Real-time detection of the early event of cytotoxicity of herbal ingredients on single leukemia cells studied in a microfluidic biochip†
XiuJun
Li
,
Xiaoyan
Xue
and
Paul C. H.
Li
*
Department of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, Canada BC, V5A 1S6. E-mail: paulli@sfu.ca; Fax: 778-782-3765; Tel: 778-782-5956
First published on 12th November 2008
Abstract
A microfluidic approach has been developed for the real-time detection of drug effects, based on the quantitative measurement of calibrated cytosolic calcium ([Ca2+]i) on single cancer cells. This microfluidic method is rapid by detecting the early event of cytotoxicity of drug candidates on cancer cells, without waiting for a couple of days needed for cell seeding and drug treatment by conventional assays. The miniaturized biochip consists of a V-shaped structure for the single-cell selection and retention. Various test reagents such as the chemotherapy drug (daunorubicin), an ionophore (ionomycin), and herbal ingredients from licorice (isoliquiritigenin or IQ) were investigated for their abilities to stimulate sustained cellular [Ca2+]i elevations. The microfluidic results obtained in hours have been confirmed by conventional cytotoxicity assays which take days to complete. Moreover, any color or chemical interference problems found in the conventional assays of herbal compounds could be resolved. Using the microfluidic approach, IQ (50 μM) has been found to cause a sustained [Ca2+]i elevation and cytotoxic effects on leukemia cells. The microfluidic single-cell analysis not only reduces reagent cost, and demands less cells, but also reveals some phenomena due to cellular heterogeneity that cannot be observed in bulk analysis.
Insight, innovation, integrationThere is a need for rapid testing of drug candidates obtained from various sources, such as herbal compounds. There is also an increasing demand for drug testing using the cell-based method. The direct testing of chemotherapeutic drugs on patient cancer cell samples before treatment may open a new avenue for personalized medicine. We invented a microfluidic chip for the real-time detection of cytotoxicity of drug candidates. This is based on the quantitative measurement of cytosolic calcium ([Ca2+]i) on single cancer cells. Such a single-cell method is rapid because it readily detects the early event of drug cytotoxicity based on the sustained increase in [Ca2+]i. This method is faster than the conventional MTT assays that usually take a few days to complete. Moreover, any color or chemical interference problems interfering conventional assays for herbal compounds could be resolved. The microfluidic single-cell method not only reduces reagent cost and demands less cells, but also reveals some phenomena due to cellular heterogeneity that cannot be observed in bulk analysis. |
Introduction
Although medicinal herbs have been widely used in folk medicine with a long history, only recently has the screening of natural anticancer drugs from herbs gained much interest.1–4 Microtiter plate-based cytotoxicity assays, such as the one using MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide), are widely used for drug screening.5 These cell-based colorimetric assays have been used to test the cytotoxic effects of drug candidates on cancer cells.6–8 However, these conventional assays usually require large amounts of herbal ingredients which are often expensive and limited in amount. In addition, these assays are time-consuming, usually taking ∼4 days for each experiment.6,7 Moreover, when screening for herbal compounds, the reliability and sensitivity of some assays, e.g., MTTassay , are sometimes affected by the presence of antioxidants and colored substances that may lead to chemical and color interferences.9 Therefore, there is a need to develop new cell-based techniques to test the bioactivity of herbal compounds.It is well-known that intracellular calcium acts as a universal second messenger to regulate a diverse range of cellular functions (e.g., cell death and myocyte contraction).10 Moreover, the elevation of cytosolic Ca2+ concentration or [Ca2+]i is associated with the activation of cell membrane-bound G-protein-coupled receptors (GPCRs), which represent the drug targets of 50–60% of current therapeutic agents.10–12 Consequently, the cytosolic Ca2+ measurement is now one of the most important cell-based assays in screening for new drug candidates.13,14
The intracellular Ca2+ homeostasis is tightly regulated by the combined effects from different Ca2+ channels and pumps residing on the cell membrane, endoplasmic reticulum (ER) and mitochondria.15 Various stimuli (e.g., anti-cancer drugs) can cause sustained [Ca2+]i elevations, which disrupt the Ca2+ homeostasis and result in cytotoxicity,8,15–17 and even cell death.15,18,19 These [Ca2+]i elevations can lead to Ca2+ overload in mitochondria,15,20–22 resulting in cell death either via the release of cytochrome c from mitochondria to trigger apoptosis (programmed cell death),15,20,23,24 or via necrosis (another mode of cell death).25–27 On the other hand, cytotoxicity or cell death can be prevented by suppressing [Ca2+]i elevations using the mitochondrial Ca2+ uptake inhibitors,15Ca2+ channel antagonist (e.g.verapamil, nifedipine),28–30 and Ca2+chelators.16,17 Therefore, the [Ca2+]i elevations are believed to be an early event of cytotoxicity.15,17,31–33 The [Ca2+]i measurement on cells as stimulated by antitumor herbal compounds can be rapidly achieved, thus alleviating the need of waiting for 4 days in conventional assays.
Recently, it is shown that the use of miniaturized microfluidic devices leads to low reagent consumption when compared with the traditional microtiter plate-based assays. In addition, the small dimensions of microfluidic devices, which are compatible with the sizes of biological cells and also allow for the study of a small number of cells,34 have made the cell-based assay a popular micro total analysis system (μTAS) application.35 Among the microfluidics-based cellular applications, much emphasis has been placed on single-cell analysis, as summarized by recent review articles.36,37 Microfluidic single-cell analysis is preferred to traditional bulk cellular analysis, because the former does not overlook cellular heterogeneity and can provide information about cell-to-cell variations.38,39 Using microfluidic single-cell analysis approaches, Wheeler et al.,39 Mathies and et al.,40 Peng and Li,41 Yin et al.,42,43 Yang et al.,44 and Zhang et al.45 have measured the [Ca2+]i flux of spherical cells. Furthermore, [Ca2+]i flux of single cylindrically shaped cardiomyocytes (heart muscle cells) have been measured by Li et al.,46–48 Kaji et al.,49 Klauke and Cooper et al.50–53 Among these microfluidic studies on [Ca2+]i flux, we have for the first time quantified [Ca2+]i to study the contraction of cardiac myocytes,46 showing that only [Ca2+]i, but not the fluorescence intensity, can accurately represent the [Ca2+]i variations of muscle cells in response to drug stimulations.
