was maintained for another 5 min after the removal of cell reservoir to assure that all the cell lysate in the device reaches the silica column. After 5 min, the flow was stopped, the reservoirs were removed from the device inlets and the valve at inlet 2a was opened (Fig. 1B). The temperature of the entire chip was set to room temperature (24 °C) by turning off the heating and cooling elements. Air was pushed through in order to dry the silica column and the fluidic network. A volume of 20 μL of washing buffer (0.01% Triton in ethanol, Sigma) was then flushed over the silica column by suction from the outlet, followed by air to remove any traces of the washing buffer. Subsequent cleaning of the contaminant DNA was performed by releasing and then recapturing the nucleic acid on a second column. For these steps, the fluidic configuration of the device was changed by using three valves placed outside the microfluidic device. These valves were closed during cell processing and nucleic acid capture on the first silica column and consecutively opened during the second sequence of steps for removing contaminant DNA. One valve was opened upstream of the first column and used to wash the salt from the silica with ethanolbuffer. A DNase solution, 5 μL in proper buffer (Sigma), was then introduced and flowed across the silica column at 0.7 μL min−1. The DNase solution cannot flow backwards into the device because of the hydrophobic character of the channel walls and their size being smaller than the silica column. After 7 min DNase was replaced by distilled water and the inlet 2b (Fig. 1B) opened. Suction was applied at the outlet using a syringe pump set-up for a total flow rate of 3.2 μL min−1, in the same manner as for the first column. The silica column on the second inlet plays only the role of balancing the hydrodynamic pressures in the system. After 40 min, the second silica column was washed with washing buffer and dried. Following this step, inlet 3a was opened and 20 μL RNase free water (Ambion) was used to elute the RNA into a 20 cm long Teflon tubing attached to a syringe. The water was pulled into the Teflon tubing, eluting the RNA from the column. The total RNA solution was then ejected from the tubing into a 1.5 mL RNase free tube (Ambion) and frozen immediately in a deep freezer. Frozen samples were packed in dry ice and then shipped overnight to Stanford, CA.
NuGEN® RNA amplification and Affymetrix® gene chips
Samples from microfluidic RNA separation were thawed quickly and concentrated to the appropriate volume using Speed Vac. Synthesis of cDNA from total RNA was performed using a pre-commercial prototype of a low-input whole transcriptome amplification assay and labeled cDNA target was generated using the FL-Ovation™ cDNA Biotin Module V2 system. (NuGEN Technologies Inc., San Carlos, CA) Biotin-labeled amplified cDNA targets were generated starting from 1 ng and 0.3 ng total RNA, as well as the total RNA isolated from the 150 cells. The amplified, fragmented and biotin labeled cDNA was hybridized onto GeneChip® Human Genome U133A v2 arrays. The arrays were washed, and scanned as recommended by the FL-Ovation™ cDNA Biotin Module V2 System User Guide.Some of the samples were used as controls and the amount of RNA estimated using qPCR (iQ SYBR Green Supermix, Catalog # 170-8882, Bio-Rad Laboratories, CA) for quantification of the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcript in the total RNA or amplified cDNA products. GAPDH quantification was carried out using two primer sets located at the 3′ or 5′ portion of the transcript. Detailed sequence information for the two primer sets is given in Table 1. To estimate the amount of contaminating DNA, amplification was performed with and without reverse transcriptase.
