Succinic acid from renewable resources as a C4 building-block chemical—a review of the catalytic possibilities in aqueous media
Clara Delhomme*a, Dirk Weuster-Botza and Fritz E. Kühnb
aInstitute of Biochemical Engineering, Technische Universität München, Boltzmannstr. 15, D-85748, Garching b. München, Germany. E-mail: C.Delhomme@lrz.tum.de
bChair of Inorganic Chemistry and Molecular Catalysis, Faculty of Chemistry, Technische Universität München, Lichtenbergstr. 4, D-85747, Garching b. München, Germany. E-mail: fritz.kuehn@ch.tum.de.
First published on 4th November 2008
Abstract
Aqueous hydrogenation of bio-based succinic acid has been reported for the production of value added chemicals, e.g. 1,4-butanediol, tetrahydrofuran, γ-butyrolactone, 2-pyrrolidone or N-methyl-2-pyrrolidone. A variety of heterogeneous metallic catalysts, active under quite severe conditions have previously been studied, whereas research into organometallic complexes is thus far limited to solvent reactions or to aqueous reactions producing succinic acid.
1. Introduction
Prior to the end of the 18th century, the economy was largely based on agriculture; however, the industrial revolution changed this, establishing fossil fuels (petroleum, coal and natural gas) as the main resources. In continuation of this development, the chemical industry nowadays consumes more than 1 billion barrels of oil per year.1 Considering this consumption and the limited nature of the fossil fuels, it is not astonishing that the economy faces several problems—a continuously rising price of oil, political and economic issues related to the unequal distribution of the remaining oil stocks and increasingly severe environmental impacts due to the use of these resources and the by-products generated during their consumption.These problems have motivated the industry to find alternatives to fossil fuels. Considerable effort is being invested in biotechnology and “green chemistry” (among others) to develop a chemical industry based on renewable resources as at least (a partial) substitute for the dwindling fossil fuels. To that end, fermentation of biomass for the production of renewable building-block chemicals is currently being developed. The so-called “green chemistry” deals to a considerable extent with the optimization of chemical processes, reducing the negative environmental impact of the chemical industry, e.g. by lowering energy consumption, with more efficient catalysts and by enabling use of solvents with a lower environmental impact. Furthermore, bio-based feedstock (produced by fermentation) must be transformed into valuable chemicals. Since the output of fermentations consists usually of dilute aqueous solutions of organic products, new water-stable catalysts need to be designed for such applications.
Succinic acid is among the new bio-derived building-block chemicals that could replace the current maleic anhydride C4 platform. The main interest in succinic acid lies in its derivatives,1 since it can be transformed into a lot of interesting products: 1,4-butanediol (BDO), γ-butyrolactone (GBL), tetrahydrofuran (THF), N-methyl-2-pyrrolidone (NMP), 2-pyrrolidone (2-Pyrr), succinimide, succinic esters, maleic acid (M.A.)/maleic anhydride (M.Anh.) and several others (cf.Fig. 1).
 | ||
| Fig. 1 Succinic acid derivatives.2 | ||
The optimization of the fermentation process for succinic acid production is currently underway. To that end, various natural succinate producing strains of bacteria (e.g. Anaerobiospirillum succiniciproducens, Actinobacillus succinogenes and Mannheimia succiniciproducens) and engineered Escherichia coli strains have been investigated.3
However, among the bottleneck problems of the industrial production of succinic acid from renewable resources remain the costs related to purification.4 Because the fermentation must be done at neutral or low acidic pH, the fermentation broth must be acidified upon conclusion of the reaction to create the free acid. Furthermore, other organic acids produced as side products during the fermentation can unfavorably affect the recovery of succinic acid.4 Therefore, the direct downstream catalysis of succinic acid in the filtered aqueous fermentation broth allows the production of valuable derivatives without the need to isolate pure succinic acid.
In the past century, the utilization of water as a reaction medium for organic synthesis was quite limited. The low water-solubility of most organics and the water-sensitivity of some reagents or intermediates5 were serious disadvantages. However, the high waste production due to organic solvents, their volatility and the high energy consumption for gas phase reactions are two major environmental drawbacks for the chemical industry that must adapt to new environmental restrictions and to growing consumer awareness. Furthermore, the handling of flammable, explosive and carcinogenic organic solvents is a major safety problem. Water—an abundant and non-toxic solvent—could be a greener alternative as well as having other advantages: it favors ionic reactions, solvates cations and anions, is an ideal solvent for radical reactions, can facilitate the control of exothermic reaction due to its high phase change enthalpies and heat capacity6,7 and, finally, water has been reported to suppress the coke and tar formation for the vapor hydrogenation of maleic acid with metal containing catalysts.8
Furthermore, in opposition to liphophilic oil, coal or natural gas derived compounds, the chemicals produced from renewable resources are often hydrophilic. Hence, a new class of catalysts must be developed. These new catalysts must fulfill the following requirements: (1) water-stability, (2) high selectivity towards substrate and product, (3) no inhibition due to or degradation by fermentation side products, (4) immobilizability to enable easy recovery, (5) high activity at low pressure and temperature (ideally atmospheric pressure and room temperature so as to minimize energy requirements). Furthermore, the chemicals produced from the fossil fuels are mostly in a low oxidation state and must therefore be oxidized, whereas the compounds produced from renewable resources are often in a high oxidation state and must therefore be reduced. Current catalysts must therefore be substantially modified so that they are able to catalyze reductions of bio-based chemicals in aqueous media.
In the case of succinic acid, most derivatives presented in Fig. 1 are produced through hydrogenation or reductive amination (in the presence of hydrogen), hence, this review will focus on these types of reactions for production of succinic acid derivatives in presence of water. Additionally, some space is dedicated to the synthesis of succinic acid by the hydrogenation of maleic and fumaric acid with the help of aqueous organometallic catalysts. Although at first glance, this approach seems not to be relevant to succinic acid derived from renewable feedstock, it might nevertheless be helpful in the design of water-stable homogeneous or immobilized catalysts that are able to hydrogenate the renewable succinic acid in an aqueous environment. A summary of achievements in the area of organometallics for solvent hydrogenation of succinic anhydride has been added as further information and for comparative purposes.