In this paper, we apply our previously developed microfluidic [Ca2+]i measurement method to single cancer cells to study the cytotoxicity of drug candidates for drug discovery. This method is rapid because it quickly detects the sustained [Ca2+]i increase, which is an early event of cytotoxicity, caused by different reagents on leukemia cancer cells. These reagents tested in this work include chemotherapy drugs and ionophores as positive controls, and herbal licorice compounds.
Experimental
Chip layout and fabrication
The layout of the microfluidic chip is shown in Fig. 1A. It consists of three channels, three reservoirs and one chamber containing a V-shaped cell retention structure with a central stretch. The right reservoir is the cell inlet, the left one is the waste outlet, and the reagent reservoir (reservoir 3) is used for drug delivery. The glass chip was fabricated by Canadian Microelectronic Corporation (CMC, Protolyne® Chip) by the standard microfabrication procedure. The etch depth was 20 μm on the bottom plate, and through-holes (2 mm in diameter) were drilled on the 600-μm thick cover plate to serve as solution reservoirs. The cell retention structure was shown in the inset of Fig. 1A. | ||
| Fig. 1 Layout of the microfluidic chip and the instrument setup. (A) The schematic of the microfluidic chip consisting of three solution reservoirs and a cell retention structure, which is zoomed out as shown in the inset. (B) An image of the instrument setup. (C) A single RAW 264.7 cell was retained in the cell retention structure. The scale bar is 50 μm. The inset shows the fluorescence image of a RAW cell (diameter, 7 μm) after the stimulation by 10 μg mL−1ionomycin. | ||
Reagents
A fluorescent calcium probe, Fluo-4 AM ester (50 μg, special packaging) was purchased from Molecular Probes (Eugene, OR). It was first dissolved in 50 μL of dimethyl sulfoxide (DMSO, >99.9%, Sigma-Aldrich, St. Louis, MO) to make a stock solution (1 μg μL−1). It was freshly diluted in Hanks’ balanced salt solution (HBSS, Invitrogen Corp., Grand Island, NY) to make a 5-μM working solution. Ionomycin (IM), daunorubicin (DNR), isoliquiritigenin (IQ), glycyrrhizin (GL), 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) and penicillin were also obtained from Sigma-Aldrich. Both IM and IQ were dissolved in DMSO to make stock solutions. DNR, GL and IQ were finally diluted in HBSS containing 1 mM CaCl2 to make working solutions. DMEM (Dulbecco’s modified eagle’s medium) cell culture medium and fetal bovine serum (FBS) were purchased from ATCC (Manassas, VA). Note that Fluo-4 AM is light-sensitive and must be stored in the dark at −20 °C. The MTT solution is also light-sensitive and must be stored in the dark at 4 °C. All reagents were of analytical grade.Cell culture
The mouse leukemic monocytemacrophagecell line (RAW 264.7) was obtained from ATCC. The RAW cells were maintained in the DMEM medium, supplemented with 10% FBS and 50 U mL−1penicillin in a 5% CO2 atmosphere at 37 °C. The cells were passaged twice a week.Instrument
An inverted microscope (TE300, Nikon, Mississauga, ON, Canada), equipped with a photometric system (PTI International, London, ON, Canada), was used for the optical measurement. This optical detection system was employed for simultaneous fluorescence measurement and bright-field observation, as previously described.34,46 A photo of the instrumentation setup is shown in Fig. 1B. A digital camera (Nikon, D50) was employed to obtain fluorescent cell images. A microplate reader (Sunrise, Tecan, Austria) was used to conduct the MTT cytotoxicity assay .Procedures
On-chip dye loading
Once a cell was retained in the cell retention structure, the cell medium in all the reservoirs was removed. Then the reagent reservoir and cell inlet were topped up with the Fluo-4 AM (5 μM) solution for on-chip loading of the dye into the cell, for ∼30 min in the dark. The loaded Fluo-4 AM dissociated after hydrolysis by cellular esterases to give Fluo-4, which would form a fluorescent complex with the Ca2+ ion. The inset of Fig. 1C shows the fluorescence image of a RAW cell after the stimulation by ionomycin. Afterwards, the Fluo-4 AM solution was washed away by HBSS introduced from the cell inlet and reagent reservoir. This on-chip dye loading method has been proven to minimize the cell damage that would result from the use of a centrifuge in the conventional off-chip dye loading procedure.46Fluorescent measurement and calibration of [Ca2+]i of a single cell
After Fluo-4 loading, fluorescent measurement was started to monitor [Ca2+]i in the cell at room temperature. The chip was translated back and forth so that the detection aperture window observed the cell or its surrounding region in turn. When the cell was inside or outside of the detection aperture, the cellular fluorescence signal or the background was measured, respectively. The test compounds were continuously introduced through the reagent reservoir to stimulate the retained cell. At the end of each experiment, a 10 μg mL−1ionomycin solution containing 50 μM CaCl2 was introduced to saturate the cellular fluorescence. The maximum fluorescence, Fmax, resulted from 10 μg mL−1ionomycin addition, was used in eqn (1) for [Ca2+]i calibration, as previously described.46 As defined by eqn (1),54 [Ca2+]i was quantified as follows,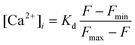 | (1) |