| Assay name | Forward primer sequence (5′–3′) | Reverse primer sequence (5′–3′) |
|---|---|---|
| GAP330 | TCCACCTTTGACGCTGG | TCATACCAGGAAATGAGCTTGACA |
| GAP1.0 | GAGTCAACGGATTTGGTCGTATT | GAATTTGCCATGGGTGGAAT |
Cell stimulation
Human monocytecell line cells (THP1, ATCC, Manassas, VA) were cultured according to the standard protocol in RPMI media supplemented with 2 mM L-glutamine, 1 mM pyruvate, 0.05 mM mercaptoethanol, 10% fetal bovine serum and 10 mM HEPESbuffer. Before experiments cells were resuspended in Ca free PBS. For stimulation, cells were re-suspended in medium with phorbol 12-myristate 13-acetate, 12 ng mL−1, (PMA, Sigma) and cultured for 6 h. Cells were then resuspended in PBS and used for experiments. Between successive runs, the cell suspension was stored at 4 °C in a refrigerator.Macroscale RNA isolation
Total RNA was isolated and DNA removed using a Qiagen RNeasy mini kit and protocol from samples of 1 million THP1 cells. The amount of RNA in the sample was quantified using a Nanodrop instrument (ThermoFisher Scientific, Waltham, MA) and RiboGreen dye (Invitrogen, Carlsbad, CA), following the procedure recommended by the manufacturer. Serial dilution series were employed to prepare samples of 1 ng RNA and 300 pg RNA.RNA quality and quantity assessment
The quality of the RNA isolated in the device was tested using Agilent pico RNA chips, specific kits, and Agilent 2100 machine and software (Agilent Technologies, Santa Clara, CA). Due to limitations in the sensitivity of the machine, only samples resulting from approximately 2000 cells were analyzed. The Agilent chip was prepared as required by the manufacturer and 1 μL of the 20 μL undiluted sample was loaded on the chip for analysis. In addition, the amount of RNA was estimated using PCR and specific primers (Table 1) and comparing with a standard curve from known amounts of RNA isolated from the same cells using standard RNA isolation techniques.Microarray data analysis
DChip34 software was used for normalization of the arrays (invariant set) and model-base expression of the probe sets (PM-only option). Quality parameters were also calculated for each array, and all the arrays passed the default quality control settings of DChip. To evaluate the performance of microfluidic isolation of neutrophils for microarray analysis, probe sets were first mapped to unique RefSeq genes according to the MicroArray Quality Control project35 (Supplementary Table 3 in ref. 35). Expression values were transformed to log2 for analysis, and differentially expressed genes between unstimulated and stimulated samples were identified with greater than or equal to two fold change and P < 0.001 thresholds, as recommended by the MAQC project.35Gene ontology analysis
Analysis of gene function was performed using the Ingenuity Pathways Knowledge Base (IPKB).4 Briefly, genes identified as differentially expressed between stimulated and unstimulated samples were overlaid on the knowledge base to identify relevant cellular and metabolic functions. The statistical significance of each biological function was then computed based on a statistical likelihood as previously described.4 Pathways with P < 0.001 were identified as significant.Results
We have designed a microfluidic device for the purification of total cellular RNA and utilized it in synergy with a powerful linear whole transcriptome amplification protocol to accomplish robust genome -wide expression analysis from as few as 150 cellsTo purify total RNA, we implemented a highly efficient protocol where total RNA is released from intact cells by chemical lysis, dissociated from proteins, and captured on a silica column where contaminant DNA is removed by enzymatic digestion (Fig. 1A–D). We evaluated the overall efficiency of the DNA removal steps and verified the conservation of the RNA through the digestion protocol by quantitative PCR (qPCR). The cycle threshold (Ct) values for the DNase treated samples are close to those obtained for “no template” samples when tested in the absence of reverse transcriptase, thus demonstrating the complete removal of genomic DNA (Fig. 2A). We have implemented additional steps to minimize the nonspecific absorption of RNA to PDMS, and to maximize the yield of RNA isolated from the column, by optimizing the size of silica particles and column pre-treatment (see ESI Fig. S1 and 2† ). To estimate the quality of the total RNA isolated from the THP1 cell line using the microfluidic device, we performed gel electrophoresis in the Agilent Bioanalyzer Pico Chip. The absence of RNA degradation and the good quality of the sample were an indication of an effective processing protocol in the microfluidic device (see ESI Fig. S3† ). In addition to quality, we estimated the average amount of RNA isolated from each cell using the microfluidic device. We compared the Ct values determined for RNA isolated from samples containing 50, 100 or 1000 cells, and calculated an average amount of total RNA recovered of 2.2 ± 0.4 pg/cell for the 50 cell samples (N = 12), 2.7 ± 0.8 pg/cell for the 100 cell samples (N = 4), and 2.6 ± 0.5 pg/cell for the 1000 cell samples (N = 4) (Fig. 2B). The total time for processing was approximately 50 min for a 150 cell sample, and 60 min for a 1000 cell sample.