2. Aqueous phase catalysis with metal containing heterogeneous catalysts
2.1. γ-Butyrolactone, tetrahydrofuran, 1,4-butanediol
Maleic anhydride is a petroleum derived C4 starting material used for the production of many chemicals. One of the possible downstream treatments of this product is its hydrogenation for the manufacture of valuable chemicals such as GBL, BDO and THF. GBL is, for example, used as the starting material for the synthesis of NMP and other pyrrolidones, in particular N-vinylpyrrrolidone and its polymer which is widely used in medicine. It can also be utilized as a solvent. Its world wide production represents 250,000 t, approximately 20% of which originates from USA based production sites. BDO is an intermediate mainly for the synthesis of THF and polybutylene terephtalate. It is sold at 0.30–0.41 $/kg into a large global market of 1.3 million t/a (with the U.S. market being ca. 25% of this). Finally, THF is used as a monomer for the production of PTMEG, as solvent a in PVC cement, in pharmaceuticals and coatings, or as a reaction solvent. It is a high-value chemical (0.70–0.77 $/kg) and is produced worldwide at a level of 439,000 t/a (with 25% of that figure coming out of the USA alone).†There has been much industrial research on the synthesis of these compounds and different research groups have tried to establish a hydrogenation approach starting from maleic anhydride or acid.9–11 In Fig. 2, one possible reaction pathway starting from maleic acid—the hydrolyzed form of maleic anhydride—is represented.11 According to all published work, the first step of the hydrogenation is the hydrogen addition on the maleic anhydride/acid double bond for the formation of succinic anhydride/acid. Publications and patents discussing the hydrogenation of maleic acid in the aqueous phase could hence be easily adapted to the aqueous hydrogenation of succinic acid.
 | ||
| Fig. 2 Maleic acid hydrogenation pathway.11 | ||
Some reviews summarize data on the hydrogenation of maleic or succinic anhydride/acid.12,13 As the large majority of the published literature focuses on metal supported catalysts for vapor or solvent hydrogenation, not much information on aqueous phase reaction can be found. In the present review, attention will hence be brought to hydrogenation in aqueous media. Since water is a relatively reactive medium, ligand exchange or redox reactions would in many cases lead to rapid catalyst deactivation.14 Accordingly, water stable derivatives of common organometallic or inorganic supported catalysts have been developed. An overview of the achievements in the domain of metal containing heterogeneous catalysts for the aqueous hydrogenation of maleic/succinic acid is presented in Table 1 and discussed in the following chapter.
| Company or Institute | Reaction | Optimal catalyst | T [°C] | P [MPa] | S [%] | Y [%] | Ref. | Year |
|---|---|---|---|---|---|---|---|---|
| a First set of conditions.b Second set of conditions. | ||||||||
| E.I. Du Pont de Nemours | M.Ac. → THF +BDO | 3%Pd–3%Re on C | 190–200 | 17 | 90 (THF) | — | 15–17 | 1985–1987 |
| 180 | 17 | 79 (BDO) | — | |||||
| S.Ac. → GBL + BDO | 1%Pd–4%Re on TiO2 | 200 | 3.5 | 90 (GBL) | 90 | 18 | 1988 | |
| 200 | 6.9 | 90 (BDO) | 89 | |||||
| S.Ac. → THF (+GBL) | 1%Ru–4%Re on C | 250 | 8.3 | 83 (THF) | 82 | 19,20 | 1995 | |
| M.Ac. → (GBL) → BDO | 7%Ru–5%Sn on ZrO2 | 225 | 14 | 96 (BDO) | 94 | 21 | 1999 | |
| M.Ac. → S.A. → BDO + THF | 1) 1% Pd on C | 1) 120 | 20.7 | — (BDO) | 82 | 22 | 1999 | |
| 2) 1%Ru–6%Re on C | 2) 175 | |||||||
| 1.5%Ru-3%Re-0.6%Sn on C | 250 | 13.8 | — (THF) | 67 | ||||
| M.Ac. → THF + GBL | 1%Ru–6%Re on C | 250 | 13.9 | 48–56 (GBL) | — | 23 | 2003 | |
| 270 | 13.9 | 72 (THF) | — | |||||
| M.Ac. → THF (+GBL) | 1%Ru–6%Re on C | 270 | 15 | 78 (THF) | — | 24 | 2003 | |
| M.Ac. → THF (+GBL) | 1%Pt–6%Re–0.8%Sn on C | 250 | 13.9 | 94 (THF) | — | 25 | 2003 | |
| M.Ac. → THF (+BDO) | 4%Ru–1.4%Sn–1.3%Mo on TiO2 | 250 | 13.9 | 89 (THF) | — | 26 | 2004 | |
| Standard Oil Company | M.Ac. → THF +BDO | 3%Pd–3%Re on C | 175 | 9 | 80 (THF) | — | 27 | 1995 |
| 3.3%Pd–3.2%Ag-6.6%Re on C | 175 | 9 | 74 (BDO) | — | ||||
| M.Ac. → BDO (+BuOH) | 3%Pd–3%Ag–6%Re on oxidized C | 140 | 17.2 | 93 (BDO) | — | 28 | 1997 | |
| M.Ac. → BDO (+THF + BuOH) | Pd–Ag–Re–Al on oxidized C | 152 | 17.2 | 91 (BDO) | — | 29 | 1999 | |
| M.Ac. → S.Ac. → BDO (+GBL + THF + BuOH) | Pd–Ag–Re on oxidized C | 1) 130 | 27.6 | 70 (BDO) | — | 30 | 2002 | |
| 2) 162 | ||||||||
| ISP Investments | M.Ac. → S.Ac. → BDO (+GBL + THF + BuOH) | 4%Pd–4%Ag–4%Re on C | — | — | — | — | 31 | 2006 |
| BP Inc. | M.Ac. → GBL | 3%Pd–6%Ag–3%Re | 230–260 | 5–8 | 98 (GBL) | — | 32,33 | 1991–1992 |
| M.Ac. → S.Ac. → BDO (+GBL + THF + BuOH) | 1) 0.5% Pd on Rutile TiO2 | 1) 110a or 100b | 17.2–27.6 | 94 (BDO)a | 91a | 34–36 | 2006 | |
| 2) 5%Re on Rutile TiO2 | 2) 165a or 177b | 89 (BDO)b | 90b | |||||
| Battelle Memorial Inst. | S.Ac. → GBL | 5%Pd–5%Zr on C | 225 | 17 | 92 (GBL) | 90 | 14,37 | 2003 |
With regard to the nature of the metal used, different interpretations can be found in the literature. For example, Tooley and Black advocated the use of ruthenium—a group VIII metal—for its effectiveness towards carboxylic functional group reduction and as well as its acid resistance.21 Campos and Strickland suggested replacing the expensive rhenium present in many catalysts by molybdenum—a lower cost and more available metal—particularly for bimetallic Ru-Re catalysts for the production of THF.26
The heterogeneous catalysts mentioned in the literature generally contain two or more metals. The reason for the presence of several metals is said to be the promoting and/or inhibition effect of the different metals and their synergetic interactions. In a complex set of reactions, such as the hydrogenation of succinic acid, metals can favor or inhibit different reactions and therefore increase the selectivity towards certain products.