MTT cytotoxicity assay
The cytotoxic effects of various test compounds on RAW cells were also determined by conventional 96-well microtiter plate assay .8 First, 100 μL cell suspension at a density of ∼5 × 104 cells mL−1 was added to seed each well which was then incubated at 37 °C. After 1 day of cell seeding, 100 μL of test compounds prepared in the medium at various concentrations were separately introduced in each well. After the cells were further incubated for 3 days, 40 μL of MTT(5 mg mL−1) was added to each well and incubated at 37 °C for 3 h. Then the supernatant solution was removed by an aspirator, and the purple formazan product formed inside the cells was solubilized by adding 100 μL of DMSO into each well. Finally, the absorbance of each well was measured at 570 nm with a microplate reader. Administrations of the medium without test compounds and of DMSO alone were used as the negative control and the positive control, respectively. All tests were carried out in triplicate.Results and discussion
Real-time monitoring of [Ca2+]i dynamics of single RAW cells
Real-time monitoring of the temporal changes of cytosolic calcium or [Ca2+]i was carried out on single RAW cells. It was observed that [Ca2+]i of most untreated RAW cells did not vary greatly, with a typical measurement shown in Fig. 2A. After calibration, [Ca2+]i at the resting status was determined to be ∼74 ± 14 nM (n = 9), consistent with the literature value of ∼100 nM.56–58 However, in some experiments, several Ca2+ spikes up to ∼400–500 nM were observed even before the test compound was added (Fig. 2B). These kinds of Ca2+ spikes may be related to some intracellular processes, such as the activation of mitochondrial Ca2+-sensitive enzymes.59 Since the traditional endpoint analysis on the bulk cell population often overlooks such information, the real-time monitoring of individual cells is necessary to reveal the dynamic and temporal changes of intracellularCa2+ among cellular heterogeneity. Therefore, we have employed such a method to study the [Ca2+]i mobilization of single cells induced by different stimuli (e.g.drug candidates).![Real-time monitoring of [Ca2+]i of two different single RAW cells in a microchip. At the end of each experiment, a 10 μg mL−1ionomycin (IM) solution containing 50 μM CaCl2 was added for the calibration of [Ca2+]i](/image/article/2009/IB/b812987h/b812987h-f2.gif) | ||
| Fig. 2 Real-time monitoring of [Ca2+]i of two different single RAW cells in a microchip. At the end of each experiment, a 10 μg mL−1ionomycin (IM) solution containing 50 μM CaCl2 was added for the calibration of [Ca2+]i | ||
Ionomycin-stimulated [Ca2+]i flux
Ionomycin has been reported to elicit cytosolic Ca2+ elevation in a variety of cells.39,60–62 Because of the ionophore property of ionomycin, high concentrations of ionomycin are often used to obtain Fmax for calcium calibrations. On the other hand, ionomycin has been reported to cause cytotoxic effects on cancer cells. For instance, ionomycin has also been found to induce apoptosis in the human prostate cancer cell line LNCaP,61 bladder cancer cell line HT1376,6 and leukemia cell line HL-60.62 In this work, we use the microfluidic method to study the effect of ionomycin on [Ca2+]i flux of single mouse leukemia cells of RAW 264.7.Fig. 3A shows the [Ca2+]i flux from different single live cells as stimulated by two different concentrations of ionomycin. In the experiment of the low concentration of ionomycin (cell 1; 2 μg mL−1), an obvious transient peak (850 nM) was observed, and the transient peak was followed by a sustained level of [Ca2+]i (∼150 nM). This sustained level was lower than the transient peak, but was higher than the resting [Ca2+]i of ∼79 nM. However, in the experiment of a higher ionomycin concentration (cell 2; 10 μg mL−1), the transient peak was not observed, and a quick increase followed by a higher sustained level were seen instead (see Fig. 3A inset). What’s more, the sustained level of [Ca2+]i was higher than the first increase in cell 2, which was different from cell 1 that the sustained level was lower than the transient peak. We explain this observation using the theory that ionomycin-stimulated [Ca2+]i flux occurs through a biphasic process.61 The occurrence of the first transient peak is due to the Ca2+ release from the internal store ER, as observed in the lower concentration of ionomycin (2 μg mL−1). This release in turn triggers the Ca2+ influx through the cell membrane, thus resulting in a subsequent sustained [Ca2+]i increase, based on the mechanism called store-regulated Ca2+ uptake (SRCU).60,61 However, this sustained increase will be modest if the ionomycin concentration is not high enough, as observed in cell 1. In cell 2, the use of 10 μg mL−1 of ionomycin induced a faster and more substantial Ca2+ influx through cell membrane, thus resulting in a higher sustained [Ca2+]i plateau following the first increase, which has veiled the appearance of the first transient peak (see curve 2′ in Fig. 3 inset). The second Ca2+ increase demands the presence of sufficient extracellularCa2+, and if there is none, only a transient Ca2+ peak (curve 4 in Fig. 4), rather than a subsequent sustained Ca2+ increase (curve 2′ in Fig. 3A inset), is observed.