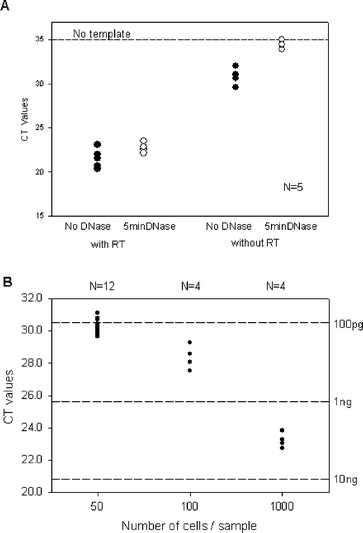 | ||
| Fig. 2 Quality of RNA isolated using the microfluidic devices. A: Removal of genomic DNA from the isolated RNA. RNA samples with and without on-chip 5 min DNase treatment, isolated from 50 cell samples using the microfluidic device, are split into two and GAPDHtranscript is quantified by qPCR using the GAP330 primer pair, in the presence and absence of reverse transcriptase. No significant amount of DNA is detected in the sample after the DNase treatment. B: Consistency of RNA amount isolated from samples of 50, 100 and 1000 cells. Total RNA was isolated using the microfluidic device and its quantity estimated using qPCR and a GAP330 primer pair. | ||
To evaluate the performance of the integrated microfluidic and amplification protocols for genome wide expression analysis, we compared the microarray data from six sets of repeated sample isolations (N = 5 for each condition): 1 ng RNA diluted from bulk isolation of control, unstimulated THP1 cells (a), 1 ng RNA from stimulated THP1 cells (b), 300 pg RNA diluted from bulk isolation of unstimulated (c) and stimulated (d) THP1 cells, microfluidic isolated 150 cells from unstimulated (e) and stimulated (f) THP1 cells. As shown in Fig. 3A, the average number of probe sets and percent of present calls from 1 ng RNA samples are somewhat higher than that from 0.3 ng (N = 5) and the results are comparable between the 0.3 ng RNA and 150 cell samples. Similarly, the concordance of five repeats of each condition, 1 ng, 300 pg total RNA and total RNA isolated from 150 cells was greater than 0.98. These results suggest that microfluidic purification of total RNA from 150 cells introduces little additional variation in gene expression as compared to results from a comparable amount of RNA—0.3ng—diluted from bulk purification. The concordance between samples after 6 h PMA stimulation was slightly better than that obtained for the non-stimulated cell samples, probably due to the concerted stimulation of a significant number of genes and pathways by PMA (Fig. 3B).
 | ||
| Fig. 3 Genome wide gene expression analysis; performance and reproducibility. A: Average number of probe sets (first y axis) and percent call (secondary y axis) of six sets of conditions (N = 5). B: Concordance of five repeats of each condition (a = 1 ng diluted RNA from control unstimulated THP1 cells, b = 1 ng RNA from stimulated THP1 cells, c = 300 pg RNA from unstimulated control THP1 cells, d = 300 pg RNA from stimulated THP1 cells, e = 150 unstimulated control THP1 cells, f = 150 stimulated THP1 cells). C: Gene expression profile using total RNA isolated from 5 unstimulated (N1–5) and 5 stimulated (Y1–5) samples generated using the microfluidic device and the linear whole transcriptome amplification system from 150 THP1 cells. A number of 312 genes that are differentially expressed between stimulated and non-stimulated cells are compared (standard deviation >1, P-call >20%, signal >5 in at least 50% samples). | ||
An unsupervised hierarchical clustering of the gene expression profile measured from the microfluidic isolated cell samples can clearly distinguish stimulated and non-stimulated cells (Fig. 3C). We have identified 776 genes as significantly changed in expression between the stimulated and non-stimulated cells (P value ⩽0.001, ![[greater than or equal, slant]](https://www.rsc.org/images/entities/char_2a7e.gif) 2 fold change, MAQC, 2007) in 1 ng RNA diluted from bulk isolation. At the same time, 445 genes were identified as significantly changed from 0.3 ng RNA and 312 genes from 150 cell microfluidic isolations. To validate the information attained from small samples, we then compared the biological information identified using bulk isolations (1 ng and 0.3 ng RNA) and microfluidic isolations (150 cells) (Fig. 4). Among the total of 23 cellular and metabolic functions and pathways significantly stimulated by phorbol 12-myristate 13-acetate (PMA) in data from bulk isolations, 20 (or 87%) were identified in data from 150 cell samples after microfluidic isolation. This result suggests that accurate biological information can be extracted using the integrated microfluidic and amplification protocols presented in this report.