The behavior of various metals for the hydrogenation of succinic acid in dioxane—a polar solvent—using a bimetallic ruthenium–cobalt catalyst was highlighted by Deshpande et al.11 It was possible to show that ruthenium promotes the reactions 1, 3, 4, 6 presented in Fig. 2, whereas the activity for hydrogenation steps 2 and 5 were attributed to cobalt.11
The addition of a third metal can have an additional beneficial impact on the reactivity of the heterogeneous catalyst. For example, Budge et al. claimed that the addition of silver to a palladium–rhenium catalyst was increasing the BDO selectivity.27 Certain patents21,22,25 also reported that the incorporation of small amounts of tin into bimetallic catalysts improved the selectivity towards BDO, reducing the formation of side products such as n-butanol. Campos and Sisley made the assumption that tin moderated the high catalytic activity of the other two metals, limiting the over-hydrogenation of the products of interest. The selectivity is therefore favored at the expense of the overall hydrogenation rate and hence higher catalyst loading is necessary to compensate for this loss of activity.25
The nature of the metals is not the only parameter influencing the performance of the process. The dispersion and the repartitioning of the metals on and in the catalyst support are also considered to be of great importance. The formation of unwanted microstructures over time can lead to catalyst aging and activity losses. Schwartz highlighted the need for catalysts with a very high degree of metal dispersion that would remain constant throughout the many repetitive runs. In bimetallic catalysts, the high degree of dispersion of the metals can indeed prevent the formation of unwanted microstructures. Schwartz thus proposed a co-deposition/co-reduction procedure for the production of highly dispersed metal catalysts.19,20 Later, Werpy et al. developed a new type of catalysts called “textured catalysts”. These catalysts were prepared on high surface area carbon supports that can maintain their integrity under aqueous, acidic or basic conditions. The use of texturing agents during the metal deposition procedure led to a better distribution of the active metals on the catalyst's surface, particularly in the large pores that are more accessible to substrate. Such distribution enhanced the activity and the selectivity. Furthermore, less metal was used to achieve equivalent catalytic activity. Metal was deposited preferentially in the more accessible areas and little or no metal was “lost” to smaller inaccessible pores.14
Another important parameter in the design of metallic heterogeneous catalysts is the support itself. Carbon has generally been used as supporting material for heterogeneous hydrogenation catalysts, largely due to its inert behavior with respect to the hydrogenation reaction itself, its high surface area and its low cost. However, the main disadvantage of such a support is the formation of carbon fines during the reaction. These fines can plug void spaces in the catalyst and thereby cause a tremendous decrease in the catalyst activity. They can possibly be minimized but never completely avoided.34 In some patents, investigations into the impact and the properties of the different supports were presented. For example, Perdersen et al. investigated on the impact of the support on the catalyst selectivity and activity. They noted that oxidized carbon supports could increase the selectivity and activity in comparison to normal carbon supports.28,29 A procedure for the production of such catalysts that consists of the treatment of activated carbon with nitric acid at elevated temperature was developed.28 However, this work did not focus on the catalyst aging and its stability in acidic media. To solve the previously mentioned problems, Tooley and Black suggested the use of refractory oxides as supports. TiO2 and ZrO2 were considered to more favourable candidates with respect to SiO2 and Al2O3, which were too soluble in aqueous acidic media.21 However, Bhattacharyya and Manila cautioned that TiO2 in its normal form (anatase) was not resistant enough for such a process, due to its low crush strength, its tendency to disintegrate and to produce particles that can clog the catalyst pores thereby reducing the reaction efficiency. They proposed instead the use of the rutile form of TiO2 to overcome the disadvantages of flaking, large pressure increases, and low crush strength found in the other catalyst supports. Moreover, they claimed that rutile as a support presented the advantage of being more stable at highly acidic conditions in contrast to the anatase form of TiO2. Finally, they asserted that, using this particular support, the amount of some additionally added metals could be reduced or totally eliminated (e.g. silver).34
In conclusion, many different metals, supports and metal deposition procedures have been examined for the production of water-stable heterogeneous hydrogenation catalysts. However, no highly systematic testing procedure has been reported to date. It is therefore difficult to infer the impact of the different metals and supports.
Aside from the catalyst itself, the reaction parameters such as temperature, pressure, flow rates, etc. usually have a significant influence on the product selectivity and yield.