![Effects of ionomycin (IM) on cancer cells. (A) [Ca2+]i flux stimulated by different concentrations of IM solutions containing 50 μM CaCl2. Curve 1, 2 μg mL−1IM; Curve 2, 10 μg mL−1IM; their corresponding fluorescence intensity traces are shown in the inset. For easy comparison, the fluorescence intensities have been normalized and vertically offset, as shown in the inset. The image in the inset shows that the cell was stained by Trypan Blue, which confirmed the cell death. (B) Dose-dependent cytotoxicity of IM on cancer cells by the MTTassay. Treatment by DMSO alone, and cell culture medium alone (without test compounds) were used as the positive and negative controls, respectively.](/image/article/2009/IB/b812987h/b812987h-f3.gif) | ||
| Fig. 3 Effects of ionomycin (IM) on cancer cells. (A) [Ca2+]i flux stimulated by different concentrations of IM solutions containing 50 μM CaCl2. Curve 1, 2 μg mL−1IM; Curve 2, 10 μg mL−1IM; their corresponding fluorescence intensity traces are shown in the inset. For easy comparison, the fluorescence intensities have been normalized and vertically offset, as shown in the inset. The image in the inset shows that the cell was stained by Trypan Blue, which confirmed the cell death. (B) Dose-dependent cytotoxicity of IM on cancer cells by the MTTassay . Treatment by DMSO alone, and cell culture medium alone (without test compounds) were used as the positive and negative controls, respectively. | ||
The first transient Ca2+ release from ER due to ionomycin can result in ER stress. In addition, the sustained cytosolic Ca2+ increase can also lead to Ca2+ overload in mitochondria. Both processes can result in cell death.15 For instance, 10 μg mL−1 of ionomycin quickly caused the sustained [Ca2+]i increase of more than 1 μM. The rapid sustained cytosolic Ca2+ increase is highly cytotoxic to cells, especially when [Ca2+]i reaches above 1 μM which could promptly trigger cell death.20,63 In Fig. 3A, the cell death was manifested as the disintegration of the cell membrane, and a sharp fluorescence decrease due to the loss of the intracellularFluo-4-Ca2+ complex, as observed at the latter parts of the curves of 10 μg mL−1ionomycin, in Fig. 3A inset (curve 2′) and in Fig. 4. The loss of the intracellular Fluo-4-Ca2+ complex from a RAW cell due to the disintegration of the cell membrane was imaged in Fig. 4 inset. Cell death was also confirmed by Trypan Blue staining, as shown in Fig. 3A inset.
![Different patterns of [Ca2+]i flux from different cells stimulated by the same concentration of IM (10 μg mL−1). Curves 1–3, different [Ca2+]i patterns elicited by IM solutions containing 50 μM CaCl2 which were introduced at 2998 s separately to three different cells. The fluorescence image in the inset shows the occasion when the intracellularFluo-4-Ca2+ complexes were released into the extracellular solution due to the disintegration of the cell membrane. In Curve 4 of another inset, cell 4 was treated with a 10 μg mL−1IM solution without 50 μM of CaCl2 at 3050 s, and then with another 10 μg mL−1IM solution containing 50 μM of CaCl2 at 3786 s. The fluorescence intensities have been normalized and vertically offset for comparison.](/image/article/2009/IB/b812987h/b812987h-f4.gif) | ||
| Fig. 4 Different patterns of [Ca2+]i flux from different cells stimulated by the same concentration of IM (10 μg mL−1). Curves 1–3, different [Ca2+]i patterns elicited by IM solutions containing 50 μM CaCl2 which were introduced at 2998 s separately to three different cells. The fluorescence image in the inset shows the occasion when the intracellularFluo-4-Ca2+ complexes were released into the extracellular solution due to the disintegration of the cell membrane. In Curve 4 of another inset, cell 4 was treated with a 10 μg mL−1IM solution without 50 μM of CaCl2 at 3050 s, and then with another 10 μg mL−1IM solution containing 50 μM of CaCl2 at 3786 s. The fluorescence intensities have been normalized and vertically offset for comparison. | ||
To examine the cytotoxicity of ionomycin to cancer cells, the conventional MTT cytotoxicity was carried out on RAW cells. Fig. 3B shows that ionomycin has a dose-dependent antiproliferative effect on the RAW 264.7 cells. As the ionomycin concentration increased, more cytotoxicity occurred to cells, resulting in less live cells, as indicated by the low measured absorbance as shown in Fig. 3B. The IC50 (the half maximal inhibitory concentration) of ionomycin was found to be ∼2 μg mL−1 (or 2.7 μM). It was observed that 10 μg mL−1ionomycin led to a similar absorbance as the positive control (DMSO-treated cells). On the other hand, the absorbance obtained from 0.8 μg mL−1 was similar to the negative control (no drug treatment), thus indicating this concentration was not cytotoxic to RAW cells, which is consistent with a previous report that a concentration of ionomycin lower than 1 μM neither induced apoptosis nor produced the biphasic increase in [Ca2+]i in prostate cancer cells.61
Our microfluidic results show that the possible antiproliferative effect of ionomycin on RAW 264.7 cells was determined using only a 1 h single-cell measurement of the disruptive [Ca2+]i elevation, as compared to the conventional 4 day MTT cytotoxicity assay . In addition, in the testing of antioxidants or colored reagents, these compounds would interfere chemically or colorimetrically with the MTTassay . Nevertheless, these problems could be readily resolved by the microfluidic [Ca2+]i measurement method that we present here.
During the single-cell experiments, it was found that different cells responded to the stimulus of 10 μg mL−1ionomycin differently, as shown by the different patterns of [Ca2+]i flux in Fig. 4. Cell 1 shows a ‘two-step stair’ pattern, while cell 3 shows a pattern of a transient peak followed by a slow sustained increase. Although the patterns are different, we can still tell they are biphasic increases. Nevertheless, cell 2 shows a different pattern of [Ca2+]i flux, and it is hard to discern the biphasic feature from curve 2. We believe these different patterns are resultant from the time lag between the two phases of ionomycin-stimulated [Ca2+]i flux, i.e. (1) the transient Ca2+ release from ER and (2) the sustained influx from extracellularCa2+ through the cell membrane.61 If the sustained Ca2+ influx through the cell membrane occurs very quickly, the time lag between the two phases is very short, a pattern like curve 2 will be observed, i.e. without obvious biphasic changes. If the Ca2+ influx through the cell membrane occurs more slowly, the first transient Ca2+ peak will be well separated from the second sustained Ca2+ increase, resulting in a pattern like curve 3. If extracellularCa2+ was intentionally removed, but was added in a much later time to allow Ca2+ influx to occur, the two phases were even more spread out, as depicted by curve 4. These different cellular responses are the different manifestations of the biphasic Ca2+ flux process due to cellular heterogeneity, which is overlooked in the conventional bulk analysis. Therefore, we believe that the microfluidic single-cell measurement can help reveal cellular heterogeneity, and help identify the underlying mechanism of the biphasic cell responses.