2 fold change, MAQC, 2007) in 1 ng RNA diluted from bulk isolation. At the same time, 445 genes were identified as significantly changed from 0.3 ng RNA and 312 genes from 150 cell microfluidic isolations. To validate the information attained from small samples, we then compared the biological information identified using bulk isolations (1 ng and 0.3 ng RNA) and microfluidic isolations (150 cells) (Fig. 4). Among the total of 23 cellular and metabolic functions and pathways significantly stimulated by phorbol 12-myristate 13-acetate (PMA) in data from bulk isolations, 20 (or 87%) were identified in data from 150 cell samples after microfluidic isolation. This result suggests that accurate biological information can be extracted using the integrated microfluidic and amplification protocols presented in this report.
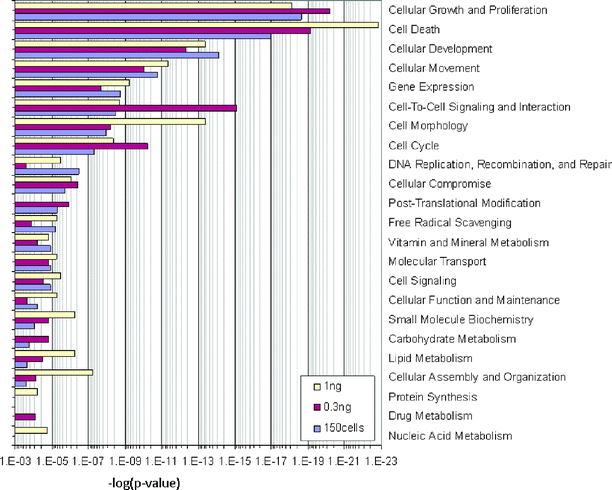 | ||
| Fig. 4 Comparison of the cellular and metabolic pathways identified as differentially expressed between THP1 cells stimulated with phorbol myristate acetate (PMA) and unstimulated THP1 cells (control). Genome wide expression analysis was performed for amplified whole transcriptome derived from samples prepared using different RNA isolation methods and various amounts of total RNA as starting material (yellow bar = 1 ng total RNA from larger THP1 cell sample isolated using the Qiagen RNeasy protocol, red bar = 300 pg total RNA from larger THP1 cell sample isolated using the Qiagen RNeasy protocol, light blue bar = total RNA isolated from 150 cells using the microfluidic device). | ||
Discussion
We have developed a method to investigate global gene expression in biological samples as small as 150 cells. The method employs microfluidic devices for the purification of total RNA from mammalian cells and ultra-sensitive whole transcriptome amplification techniques. This new method has the ability to interrogate the whole transcriptome at once and uncover complex patterns of gene expression in small samples relevant for biology and clinical applications. Previous studies have shown that total RNA is the preferred starting material for high-quality amplification20 and that a single order of magnitude smaller amount of total vs.messenger RNA is needed to access 80% of the genomic information.36In response to limitations of reproducibility of current RNA isolation techniques from small samples, we focused our efforts on the careful purification of the total RNA which appears to be of critical importance for the accuracy and reproducibility of subsequent amplification steps. We chose to implement a protocol based on the use of chaotropic salts (GTC) and nucleic acid capture on a silica column. The protocol does not require centrifugation, an advantage for implementation in microfluidic format, and is widely used for isolating of good integrity and high purity total RNA from large samples.37–39 To accomplish the sequence of reactions necessary for cell and RNA processing, we designed a network of microfluidic channels, in which the size and position of branching channels serves to hard-wire the precise temporal sequence, timing, and stoichiometry of the chemical reactions on the chip. We achieved homogenous conditions for the lysis of individual cells for the entire sample by serially lysing each of the cells as they enter the device. In addition to the fast cell lysis, this allowed us to also achieve reproducible cell lysate processing and adequate control over the endogenous ribonucleases released from the cells during lysis. While the high salt concentration takes care of the enzymes in the first few seconds, enzymatic degradation of the RNases by proteinase K takes care of the RNase enzymes for the rest of the sample preparation reactions. We have further optimized the conditions for nucleic acid capture on the first silica column by the addition of supplementary salt after the proteinase K treatment. We have also implemented a second silica column on the chip in order to recapture the RNA after releasing the nucleic acids from the first column and enzymatic degradation of the DNA. Addition of salt after the DNase treatment and pre-conditioning of the second column with magnesium chloride were critical steps for the overall efficacy of the DNA cleaning steps. Overall, the precise implementation of the multiple steps for RNA processing enabled by microfluidic technologies resulted in high quality and purity of the isolated RNA and the good reproducibility in subsequent microarray analysis.