Various patents and articles describe the investigations into the influence of reaction parameters on the product distribution. In particular, Takar et al. performed a simulation of a bubble–column–slurry reactor for the conversion of maleic acid into THF.24 The results of this simulation gave useful information on the optimal reaction conditions. They showed for instance that maleic acid and succinic acid conversion increased with catalyst loading but decreased with the liquid velocity and that maleic acid and succinic acid displayed substrate inhibitory effects. It was also found that the selectivity towards the three useful products (GBL, THF and BDO) was much higher than the selectivity towards n-butanol and other unwanted end-products, except at high catalyst loadings and residence times, where the desirable products were further hydrogenated.24
A lot of patents focus on the production of THF as it is a high-value product that can be easily purified, due to its volatility. The studies on the effect of reactions parameters on THF selectivity revealed that selectivity towards it increases with catalyst loading, pressure and temperature15,16,23 and is favored at lower liquid velocities or long liquid residence times.24 Furthermore, continuous vapor removal of the product from the hydrogenation promotes the production of THF at the expense of BDO. Therefore, Campos and Sisley pointed out that slurry reactors or constantly stirred reactors are optimal for the production of THF.25 Campos and Strickland optimized the THF formation by maintaining appropriate acidity in the reactor to favor ring closure and cyclic ether production at the expense of diol formation.26
In contrast, BDO formation is favored at lower temperature and with low temperature liquid removal. Campos and Sisley recommended therefore the use of a fixed bed catalyst reactor for the production for BDO.25
Finally, the hydrogenation of maleic acid or succinic acid to GBL is generally difficult to accomplish because GBL can be further hydrogenated. The metals must be carefully selected to decrease the rate of the unwanted reactions.32
Apart from the impact of the reaction parameters on the product distribution and the yield, optimization has also to concentrate on the safety and economics of the process. One major problem with the hydrogenation process of maleic acid is indeed the highly corrosive impact of this product on equipment at temperatures exceeding 140 °C22,31,34 and especially in the temperature range required for the conversion of succinic acid into its valuable derivatives. Since succinic acid is much less corrosive at elevated temperature, a two-step process has been considered for the hydrogenation reaction starting from maleic acid. In the first step, maleic acid is converted to succinic acid at temperatures lower than 130 °C, because this reaction is much faster than the other hydrogenations. Then succinic acid is converted into THF, BDO or GBL at higher reaction temperatures. Accordingly, the corrosive impact of maleic acid is reduced. Since the hydrogenation of the double bond of maleic acid is exothermic, the heat of reaction can be used to preheat the feed to the second reactor,22 thereby making better use of energy and prolonging reactor life, leading to an improvement in the overall process economics (capital, operating and maintenance costs).31 Finally, the use of bio-derived (i.e. succinic acid), instead of oil derived building-block chemical (i.e. maleic acid) has the advantage of avoiding the hazardous handling of the latter.
In summary, optimizing the reaction conditions and using the optimal reactor type can have a crucial impact on the selectivity towards the desired products. However, considering the data presented in Table 1, the optimal reaction conditions reported are generally relatively severe (temperatures above 150 °C and pressures above 10 MPa). This has a major (negative) impact on the operating costs and the required equipment. Therefore, developing novel types of catalysts, which are active under milder conditions, is of great interest.
2.2. 2-pyrrolidone and N-methyl-2-pyrrolidone
1,4-Butanediol (BDO), γ-butyrolactone (GBL) and tetrahydrofuran (THF) are not the only products that can be produced by hydrogenation of succinic acid. Using amine, ammonium or ammonia and optionally alcohol, pyrrolidones and pyrrolidone derivatives can also be synthesized through reductive amination. Those products are C5 lactams, i.e. cyclic amides.2-Pyrrolidone (2-pyrr) is particularly useful, e.g. as an intermediate in the preparation of nylon-4 type polymers or in the synthesis of pharmaceuticals, medicines and agrochemicals.41N-methyl-2-pyrrolidone (NMP) is a solvent, used for instance for polyurethanes, polyacrylonitriles and heterocyclic polymers of high melting points.52 It is also used as an extracting solvent for acetylene and butadiene. NMP can also be used as a replacement for chlorinated solvents as its low volatility results in lower VOC emissions.53
Pyrrolidones were produced originally by BASF and ISP from acetylene, using the Reppe process derived in the 1930s by W. Reppe and IG Farben. Because of the risks associated with the handling of acetylene under pressure, new routes were developed in the following decades. In the late 1970 s, Mitsubishi Chemical described a process starting from butadiene with 1,4-butanediol as intermediate. In 1990, ARCO Chemical transformed propylene oxide into pyrrolidone through the formation of 1,4-butanediol. However, these two processes were using old techniques for the downstream production of pyrrolidones. More recently developed technologies are based on butane or maleic anhydride with GBL as an intermediate. Maleic anhydride is indeed converted to GBL by the solvent hydrogenation process mentioned in the previous paragraph. GBL is then converted to pyrrolidones by conventional ammonolysis.54 In recent decades, however, direct conversions of maleic anhydride/acid or succinic acid have also been reported. This reaction pathway in water will be the subject of paragraphs to come.