Daunorubicin-induced [Ca2+]i mobilization
Daunorubicin (DNR) is a highly effective chemotherapy drug of the anthracycline family that is used to treat a variety of cancers (e.g. leukemia, breast cancer, and small-cell lung cancer).64 For example, it has been reported that DNR caused apoptosis in leukemic cell lines, such as U937 and HL60,65,66 but there are no reports about the effect of DNR on RAW cells. Again, monitoring the [Ca2+]i mobilization may provide a fast way to measure the drug candidate’s cytotoxic effect. Moreover, since the [Ca2+]i mobilization precedes the caspase activation in the apoptosis pathway,66 this measurement method will reveal the early-stage information of cell death.Fig. 5 shows the [Ca2+]i mobilization of a single live RAW cell (cell 1) by the DNR treatment. It can be seen that once DNR was added, [Ca2+]i did not increase much at the first 1200 s (see curve 1). Then a sustained [Ca2+]i increase up to ∼420 nM was observed after ∼3800 s of the DNR treatment. A control experiment on another individual cell (cell 2) without Fluo-4 loading was performed to ensure that the cellular accumulation of DNR, which is intrinsically fluorescent,34 did not interfere with the fluorescent measurement of [Ca2+]i. It could be seen that there was no obvious cellular fluorescence increase observed due to the DNR (3.5 μM) accumulation into the cell as shown in curve 2, which confirmed that 3.5 μM DNR did not interfere with the fluorescent measurement of [Ca2+]i. This is mainly due to the different emission wavelengths of DNR (585 nm) and Fluo 4-Ca2+ (525 nm).34,46 At a higher concentration of DNR (35 μM), the [Ca2+]i increase was even higher, and it was observed that some RAW cells died within ∼4500 s, confirmed by Trypan Blue staining (data not shown).
![DNR-induced [Ca2+]i mobilization on single leukemia RAW cells. Curve 1, [Ca2+]i flux induced by DNR solution (3.5 μM). Curve 2, the fluorescence intensity versus time curve due to DNR accumulation without Fluo-4 loading (control experiment). At 2361 s, DNR solutions were introduced to these two different cells separately.](/image/article/2009/IB/b812987h/b812987h-f5.gif) | ||
| Fig. 5 DNR-induced [Ca2+]i mobilization on single leukemia RAW cells. Curve 1, [Ca2+]i flux induced by DNR solution (3.5 μM). Curve 2, the fluorescence intensity versus time curve due to DNR accumulation without Fluo-4 loading (control experiment). At 2361 s, DNR solutions were introduced to these two different cells separately. | ||
The MTT assay results shown in Fig. 6A confirmed the effect of DNR on RAW cells. There is no obvious antiproliferative effect observed on RAW cells when DNR concentration is lower than ∼0.10 μM. When its concentration increased above ∼ 1 μM, DNR killed all the RAW cells during the 3-day drug treatment time, resulting in a similar absorbance with that of DMSO. The IC50 of DNR on RAW cells is ∼0.35 μM, which is similar to the value on U937 cells (IC50 = 0.20 μM).65
 | ||
| Fig. 6 Dose-dependent cytotoxicity on RAW cells by (A) DNR, (B) GL and (C) IQ using the conventional MTTassay . | ||
Effects of licorice compounds on RAW 264.7 cancer cells
Glycyrrhizin (GL), which is a major ingredient of licorice, has various desirable pharmacological properties such as anti-viral,67,68 anti-inflammatory.69 Therefore, we studied to see whether this ingredient has anticancer or cytotoxic effects on leukemia cells.Similarly, the effect of GL on the [Ca2+]i level was measured. However, the increase of [Ca2+]i on single RAW cells from GL was not observed, see Fig. 7A. From the conventional MTTassay (Fig. 6B), it can seen that GL does not show antiproliferative effect on RAW cells. This is consistent with the microfluidic results, indicating that GL does not cause a sustained [Ca2+]i increase on RAW cells.
![[Ca2+]i mobilization by licorice compounds on single leukemia RAW cells. (A) The effect of GL on [Ca2+]i. At 2913 s, 100 μM GL was added. IM was added at the end of the experiment to quantify [Ca2+]i values (B) IQ-induced [Ca2+]i mobilization as shown as Curve 1: At 3006 s, 50 μM of IQ was introduced to a single RAW cell after dye loading. Curve 2: fluorescence intensity of IQversus time. At 3006 s, IQ was introduced to another RAW cell without dye loading (a control experiment). The fluorescence increase upon the introduction of IQ (100 μM) in curve 2 is attributed to the fluorescence background increase due to the intrinsic fluorescence of IQ, not cellular fluorescence due to the IQ accumulation in the cell, as confirmed by checking the difference between cellular fluorescence and the background (see ESI ).](/image/article/2009/IB/b812987h/b812987h-f7.gif) | ||
| Fig. 7 [Ca2+]i mobilization by licorice compounds on single leukemia RAW cells. (A) The effect of GL on [Ca2+]i. At 2913 s, 100 μM GL was added. IM was added at the end of the experiment to quantify [Ca2+]i values (B) IQ-induced [Ca2+]i mobilization as shown as Curve 1: At 3006 s, 50 μM of IQ was introduced to a single RAW cell after dye loading. Curve 2: fluorescence intensity of IQversus time. At 3006 s, IQ was introduced to another RAW cell without dye loading (a control experiment). The fluorescence increase upon the introduction of IQ (100 μM) in curve 2 is attributed to the fluorescence background increase due to the intrinsic fluorescence of IQ, not cellular fluorescence due to the IQ accumulation in the cell, as confirmed by checking the difference between cellular fluorescence and the background (see ESI† ). | ||
Isoliquiritigenin (IQ), a flavonoid ingredient from licorice, was found to exhibit cytotoxic effects on human gastric,70 prostate,2 heptoma71 and breast cancer cells,72 and also on mouse renal73 and melanoma cells.74 However, as far as we know, there are no reports about its effect on leukemia cells. As it was reported that the cytotoxic effect of IQ on gastric cancer cells may involve a calcium-dependent pathway,70 we decided to monitor [Ca2+]i mobilization of leukemia RAW cells as induced by IQ.