The microfluidic device for total RNA isolation and purification was designed as a research tool and implemented using prototyping techniques. A number of distinct features could make this approach also attractive for scaled up production and automation of operation. One example is the simple handling of more than five different reagents. All flow rates are controlled from the outflow conditions and the fixed geometry of channels on the chip. The pipette tips used in the prototype could easily be replaced by reservoirs on the chip, to which reagents could be delivered directly. Another example of a scalable design feature is the implementation of valves and flow controllers off the chip. By avoiding the need for elastomeric valves on the chip, it is possible to fabricate the chips in different plastic materials and use high-throughput fabrication technologies e.g. hot embossing or injection molding. The choice of a plastic material with low RNA absorption would in addition eliminate the need for additional surface modification steps, critical in the case of polydimethylsiloxane elastomeric material. Moreover, the implementation of the valves outside the chip allows for greater flexibility in automating the device using standard components and valves, reducing the costs for operation. Finally, further evaluation of silica particles for higher RNA yield,27 or alternative fabrication technologies of silicon oxide coated microscale posts29 may provide additional benefits. Other types of capture columns could be implemented as well, e.g. silica gels, for capturing smaller RNA below the 100 bp cut-off of regular silica particles.
The THP1 monocytecell line used in our study is an established model for monocyte function, exhibiting several of the monocyte characteristics40 and, following PMA stimulation, morphological similarities to macrophages.41 THP1 cells were suspended in the media in the absence of stimulation, and more than 80% of them adhered to the glass substrate with marked morphological change after 6 h of PMA treatment. The adherent cells were flat and amoeboid in shape, and the dramatic changes in morphology were matched by significant modifications in the level of gene expression. Pathway analysis showed that not only individual genes but entire pathways are regulated. The changes in pathway activity that we could identify in large and small samples were correlated to changes previously identified in THP1 cells using microarray technologies.42,43
Conclusions
We designed a microfluidic system for the purification of total cellular RNA and a powerful linear, whole transcriptome amplification protocol, and validated the genome wide expression analysis from as few as 150 cells. We verified that the RNA integrity and amount is preserved through the isolation process and accomplished high correlation between repeated isolations of 150 cells using microfluidics from the same cell culture sample. We validated the technology by demonstrating that the combined microfluidic and amplification protocols are capable to identifying biological pathways perturbed by stimulation, which are consistent with the information recognized in bulk-isolated samples. The new technology has the potential to bridge current knowledge of expression from large and small samples, a challenge incompletely addressed by current techniques. The same device could be used to process samples from 50 to 2000 cells without any modification other than the total time required to process the sample. One immediate application for the combined protocols for microfluidic isolation and linear amplification could be in cancer research. Recently, our group reported the isolation with good yield and purity of tumor cells from the blood of cancer patients.13 The number of isolated cells is of the order of hundreds from millilitre amounts of blood in patients with metastatic cancer, a sample too small for whole genome analysis using standard technologies. Analysis of these cells could enable the elucidation of metastatic signatures and help in identifying the genomic landscape in cancer. Other areas of clinical and biological research in cancer, metastasis, leukocyte subpopulations, development, stem cell biology, or neurology could benefit as well, as they are increasingly targeting much smaller samples.Acknowledgements
This work was supported by grants from the National Human Genome Research Institute (P01HG000205) to RWD, National Institutes of General Medical Sciences (U54GM62119) to RGT, and National Institute of Biomedical Imaging and Bioengineering (P41 EB002503) to MT.References
- M. Schena, D. Shalon, R. W. Davis and P. O. Brown, Science, 1995, 270, 467–470 CrossRef CAS.
- M. Chee, R. Yang, E. Hubbell, A. Berno, X. C. Huang, D. Stern, J. Winkler, D. J. Lockhart, M. S. Morris and S. P. Fodor, Science, 1996, 274, 610–614 CrossRef CAS.
- R. A. Heller, M. Schena, A. Chai, D. Shalon, T. Bedilion, J. Gilmore, D. E. Woolley and R. W. Davis, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2150–2155 CrossRef CAS.