The direct aqueous reaction of maleic/succinic acid to pyrrolidones without producing GBL as intermediate is mainly described in patents. All these patents report different metallic heterogeneous catalysts using diverse reaction conditions. For better readability, all this information is presented in Table 2. As the information is mainly held in patents, no reliable kinetic data and only limited information concerning the reaction pathway can be found. Possible side products are often mentioned in the literature but only one patent tried to establish a reaction pathway for the synthesis of NMP with N-methyl-succinimide (NMS) as intermediate.51 A reaction scheme for the production of NMP from diammonium succinate with addition of methanol is presented in Fig. 3. Side products mentioned in the literature are also integrated into the scheme. If 2-pyrrolidone is the desired product, no methanol is added to the reactor and the right hand side of the reaction diagram does not occur. As the reaction pathway from succinic/maleic acid to NMP or 2-pyrrolidone is relatively complex, forcing the reaction into a particular pathway seems to be a matter of carefully choosing the catalyst metals, the reaction conditions and especially the reactant ratios.40,42
| Product | Company or Institute | Reaction | Other reactants/Solvent | Reactant ratios | Optimal catalyst | T [°C] | P [MPa] | S [%] | Y [%] | Ref. | Year |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a NMS yield.b NMP yield. | |||||||||||
| 2-Pyrr | FMC Corp. | M.Anh./M.Ac.→ 2-Pyrr | H2 + NH3 (liq) + water | Am/Ac = 3.1:1 | Raney Cobalt | 238–258 | 9–11 | — | 61 | 38 | 1963 |
| Anilin- & Soda-Fabrik | DAS (or DAM) → 2-Pyrr | H2 + water | Am/Ac = 2:1 | Co | 300 | 25 | — | 79 | 39 | 1965 | |
| Sun R & D and Co. | M.Anh. → 2-Pyrr | H2 + NH4OHaq | Am/Ac = 1.5:1 | 5% Ru on Al2O3 | 252 | 11.7 | — | 92 | 40 | 1975 | |
| Standard Oil Company | M.Anh./M.Ac.→ 2-Pyrr | H2 + NH3 (liq) + water | Am/Ac = 2:1 | RuFeNiOx | 250 | 6.9 | 95 | 77 | 41 | 1981 | |
| Phillips Petroleum Company | S.Anh → 2-Pyrr | H2 + NH3 | Am/Ac = 4.4:1 | 5% Pd on Al2O3 | 270 | 17.2 | — | 77 | 42 | 1990 | |
| NMP | Huels Aktiengesellschaft | S.Anh./NMS → NMP | methylamine + H2 + water | Am/Ac = 1.2:1 | Ni, Ca, Mg, Fr, Cr, Si, Al oxides | 200 | 30 | — | 88 | 43 | 1988 |
| BASF | DAM → NMP | methanol + H2 + water | Am/Ac = 2:1 | Co,Cu,Mn,Mo,P,Na, catalyst | 230 | 20 | — | 89 | 44 | 1992 | |
| Al/Ac = 4.5:1 | |||||||||||
| BASF | M.Anh./M.Ac. → NMP | methylamine + H2 + water | Am/Ac = 1.5:1 | Co,Cu,Mn,Mo,P,Na, catalyst | 250 | 20 | — | 91 | 45,46 | 1992, 1995 | |
| Battelle Memorial Inst. | DAS → NMP | methanol + water + H2 | Am/Ac = 2:1 | 2.5%Rh–2.5%Re on C | 265 | 13 | 68b | 66 | 47–50 | 2003 | |
| Al/Ac = 2:1 | |||||||||||
| Battelle Memorial Inst. | DAS → NMS → NMP | 1) methanol + water - 2) H2 | Am/Ac = 2:1 | 1) — | 1) 300 | 1) — | 1) 83a | 1) 83 | 51 | 2007 | |
| 2) 2.5%Rh–2.5%Re on C | 2) 200 | 2) 13 | 2) 92b | 2) 88 | |||||||
| Al/Ac = 2:1 | |||||||||||
| 1) methanol + water - 2) H2 | Am/Ac = 1.2:1 | 1) — | 1) 280 | 1) — | 1) 75a | 1) 70 | |||||
| Al/Ac = 1.5:1 | 2) Rh cat. | 2) 220 | 2) 10.3 | 2) 95b | 2) 91 |
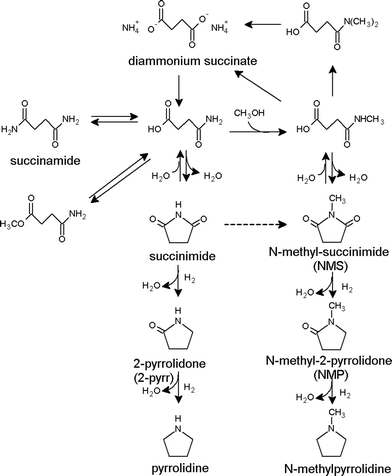 | ||
| Fig. 3 Pyrrolidone synthesis pathway from succinic acid.51 | ||
For the production of 2-pyrrolidone, Liao stated that suitable metals were cobalt, nickel, ruthenium and palladium.38 He further claimed that Raney cobalt and nickel were the optimal catalysts for the formation of 2-pyrrolidone. However, two years later, Walldorf and von Kutepow asserted that the use of those catalysts was leading to the side formation of pyrrolidines and proposed a catalyst with cobalt, nickel, copper, iron and (optionally) manganese, silver and chromium.39 As an alternative to cobalt catalysts, Hollstein and Butte highlighted the advantages of supported ruthenium catalysts, particularly because of their insensibility to catalyst poisons.40 Pesa and Graham also studied ruthenium catalysts but with the addition of rhenium, iron and nickel.41 Finally, palladium catalysts on alumina were also investigated for the production of 2-pyrrolidone by the Phillip Petroleum Company.42
For the production of NMP, mainly cobalt and rhenium catalysts were developed. The cobalt catalyst patented by BASF was doped by several other metals (copper, manganese, molybdenum, phosphate and sodium) to increase the NMP yield,44,45 whereas the Battelle Memorial Institute focused on simpler rhodium and rhenium catalysts. However, for these, lower yields were achieved when using a one-step process. Nickel catalysts with alkaline earth metal oxides in combination with iron and chromium oxides have also been reported earlier by zur Hausen and Otte for the reductive amination of succinic anhydride or the hydrogenation of NMS.43
In summary, various cobalt, ruthenium, palladium, rhenium or nickel catalysts have been studied for the synthesis of pyrrolidones. However, only very limited information can be found in the literature regarding the impact of the various metals on the activity and selectivity of the heterogeneous reductive amination of succinic acid.
The formation of pyrrolidones from succinic/maleic acid is conducted in the presence of ammonia (in the gaseous or liquid phase), ammonium ions (from NH4OH or diammonium salts of the acid)39 or primary amine in the case of NMP and other pyrrolidones production.43,45 If NMP is the desired product, ammonia/ammonium with ethanol is an alternative to primary amine. The reaction appears to be highly sensitive to the reactant ratios. It has been described in several patents that an excess both of alcohol and the nitrogen source is desirable.38,41,47 The optimal ammonia/ammonium/amine to acid ratio seems to lie in a range from 1:1 to 3:144 but more specifically between 1:1 and 1.7:1.43–47 The optimal methanol to acid ratio is approximately in the same range and according to Werpy et al. in the area of 1.5:1 to 2:1.47 An excess of alcohol is necessary for the production of NMP because the 2-pyrrolidone production is a competing pathway that does not require alcohol.47 In summary, choice of an appropriate reactant ratio is critical to achieve high yields, as a rapid decrease in yield is generally observed when deviating from the optimal ratios.40
For the pressure and temperature control, the data gathered in Table 2 show first of all that, like for the production of GBL, THF or BDO, severe reaction conditions (temperature above 200 °C, pressure above 10 MPa) are almost always necessary. In addition to this, Hollstein and Butte mentioned that the temperature control is an important parameter, particularly for the production of 2-pyrrolidone.40 Indeed, they found that at low temperature (T < 250 °C), the reaction rate was too slow for industrial application, whereas at higher temperature (T > 275 °C), the reaction proceeds to give pyrrolidines and not the desired pyrrolidones.40 In contrast, Matson observed that as the temperature increased (above 290 °C) the 2-pyrrolidone yield improved.42 However, it is difficult to compare these two statements, as Hollstein and Butte used a ruthenium catalyst and Matson a palladium one and metals might have quite different effects on the reaction rates.