Fig. 7B shows IQ has caused a sustained [Ca2+]i increase which represents an early cytotoxic event on a single RAW cell (curve 1). However, during the first 2000 s after introducing 50 μM IQ, the [Ca2+]i increase was slow. Thereafter, the increase of [Ca2+]i became faster, and finally it reached a level of ∼370 nM after 1.5 h. More experiments show that the maximum [Ca2+]i due to IQ within 1.5 h are 342 ± 39 nM (n = 3). Since IQ has an intrinsic fluorescence at λem = 525 nm as well, a control experiment was performed on a RAW cell without Fluo-4 loading, as shown in curve 2 in Fig. 7B. It was found, even at a high concentration of IQ (100 μM), no cellular fluorescence increase due to IQ accumulation in the single cell was observed, using a previously described procedure,41 see Fig. S1, ESI.† This was further confirmed by checking cellular fluorescence after washing away the IQ solution (see Fig. S2, ESI† ). So the intrinsic fluorescence of IQ did not interfere with the fluorescent measurement of IQ-induced [Ca2+]i elevation. This can be attributed to the fact that IQ cannot permeate the cell membrane because IQ is a polar molecule (See IQ chemical structure in the inset of Fig. 7B). Accordingly, the sustained [Ca2+]i increase induced by IQ may go through the interaction of IQ with cell membrane receptors, like the GPCR activation process.10
Similarly, the MTT assay confirmed the dose-dependent effect of IQ on RAW cells, see Fig. 6C. A low concentration of 5 μM IQ did not inhibit growth of RAW cells. When IQ was above 10 μM, it began to show substantial toxicity. At 100 μM of IQ, most of the cells did not survive as the absorbance of IQ was very close to that of DMSO (positive control). The IC50 value of IQ on RAW 264.7 cells is ∼35 μM, which is comparable to the IC50 value (∼11 μM) on human prostate cancer cells.2
From the microfluidic [Ca2+]i measurement and MTTassay data, we report for the first time that IQ has an antiproliferative effect on leukemia cells, suggesting that IQ would be a candidate agent for the treatment of leukemia. Although DNR is widely used in treatment of leukemia, the use of DNR is unfortunately limited by its potentially fatal cardiotoxicity.75,76 Previously, we reported that IQ has less effect on [Ca2+]i of heart muscle cells, and hence less cardiotoxicity, as compared with DNR.47 Therefore, IQ can be a potential drug candidate for the treatment of leukemia with less cardiotoxicity. But the findings of these cell-based in vitro tests are subject to the confirmation by in vivo assays.
Conclusion
A microfluidic approach of single-cell analysis on cancer cells has been developed for drug discovery. This is based on the real-time measurement of the sustained increase in [Ca2+]i, which could be rapidly measured, without the 4 day wait when using the conventional assays. Therefore, this approach is suitable for preliminary drug screening of traditional medicinal herbs using less reagents to reduce the cost in drug discovery, and especially for colored compounds and antioxidants that usually interfere with the cytotoxicity assay . Using this approach, IQ, a herbal ingredient of licorice has been found to have an antiproliferative effect on leukemia cells, suggesting it is a drug candidate for leukemia. This microfluidic method with the real-time measurement also has great potentials in the dynamic study of drug actions on rare cells (e.g. cancer stem cells) or patient samples because of only a small number of cells needed, and in the mechanistic study of cellular functions at the single-cell analysis level.Acknowledgements
Financial support from Natural Science and Engineering Research Council of Canada (Idea-to-Innovation) and Canadian Microelectronic Corporation (Protolyne) is acknowledged. We are also grateful to Dr Joseph Tai (UBC Child and Family Research Institute, Vancouver, BC, Canada) for providing the cell samples, and Michael Sung, Derek Chew and Peter Chou for the help in cell culture and MTT assay procedure. We also thank Samar Haroun for the help in the manuscript preparation.References
- C. K. Lee, K. K. Park, S. S. Lim, J. H. Park and W. Y. Chung, Biol. Pharm. Bull., 2007, 30, 2191–2195 CrossRef CAS.
- M. Kanazawa, Y. Satomi, Y. Mizutani, O. Ukimura, A. Kawauchi, T. Sakai, M. Baba, T. Okuyama, H. Nishino and T. Miki, Eur. Urol., 2003, 43, 580–586 CrossRef CAS.
- Q. Wang, L. M. Wu, Y. Zhao, X. L. Zhang and N. P. Wang, Acta Pharm. Sinica, 2002, 37, 477–478 Search PubMed (Yao Xue Xue Bao).
- J. K. Srivastava and S. Gupta, J. Agric. Food Chem., 2007, 55, 9470–9478 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- H. Miyake, I. Hara, E. Yamanaka, S. Arakawa and S. Kamidono, J. Urology, 1999, 162, 916–921 Search PubMed.
- C. Dell'Erba, B. Chiavarina, C. Fenoglio, G. Petrillo, C. Cordazzo, E. Boncompagni, D. Spinelli, E. Ognio, C. Aiello, M. A. Mariggio and M. Viale, Pharmacol. Res., 2005, 52, 271–282 CrossRef CAS.
- G. M. Jow, C. J. Chou, B. F. Chen and J. H. Tsai, Cancer Lett., 2004, 216, 165–173 CrossRef CAS.
- X. Wang, J. Ge, K. Wang, J. Qian and Y. Zou, Assay Drug Dev. Technol., 2006, 4, 203–207 CrossRef CAS.