- S. E. Calvano, W. Xiao, D. R. Richards, R. M. Felciano, H. V. Baker, R. J. Cho, R. O. Chen, B. H. Brownstein, J. P. Cobb, S. K. Tschoeke, C. Miller-Graziano, L. L. Moldawer, M. N. Mindrinos, R. W. Davis, R. G. Tompkins and S. F. Lowry, Nature, 2005, 437, 1032–1037 CrossRef CAS.
- J. P. Cobb, M. N. Mindrinos, C. Miller-Graziano, S. E. Calvano, H. V. Baker, W. Xiao, K. Laudanski, B. H. Brownstein, C. M. Elson, D. L. Hayden, D. N. Herndon, S. F. Lowry, R. V. Maier, D. A. Schoenfeld, L. L. Moldawer, R. W. Davis, R. G. Tompkins, P. Bankey, T. Billiar, D. Camp, I. Chaudry, B. Freeman, R. Gamelli, N. Gibran, B. Harbrecht, W. Heagy, D. Heimbach, J. Horton, J. Hunt, J. Lederer, J. Mannick, B. McKinley, J. Minei, E. Moore, F. Moore, R. Munford, A. Nathens, G. O’Keefe, G. Purdue, L. Rahme, D. Remick, M. Sailors, M. Shapiro, G. Silver, R. Smith, G. Stephanopoulos, G. Stormo, M. Toner, S. Warren, M. West, S. Wolfe and V. Young, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4801–4806 CrossRef CAS.
- Q. T. Wang, K. Piotrowska, M. A. Ciemerych, L. Milenkovic, M. P. Scott, R. W. Davis and M. Zernicka-Goetz, Dev. Cell, 2004, 6, 133–144 CrossRef CAS.
- L. D. Wood, D. W. Parsons, S. Jones, J. Lin, T. Sjoblom, R. J. Leary, D. Shen, S. M. Boca, T. Barber, J. Ptak, N. Silliman, S. Szabo, Z. Dezso, V. Ustyanksky, T. Nikolskaya, Y. Nikolsky, R. Karchin, P. A. Wilson, J. S. Kaminker, Z. Zhang, R. Croshaw, J. Willis, D. Dawson, M. Shipitsin, J. K. Willson, S. Sukumar, K. Polyak, B. H. Park, C. L. Pethiyagoda, P. V. Pant, D. G. Ballinger, A. B. Sparks, J. Hartigan, D. R. Smith, E. Suh, N. Papadopoulos, P. Buckhaults, S. D. Markowitz, G. Parmigiani, K. W. Kinzler, V. E. Velculescu and B. Vogelstein, Science, 2007, 318, 1108–1113 CrossRef CAS.
- T. Sorlie, C. M. Perou, R. Tibshirani, T. Aas, S. Geisler, H. Johnsen, T. Hastie, M. B. Eisen, M. van de Rijn, S. S. Jeffrey, T. Thorsen, H. Quist, J. C. Matese, P. O. Brown, D. Botstein, P. Eystein Lonning and A. L. Borresen-Dale, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10869–10874 CrossRef CAS.
- H. Y. Chen, S. L. Yu, C. H. Chen, G. C. Chang, C. Y. Chen, A. Yuan, C. L. Cheng, C. H. Wang, H. J. Terng, S. F. Kao, W. K. Chan, H. N. Li, C. C. Liu, S. Singh, W. J. Chen, J. J. Chen and P. C. Yang, N. Engl. J. Med., 2007, 356, 11–20 CrossRef CAS.
- B. Ryu, D. S. Kim, A. M. Deluca and R. M. Alani, PLoS One, 2007, 2, e594 CrossRef.
- A. A. Alizadeh, M. B. Eisen, R. E. Davis, C. Ma, I. S. Lossos, A. Rosenwald, J. C. Boldrick, H. Sabet, T. Tran, X. Yu, J. I. Powell, L. Yang, G. E. Marti, T. Moore, J. Hudson, Jr, L. Lu, D. B. Lewis, R. Tibshirani, G. Sherlock, W. C. Chan, T. C. Greiner, D. D. Weisenburger, J. O. Armitage, R. Warnke, R. Levy, W. Wilson, M. R. Grever, J. C. Byrd, D. Botstein, P. O. Brown and L. M. Staudt, Nature, 2000, 403, 503–511 CrossRef CAS.