The current industrial process consists of the transformation of maleic acid/anhydride to GBL followed by its conversion into pyrrolidones. Werpy et al. proposed an alternative two-step process for the production of NMP with N-methyl-succinimide (NMS) as an intermediate, increasing the yield and selectivity of NMP.47 They indicated that the yield of pyrrolidones diminishes at long reaction times in the presence of hydrogen and the hydrogenation catalyst, most probably due the over-reduction of the products. They therefore designed a two-step process where the first step was performed without hydrogen and catalyst and was followed by a quench or flash cooling to avoid ring opening. This first step was then completed by the purification or solvent extraction of NMS. Werpy et al. claimed that the second step–the hydrogenation of NMS into NMP using the designed catalyst—was advantageously done with little or no water. By doing so, the authors affirmed that catalyst poisoning could be avoided in the second step.47
The possible application of a two-step process for the production of 2-pyrrolidone with improved selectivity and energy use was also reported by Weyer et al.45 Unfortunately, no details of the reaction set-up or pathway are given.
In summary, the pyrrolidone synthesis has been less intensively studied than the hydrogenation of succinic acid to yield THF, BDO or GBL. The first attempts in aqueous phase were reported much earlier. However, the current process utilizing GBL as intermediate seems to remain the simplest pathway so far. One major problem of the direct pathway from succinic acid is the formation of many side products. A more selective process clearly needs to be developed. Accordingly, other types of catalysts have to be examined. Homogeneous organometallic catalysts are generally regarded as being more selective than classical heterogeneous catalysts. Furthermore, recent research has focused on developing water stable organometallic complexes. The use of such catalysts for the hydrogenation of succinic acid will be described in the next section.
3. Homogeneous aqueous phase catalysis utilizing organometallic catalysts
As mentioned above, the hydrogenation or the reductive amination of succinic or maleic acid has been mainly performed using heterogeneous catalysts. However, those catalysts have the drawback of being active only under high pressure and at elevated temperatures. Furthermore, the selectivity is not always optimal. Additionally, the presence of acidic aqueous solutions can degrade the catalyst rather quickly and thereby reduce its lifetime significantly. Organometallic complexes, being able to operate generally under milder conditions and being reputed for good selectivity, are therefore promising. However, so far this research field has been largely limited to reactions in organic solvents. As a matter of fact, the only available information in the literature on organometallic catalyzed aqueous reactions relating to the C4 succinic or maleic platform concerns the hydrogenation of the double bonds of maleic and fumaric acid under formation of succinic acid. The hydrogenation of succinic acid itself has been studied solely in organic solvents. The information available from the literature is summarized in Tables 3 and 4. Given the similarities between the complexes reported in Tables 3 and 4, information on organometallic complexes for aqueous catalysis of maleic or fumaric acids and for solvent hydrogenation of succinic acid could be useful for the design of new organometallic catalysts for the aqueous hydrogenation of succinic acid, the reaction of interest. This is why the remainder of this review is dedicated to these two related subjects.| Catalyst | T [°C] | P [MPa] | TOF [mol H2/h mol metal] | Ref. | Year |
|---|---|---|---|---|---|
| [RhCl(TPPMS)3] | 60 | 0.08 | M.Ac: 53 | 55 | 1984 |
| F.Ac.: 1270 | |||||
| [RhCl(TPPMS)3] | 60 | 0.1 | — | 56 | 1993 |
| [RhCl(TPPMS)3] | 60 | 0.1 | M.Ac: ∼60–370 | 57 | 2001 |
| F.Ac: ∼300–1800 | |||||
| trans-[IrCl(CO)(TPPMS)2] | 60 | 0.1 | M.Ac: 11.3 | 58 | 2000 |
| F.Ac: 7.8 | |||||
| [RhCl(PTA)3] | 37 | ? | M.Ac: 69 | 59 | 1996 |
| F.Ac: 342 | |||||
| [RhI(CO)(MTPA+I)3·4H2O | 20 | 0.1 | M.Ac: 110 | 60 | 1998 |
| F.Ac: 210 | |||||
| [Ru4(η6-C6H6)4H6]Cl2 | 50 | 5.8 | F.Ac: 38 | 61 | 1994 |
| Product | Reaction | Reactants/Solvent | Catalyst | T [°C] | P [MPa] | X [%] | S [%] | Y [%] | TOFa | Ref. | Year |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a TOF (turn over frequency): in moles of H2 or moles of product per hour and per mole of metal.b RT: room temperature. | |||||||||||
| Succinic anhydride | M.Anh. → S.Anh. | H2 + DME | [RhCl(PPh3)3] + PPh3 | 100 | 2 | 86 | — | — | 190.6 | 62 | 1999 |
| GBL | S.Anh. → GBL | H2 + toluene | [RuCl2(PPh3)3] | 100 | 1 | 100 | 50–58 | 50 | 2.4 | 63 | 1975 |
| S.Ac. (or Anh.) → GBL | H2 + dioxane | H4Ru4(CO)8(PBu3)4 | 180 | 13 | 100 | 100 | 100 | 1.7 | 64,65 | 1980, 2007 | |
| S.Anh. (or Ac.) → GBL | H2 + TGM | [Ru(acac)3], P(octyl)3 and p-TsOH | 200 | 1–3 | 97 | 98 | 95 | 257.0 | 66–68 | 2000–2002 | |
| Asym GBL | Unsym S.Ac. → Unsym GBL | H2 + toluene | H2-[RuCl2(PPh3)3] (or LiAlH4 or Na-EtOH) | 100 | 2.1 | — | — | 70–75 | — | 69 | 1976 |
| Unsym S.Ac. → Unsym GBL | H2 + triethylamine | [RhCl2(PPh3)3] or Ru2Cl4(DIOP)3 or RuCl2(TTP) | 120 | ∼1 | — | — | 52–62 | 1.3 | 70 | 1984 | |
| Asym Lactone | Unsym substrate → Unsym lactone | H2 + toluene | [RuCl2(PPh3)3] or [RuH2(PPh3)4] or [RhCl(PPh3)3] | 180 | 1 | — | — | 56–99 | — | 71 | 1982 |
| Alcohol | Carboxylic acid → Alcohol | Diphenylsilane + THF (solvent) | [RhCl(COD)]2/4PPh3 | RTb | — | — | — | 47–99 | — | 72 | 2008 |
| [RhCl(PPh3)3] | 50 | — | — | — | 62–95 | — |
3.1. Aqueous catalysis with organometallic complexes