- R. T. Dorsam and J. S. Gutkind, Nat. Rev. Cancer, 2007, 7, 79–94 CrossRef CAS.
- A. D. Howard, G. McAllister, S. D. Feighner, Q. Liu, R. P. Nargund, L. H. Van der Ploeg and A. A. Patchett, Trends Pharmacol. Sci., 2001, 22, 132–140 CrossRef CAS.
- K. L. Pierce, R. T. Premont and R. J. Lefkowitz, Nat. Rev. Mol. Cell Biol., 2002, 3, 639–650 CrossRef CAS.
- G. R. Monteith and G. S. Bird, Trends Pharmacol. Sci., 2005, 26, 218–223 CrossRef CAS.
- J. F. Worley 3rd and M. J. Main, Receptor Channel, 2002, 8, 269–282 CrossRef.
- S. Orrenius, B. Zhivotovsky and P. Nicotera, Nat. Rev. Mol. Cell Biol., 2003, 4, 552–565 CrossRef CAS.
- P. R. Mayeux and S. V. Shah, J. Pharmacol. Exp. Ther., 1993, 266, 47–51 CAS.
- L. Virag, G. S. Scott, P. Antal-Szalmas, M. O'Connor, H. Ohshima and C. Szabo, Mol. Pharmacol., 1999, 56, 824–833 CAS.
- Y. Furuya, P. Lundmo, A. D. Short, D. L. Gill and J. T. Isaacs, Cancer Res., 1994, 54, 6167–6175 CAS.
- A. R. Boobis, D. J. Fawthrop and D. S. Davies, Trends Pharmacol. Sci., 1989, 10, 275–280 CrossRef CAS.
- G. R. Monteith, D. McAndrew, H. M. Faddy and S. J. Roberts-Thomson, Nat. Rev. Cancer, 2007, 7, 519–530 CrossRef CAS.
- R. Rizzuto, M. Brini, M. Murgia and T. Pozzan, Science, 1993, 262, 744–747 CrossRef CAS.
- G. A. Rutter, J. M. Theler, M. Murgia, C. B. Wollheim, T. Pozzan and R. Rizzuto, J. Biol. Chem., 1993, 268, 22385–22390 CAS.
- D. Boehning, R. L. Patterson, L. Sedaghat, N. O. Glebova, T. Kurosaki and S. H. Snyder, Nat. Cell Biol., 2003, 5, 1051–1061 CrossRef CAS.
- M. R. Duchen, J. Physiol., 2000, 529 Pt 1, 57–68.
- J. S. Armstrong, Br. J. Pharmacol., 2006, 147, 239–248 CrossRef CAS.
- N. Festjens, T. Vanden Berghe and P. Vandenabeele, Biochim. Biophys. Acta, 2006, 1757, 1371–1387 CAS.
- M. Diaz-Munoz, M. A. Alvarez-Perez, L. Yanez, S. Vidrio, L. Martinez, G. Rosas, M. Yanez, S. Ramirez and V. C. de Sanchez, Mol. Cell Biochem., 2006, 289, 125–136 CrossRef CAS.
- B. B. Burkey, G. M. Omann and G. T. Wolf, Arch. Otolaryngol. Head Neck Surg., 1991, 117, 1281–1286 Search PubMed.
- K. Koike, N. Masumoto, K. Kasahara, M. Yamaguchi, K. Tasaka, K. Hirota, A. Miyake and O. Tanizawa, Endocrinology, 1991, 128, 2785–2790 CrossRef CAS.
- A. M. Hurne, C. L. Chai, K. Moerman and P. Waring, J. Biol. Chem., 2002, 277, 31631–31638 CrossRef CAS.
- D. M. Gurfinkel, S. Chow, R. Hurren, M. Gronda, C. Henderson, C. Berube, D. W. Hedley and A. D. Schimmer, Apoptosis, 2006, 11, 1463–1471 Search PubMed.
- C. R. Jan, C. H. Chen, S. C. Wang and S. Y. Kuo, Cell. Signal., 2005, 17, 847–855 CrossRef CAS.
- M. J. Berridge, Cell Calcium, 2002, 32, 235–249 CrossRef CAS.
- X. J. Li, V. Ling and P. C. H. Li, Anal. Chem., 2008, 80, 4095–4102 CrossRef CAS.
- P. S. Dittrich, K. Tachikawa and A. Manz, Anal. Chem., 2006, 78, 3887–3908 CrossRef CAS.
- C. E. Sims and N. L. Allbritton, Lab Chip, 2007, 7, 423–440 RSC.
- D. Di Carlo and L. P. Lee, Anal. Chem., 2006, 78, 7918–7925 CrossRef.
- M. N. Teruel and T. Meyer, Science, 2002, 295, 1910–1912 CrossRef CAS.
- A. R. Wheeler, W. R. Throndset, R. J. Whelan, A. M. Leach, R. N. Zare, Y. H. Liao, K. Farrell, I. D. Manger and A. Daridon, Anal. Chem., 2003, 75, 3581–3586 CrossRef CAS.
- N. M. Toriello, E. S. Douglas and R. A. Mathies, Anal. Chem., 2005, 77, 6935–6941 CrossRef CAS.
- X. Y. Peng and P. C. H. Li, Anal. Chem., 2004, 76, 5273–5281 CrossRef CAS.
- H. Yin, N. Pattrick, X. Zhang, N. Klauke, H. C. Cordingley, S. J. Haswell and J. M. Cooper, Anal. Chem., 2008, 80, 179–185 CrossRef CAS.
- H. Yin, X. Zhang, N. Pattrick, N. Klauke, H. C. Cordingley, S. J. Haswell and J. M. Cooper, Anal. Chem., 2007, 79, 7139–7144 CrossRef CAS.