- A. J. Minn, G. P. Gupta, P. M. Siegel, P. D. Bos, W. Shu, D. D. Giri, A. Viale, A. B. Olshen, W. L. Gerald and J. Massague, Nature, 2005, 436, 518–524 CrossRef CAS.
- S. Nagrath, L. V. Sequist, S. Maheswaran, D. W. Bell, D. Irimia, L. Ulkus, M. R. Smith, E. L. Kwak, S. Digumarthy, A. Muzikansky, P. Ryan, U. J. Balis, R. G. Tompkins, D. A. Haber and M. Toner, Nature, 2007, 450, 1235–1239 CrossRef CAS.
- J. M. Doherty, M. J. Geske, T. S. Stappenbeck and J. C. Mills, Stem Cells (Durham, NC, U. S.), 2008, 26, 2124–2130 Search PubMed.
- L. Luo, R. C. Salunga, H. Guo, A. Bittner, K. C. Joy, J. E. Galindo, H. Xiao, K. E. Rogers, J. S. Wan, M. R. Jackson and M. G. Erlander, Nature Med. (N. Y., US), 1999, 5, 117–122 Search PubMed.
- C. Leethanakul, V. Patel, J. Gillespie, M. Pallente, J. F. Ensley, S. Koontongkaew, L. A. Liotta, M. Emmert-Buck and J. S. Gutkind, Oncogene, 2000, 19, 3220–3224 CrossRef CAS.
- D. C. Sgroi, S. Teng, G. Robinson, R. LeVangie, J. R. Hudson, Jr and A. G. Elkahloun, Cancer Res., 1999, 59, 5656–5661 CAS.
- J. M. Levsky, S. M. Shenoy, R. C. Pezo and R. H. Singer, Science, 2002, 297, 836–840 CrossRef CAS.
- M. B. Elowitz, A. J. Levine, E. D. Siggia and P. S. Swain, Science, 2002, 297, 1183–1186 CrossRef CAS.
- M. Kenzelmann, R. Klaren, M. Hergenhahn, M. Bonrouhi, H. J. Grone, W. Schmid and G. Schutz, Genomics, 2004, 83, 550–558 CrossRef CAS.
- M. K. Chiang and D. A. Melton, Dev. Cell, 2003, 4, 383–393 CrossRef CAS.
- F. Tang, P. Hajkova, S. C. Barton, K. Lao and M. A. Surani, Nucleic Acids Res., 2006, 34, e9 CrossRef.
- K. B. Jensen and F. M. Watt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11958–11963 CrossRef CAS.
- J. S. Marcus, W. F. Anderson and S. R. Quake, Anal. Chem., 2006, 78, 3084–3089 CrossRef CAS.
- J. F. Zhong, Y. Chen, J. S. Marcus, A. Scherer, S. R. Quake, C. R. Taylor and L. P. Weiner, Lab Chip, 2008, 8, 68–74 RSC.
- J. W. Hong, V. Studer, G. Hang, W. F. Anderson and S. R. Quake, Nat. Biotechnol., 2004, 22, 435–439 CrossRef CAS.
- H. Tian, A. F. Huhmer and J. P. Landers, Anal. Biochem., 2000, 283, 175–191 CrossRef CAS.
- A. Bhattacharyya and C. M. Klapperich, Anal. Chem., 2006, 78, 788–792 CrossRef CAS.
- D. S. Park, M. L. Hupert, M. A. Witek, B. H. You, P. Datta, J. Guy, J. B. Lee, S. A. Soper, D. E. Nikitopoulos and M. C. Murphy, Biomed. Microdevices, 2008, 10, 21–33 CrossRef CAS.
- C. A. Klein, S. Seidl, K. Petat-Dutter, S. Offner, J. B. Geigl, O. Schmidt-Kittler, N. Wendler, B. Passlick, R. M. Huber, G. Schlimok, P. A. Baeuerle and G. Riethmuller, Nat. Biotechnol., 2002, 20, 387–392 CrossRef CAS.
- F. Kamme, R. Salunga, J. Yu, D. T. Tran, J. Zhu, L. Luo, A. Bittner, H. Q. Guo, N. Miller, J. Wan and M. Erlander, J. Neurosci., 2003, 23, 3607–3615 CAS.
- K. Kurimoto, Y. Yabuta, Y. Ohinata, Y. Ono, K. D. Uno, R. G. Yamada, H. R. Ueda and M. Saitou, Nucleic Acids Res., 2006, 34, e42 CrossRef.