As mentioned above, the only aqueous hydrogenation involving succinic acid catalyzed by water-soluble organometallic complexes published so far is the hydrogenation of maleic or fumaric acid under formation of succinic acid. Two research groups—one at the Institute of Physical Chemistry of the University of Debrecen and the other at the Research Group of Homogeneous Catalysis of the Hungarian Academy of Sciences but both directed by F. Joo—were particularly active in this field and indeed, in the generally field of water-soluble organometallic complexes.In this discipline, which was fairly new, novel water-soluble ligands had to be designed. The sulfonated versions of the triphenylphosphine (TPPMS or TPPTS) (cf.Fig. 4) are widely used ligands in this field. Joo et al. proposed a water-soluble analogue of Wilkinson's catalyst—[RhCl(TPPMS)3]—for the hydrogenation of maleic and fumaric acids under formation of succinic acid.55 This complex had been previously reported as catalyzing the hydrogenation of olefinic substrates under neutral or slightly acidic conditions. Later two other articles were published on this particular catalyst. These featured better characterization of the hydrogenation reaction and the behavior of this complex in aqueous solution.56,57
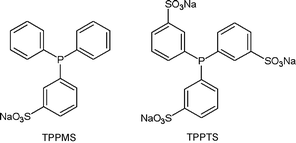 | ||
| Fig. 4 TPPMS (mono-sulfonated triphenylphosphine) and TPPTS (tri-sulfonated triphenylphosphine), two water-soluble analogues of the widely used triphenylphosphine (PPh3) ligand. | ||
Utilizing sulfonated phosphine ligands, Joo's group also designed another catalyst for this reaction: the water soluble analogue of Vaska's complex—trans-IrCl(CO)(TPPMS).58 Finally, as an alternative to [RhCl(TPPMS)3], they suggested the use of [RhCl(PTA)3], a complex with another water-soluble type of ligand.59
Other research groups came up with more complex propositions: Meister et al. synthesized [Ru4(η6-C6H6)4H6]Cl2 for the hydrogenation of fumaric acid61 and Pruchnik et al. (1998) designed [RhI(CO)(MTPA+I−)3]·4H2O for the hydrogenation of fumaric and maleic acid.60
As already mentioned, the first article describing hydrogenation of maleic or fumaric acid under formation of succinic acid was published by Joo et al. in 1984.55 The catalyst synthesized was derived from the non-water-soluble Wilkinson's catalyst [RhCl(PPh3)] by modifying the polarity of the ligands through sulfonation. This technique is widely used in the aqueous organometallic catalysis. TPPTS ligands are used, for instance, in the two phase Ruhrchemie-Rhône Poulenc propene hydroformylation process, starting from 1999.57
Studying the [RhCl(TPPMS)3] complex, Joo et al. concentrated at first on the influence of different parameters on the hydrogenation rate, namely the catalyst concentration, the partial pressure of hydrogen and the substrate concentration.55 They concluded that the initial hydrogenation rate could be characterized by the following equation:
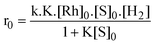
It was also observed, that at sufficiently high substrate concentrations, the influence of the ligand concentration was relatively small (no change in rate for maleic acid at 0.05 M). However, at lower substrate concentrations (0.02 M), the rate of maleic and fumaric acid dropped by as much as 75 and 78% respectively, for a [TPPMS]/[RhCl(TPPMS)3] ratio ranging from 0 to 17.6.
Efforts were then made to characterize the reaction mechanism in water so as to see whether it depended on the solvent. The role of water in organometallic catalysis has been widely discussed and it has been concluded that, in many cases, it influences the reaction significantly and that it is not merely a spectator. Water can act, for example, as a hydride donor, as a base or an acid and easily solvates ions. The impact of water was highlighted by Kovacs et al.,73 when they showed that [RhCl(TPPMS)3] or [RhCl(PTA)3] catalyzed the H–D exchange reaction, H2 + D2O ⇄ HD + HOD, in a pH range of 2–8, and 6–8 respectively. The deuteriation of olefin hydrogenation products in heavy water had also been previously reported using the catalysts mentioned above.
Further studies of the behavior of the catalysts noted proton loss while dissolving [RhCl(TPPMS)3] in water or while bubbling hydrogen through an aqueous solution of the complex. These observations were first explained by the following equilibria:56
| [RhCl(TPPMS)3] + H2O ⇄ [Rh(OH)(TPPMS)3] + H+ + Cl− |
| [Rh(OH)(TPPMS)3] + H2⇄ [RhH(TPPMS)3] + H2O |
| [RhCl(TPPMS)3] + H2⇄ [RhH(TPPMS)3] + Cl− + H+ |
After a detailed examination with 1H and 31P NMR spectroscopy, the following set of equations was suggested:57
| [RhCl(TPPMS)3] + H2O ⇄ [Rh(OH)(TPPMS)3] + H+ + Cl− |
| [RhCl(TPPMS)3] + H2⇄ [RhClH2(TPPMS)3] |
| [RhClH2(TPPMS)3] + H2O ⇄ [RhH(H2O)(TPPMS)3] + H+ + Cl− |
| [Rh(OH)(TPPMS)3] + H2⇄ [RhH(H2O)(TPPMS)3] |
Reductive elimination of HCl from transition metal complexes is a well known process, achieved in organic solvents by the addition of bases. In water the equilibria are driven by the strong solvatation of ions.57
An NMR study of the complex at various pH levels gave the following information: in acidic media, the dihydridic cis-mer-[RhClH2(TPPMS)3] (23%) (1a) and cis-fac-[RhH2X(TPPMS)] (1b) (77%, X = H2O or Cl−) were observed, the ratio of 1a to 1b being reversed by addition of a ten-fold excess of NaCl. Under basic conditions [RhH(H2O)(TPPMS)3] monohydride (2) was observed. Phosphine oxides were also detected in strong basic solution, but never exceeding 10%.57 This work claimed to have given the first evidence for the formation of rhodium(I)-monohydride in aqueous solution containing only monodentate phosphine ligands.57 The acidity of the hydrido complexes was examined by determining a pseudo-pKa (pKa∼ 8.2), i.e. the pH at which complexes 1 and 2 concentrations are equal.