- M. Yang, C. W. Li and J. Yang, Anal. Chem., 2002, 74, 3991–4001 CrossRef CAS.
- X. Zhang, H. Yin, J. M. Cooper and S. J. Haswell, Electrophoresis, 2006, 27, 5093–5100 CrossRef CAS.
- X. J. Li and P. C. H. Li, Anal. Chem., 2005, 77, 4315–4322 CrossRef CAS.
- X. J. Li, J. Huang, G. F. Tibbits and P. C. H. Li, Electrophoresis, 2007, 28, 4723–4733 CrossRef CAS.
- X. J. Li and P. C. H. Li, Methods Mol. Biol., 2006, 321, 199–225 Search PubMed.
- H. Kaji, M. Nishizawa and T. Matsue, Lab Chip, 2003, 3, 208–211 RSC.
- N. Klauke, G. L. Smith and J. Cooper, Biophys. J., 2003, 85, 1766–1774 CrossRef CAS.
- W. Cheng, N. Klauke, H. Sedgwick, G. L. Smith and J. M. Cooper, Lab Chip, 2006, 6, 1424–1431 RSC.
- N. Klauke, G. L. Smith and J. Cooper, Biophys. J., 2006, 91, 2543–2551 CrossRef CAS.
- N. Klauke, G. Smith and J. M. Cooper, Lab Chip, 2007, 7, 731–739 RSC.
- A. Takahashi, P. Camacho, J. D. Lechleiter and B. Herman, Physiol. Rev., 1999, 79, 1089–1125 CAS.
- K. R. Gee, K. A. Brown, W. N. Chen, J. Bishop-Stewart, D. Gray and I. Johnson, Cell Calcium, 2000, 27, 97–106 CrossRef CAS.
- M. R. Maurya and S. Subramaniam, Biophys. J., 2007, 93, 709–728 CrossRef CAS.
- R. H. Garrett and C. M. Grisham, in Biochemistry, Saunders College Publishing, New York, 2nd edn, 1999, pp. 540–561 Search PubMed.
- P. Failli, A. Fazzini, F. Franconi, I. Stendardi and A. Giotti, J. Mol. Cell. Cardiol., 1992, 24, 1253–1265 CrossRef CAS.
- G. Hajnoczky, L. D. Robbgaspers, M. B. Seitz and A. P. Thomas, Cell, 1995, 82, 415–424 CrossRef CAS.
- M. J. Mason and S. Grinstein, Biochem. J., 1993, 296, 33–39 CAS.
- A. A. Gutierrez, J. M. Arias, L. Garcia, J. Mas-Oliva and A. Guerrero-Hernandez, J. Physiol. (London), 1999, 517, 95–107 Search PubMed.
- H. J. Park, C. M. Makepeace, J. C. Lyons and C. W. Song, Eur. J. Cancer, 1996, 32A, 540–546 CrossRef CAS.
- T. R. Hesketh, G. A. Smith, J. P. Moore, M. V. Taylor and J. C. Metcalfe, J. Biol. Chem., 1983, 258, 4876–4882 CAS.
- J. J. Abramson, E. Buck, G. Salama, J. E. Casida and I. N. Pessah, J. Biol. Chem., 1988, 263, 18750–18758 CAS.
- J. P. Vial, F. Belloc, P. Dumain, S. Besnard, F. Lacombe, M. R. Boisseau, J. Reiffers and P. Bernard, Leuk. Res., 1997, 21, 163–172 CrossRef CAS.
- F. Durrieu, F. Belloc, L. Lacoste, P. Dumain, J. Chabrol, J. Dachary-Prigent, H. Morjani, M. R. Boisseau, J. Reiffers, P. Bernard and F. Lacombe, Exp. Cell Res., 1998, 240, 165–175 CrossRef CAS.
- R. Pompei, O. Flore, M. A. Marccialis, A. Pani and B. Loddo, Nature, 1979, 281, 689–690 CrossRef CAS.
- J. Cinatl, B. Morgenstern, G. Bauer, P. Chandra, H. Rabenau and H. W. Doerr, Lancet, 2003, 362, 293–294 CrossRef CAS.
- H. Tanaka, T. Hasegawa, M. Matsushita, H. Miichi and S. Hayashi, Ophthalmic Res., 1987, 19, 213–220 CrossRef CAS.
- J. Ma, N. Y. Fu, D. B. Pang, W. Y. Wu and A. L. Xu, Planta Med., 2001, 67, 754–757 CrossRef CAS.
- Y. L. Hsu, P. L. Kuo and C. C. Lin, Life Sci., 2005, 77, 279–292 CrossRef CAS.
- M. Maggiolini, G. Statti, A. Vivacqua, S. Gabriele, V. Rago, M. Loizzo, F. Menichini and S. Amdo, J. Steroid Biochem. Mol. Biol., 2002, 82, 315–322 CrossRef CAS.
- S. Yamazaki, T. Morita, H. Endo, T. Hamamoto, M. Baba, Y. Joichi, S. Kaneko, Y. Okada, T. Okuyama, H. Nishino and A. Tokue, Cancer Lett., 2002, 183, 23–30 CrossRef CAS.
- K. Iwashita, M. Kobori, K. Yamaki and T. Tsushida, Biosci. Biotechnol. Biochem., 2000, 64, 1813–1820 CrossRef CAS.
- R. D. Olson and P. S. Mushlin, FASEB J., 1990, 4, 3076–3086 CAS.
- R. D. Olson, X. Li, P. Palade, S. E. Shadle, P. S. Mushlin, H. A. Gambliel, M. Fill, R. J. Boucek Jr and B. J. Cusack, Toxicol. Appl. Pharmacol., 2000, 169, 168–176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: The procedure of cellular fluorescence measurement with background correction. Fig. S2: Measurement of fluorescence before IQ introduction and after washing away the IQ solution. See DOI: 10.1039/b812987h |
| This journal is © The Royal Society of Chemistry 2009 |