- N. Bontoux, L. Dauphinot, T. Vitalis, V. Studer, Y. Chen, J. Rossier and M. C. Potier, Lab Chip, 2008, 8, 443–450 RSC.
- C. Li and W. H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 31–36 CrossRef CAS.
- L. Shi, L. H. Reid, W. D. Jones, R. Shippy, J. A. Warrington, S. C. Baker, P. J. Collins, F. de Longueville, E. S. Kawasaki, K. Y. Lee, Y. Luo, Y. A. Sun, J. C. Willey, R. A. Setterquist, G. M. Fischer, W. Tong, Y. P. Dragan, D. J. Dix, F. W. Frueh, F. M. Goodsaid, D. Herman, R. V. Jensen, C. D. Johnson, E. K. Lobenhofer, R. K. Puri, U. Schrf, J. Thierry-Mieg, C. Wang, M. Wilson, P. K. Wolber, L. Zhang, S. Amur, W. Bao, C. C. Barbacioru, A. B. Lucas, V. Bertholet, C. Boysen, B. Bromley, D. Brown, A. Brunner, R. Canales, X. M. Cao, T. A. Cebula, J. J. Chen, J. Cheng, T. M. Chu, E. Chudin, J. Corson, J. C. Corton, L. J. Croner, C. Davies, T. S. Davison, G. Delenstarr, X. Deng, D. Dorris, A. C. Eklund, X. H. Fan, H. Fang, S. Fulmer-Smentek, J. C. Fuscoe, K. Gallagher, W. Ge, L. Guo, X. Guo, J. Hager, P. K. Haje, J. Han, T. Han, H. C. Harbottle, S. C. Harris, E. Hatchwell, C. A. Hauser, S. Hester, H. Hong, P. Hurban, S. A. Jackson, H. Ji, C. R. Knight, W. P. Kuo, J. E. LeClerc, S. Levy, Q. Z. Li, C. Liu, Y. Liu, M. J. Lombardi, Y. Ma, S. R. Magnuson, B. Maqsodi, T. McDaniel, N. Mei, O. Myklebost, B. Ning, N. Novoradovskaya, M. S. Orr, T. W. Osborn, A. Papallo, T. A. Patterson, R. G. Perkins, E. H. Peters, R. Peterson, K. L. Philips, P. S. Pine, L. Pusztai, F. Qian, H. Ren, M. Rosen, B. A. Rosenzweig, R. R. Samaha, M. Schena, G. P. Schroth, S. Shchegrova, D. D. Smith, F. Staedtler, Z. Su, H. Sun, Z. Szallasi, Z. Tezak, D. Thierry-Mieg, K. L. Thompson, I. Tikhonova, Y. Turpaz, B. Vallanat, C. Van, S. J. Walker, S. J. Wang, Y. Wang, R. Wolfinger, A. Wong, J. Wu, C. Xiao, Q. Xie, J. Xu, W. Yang, S. Zhong, Y. Zong and W. Slikker, Jr, Nat. Biotechnol., 2006, 24, 1151–1161 CrossRef CAS.
- M. Mahadevappa and J. A. Warrington, Nat. Biotechnol., 1999, 17, 1134–1136 CrossRef CAS.
- P. Chomczynski and N. Sacchi, Anal. Biochem., 1987, 162, 156–159 CrossRef CAS.
- R. A. Cox, Biochem. J., 1968, 106, 725–731 CAS.
- K. A. Melzak, C. S. Sherwoo, R. F. B. Turner and C. A. Haynes, J. Colloid Interface Sci., 1996, 181, 635–644 CrossRef CAS.
- J. Auwerx, Experientia, 1991, 47, 22–31 CrossRef CAS.
- S. Tsuchiya, Y. Kobayashi, Y. Goto, H. Okumura, S. Nakae, T. Konno and K. Tada, Cancer Res., 1982, 42, 1530–1536 CAS.
- T. Kohro, T. Tanaka, T. Murakami, Y. Wada, H. Aburatani, T. Hamakubo and T. Kodama, J. Atheroscler. Thromb., 2004, 11, 88–97 Search PubMed.
- Q. Chen and A. C. Ross, Exp. Cell Res., 2004, 297, 68–81 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Fig. S1–3. See DOI: 10.1039/b814329c |
| ‡ Equal contributions. |
| This journal is © The Royal Society of Chemistry 2009 |