The hydrogenation of maleic and fumaric acids as a function of pH was also investigated. The reactivity of those two compounds has been shown to be directly linked to the ratio of dissociated/undissociated forms of the acids. It was found, for example, that the fumaric HA− species was more reactive than the H2A or A2− forms (TOF of 1800 h−1 for HA− and ∼200 h−1 for H2A or A2−). In contrast, the HA− form of maleic acid led to the lowest activity (TOF of 60 h−1 for HA− and ∼360 h−1 for H2A or A2−). The rate vs. pH curves of maleic and fumaric acids are in contrast to one another: while one goes through a minimum (maleic acid) at pH 3–5, the other reaches a maximum (fumaric acid) there. Therefore, at very low pH and at pH > 7, maleic acid is hydrogenated faster than fumaric acid, whereas from pH 1.5 to 7, fumaric acid is hydrogenated faster. Increasing the pH above 7 did not lead to a further change in reactivity. That is why the dihydride ⇄ monohydride equilibrium—with the pseudo-pKa being above 7 (value: 8.2)—seems not to influence the reactivity of those two acids. Moreover, in solvents—where acids exist in their non-dissociated forms—Wilkinson's complex showed a higher hydrogenation rate of maleic acid than that of its trans-isomer. This observation is in agreement with what has been reported in water for undissociated acids. Therefore, the aqueous phase reaction proceeds most likely in a very similar manner to that in organic solvents. Finally, the pH effects on the rates in aqueous solution can be ascribed to the changes of the ionisation state of the substrate due to the dissociation/protonation in water.57
Using the sulfonated phosphine ligands, Joo's group synthesized a water-soluble analogue of Vaska's complex.58 The reactivity of this trans-[IrCl(CO)(TPPMS)2] complex was studied, starting with its behavior in water. In this situation, upon dissolution of the complex in water followed by the bubbling of hydrogen through the obtained aqueous solution, a decreasing pH was observed. The following equations have been proposed for the description of this reaction:58
| [IrCl(CO)(TPPMS)2] + H2O ⇄ [Ir(OH)(CO)(TPPMS)2] + Cl− + H+ |
| [IrCl(CO)(TPPMS)2] + H2 + TPPMS ⇄ [HIr(CO)(TPPMS)3] + Cl− + H+ |
| [IrCl(CO)(TPPMS)2] + 2H2⇄ [H3Ir(CO)(TPPMS)2] + Cl− + H+ |
In the presence of excess ligand, the presence of the monohydride complex was deemed also to be likely. Furthermore, the pH dependence of the hydrogenation rate was found to be better explained by the presence of the monohydride species. In contrast to the observations made in the presence of the [RhCl(TPPMS)3] catalyst, it was found that maleic acid was hydrogenated faster than fumaric acid at a pH of about 2.4–2.9; however, no further study of the impact of pH on the reactivity was performed and no further discussions of possible reaction mechanisms were reported.
Since sulfonated phosphine had been reported to readily link with activated olefinic bonds, leading to formation of the corresponding phosphonium salt, other ligands were investigated by Joo's group to improve understanding of the hydrogenation of maleic and fumaric acid in the absence of this side reaction;59 thus, [RhCl(PTA)3] was synthesized. PTA ligands are amino phosphane (cf.Fig. 5) displaying the following characteristics: resistance to oxidation, small size, strong binding abilities and water solubility.74 In contrast to TPPMS, this basic ligand (pKa∼ 6) does not react with olefins.59
 | ||
| Fig. 5 PTA (1,3,5-triaza-7-phosphaadamantane) ligands and modified PTA ligands: MTPA+I− (1-methyl-1-azonia-3,5-diaza-7-phosphaadamantane iodide.60 | ||
While studying the behavior of the catalyst in water, Joo's group recorded the same pH changes while dissolving the complex in water and then bubbling hydrogen through the solution as had been observed with [RhCl(TPPMS)3]. This was explained by the reaction expressed by the following equation:59
| [RhCl(PTA)3] + H2⇄ [RhH(PTA)3] + Cl− + H+ |
It was assumed that the main reaction pathway involved a monohydride complex because a strong pH dependence of the reaction rate of crotonic acid and allyl alcohol (maximum at pH 4.7) was been observed. However, the hydrogenation of maleic and fumaric acids with respect to pH was not investigated in detail. It was furthermore stated that fumaric acid is hydrogenated faster than its cis-isomer. This has been found using the rhodium sulfonated triphenylphosphine ligands at pH 1.5–7. Moreover, the hydrogenation rates recorded with this catalyst were lower than those obtained with the complex having a sulfonated ligand. Finally, it was observed that excess PTA decreases the hydrogenation rate but does not inhibit the reaction completely, probably due to the formation of other, less active catalyst complexes.
Deuteration experiments performed with the catalyst mentioned above led to partial deuteriation of the products. However, the presence of undeuterated products indicated that a dihydridic mechanism is also at work.59
Modified PTA ligands—MTPA+I− (c.f.Fig. 5)—were also evaluated for the hydrogenation of fumaric and maleic acid by Pruchnik et al.60
The complex with the MTPA+I− ligands show very strong hydrophilic properties and therefore, Pruchnik et al. concluded that this would be an optimal catalyst for two phase reactions because a certain amount of the catalyst would not be lost in the organic phase. After characterizing the complex by NMR spectroscopy, its catalytic efficiency for the hydroformylation of 1-hexene, the hydrogenation of aldehyde and the one-phase hydrogenation of maleic and fumaric acids was studied. As for the catalyst with the unmodified PTA ligands, the hydrogenation of the trans-isomer was found to be faster than that of maleic acid. The maleic acid hydrogenation rate was observed to be in the same range as the rate found with the [RhCl(TPPMS)3] catalyst, whereas the reaction rate on fumaric acid was noted to be lower than the ones recorded with the catalysts having sulfonated ligands and the [RhCl(PTA)3] complex.60
Meister et al. studied an arene ruthenium catalyst for the hydrogenation of fumaric acid.61 They mentioned that those ligands are particularly interesting for their unique structures, properties and their inherent catalytic potential.61 In the last twenty years, a lot of arene ruthenium complexes have been synthesized and tested for many applications. Under pressure (5.8 MPa) and comparatively low temperature (55 °C), Meister's arene catalyst had shown the ability to hydrogenate the olefinic bond of fumaric acid by the cycle presented in Fig. 6.
![Catalysis cycle of the hydrogenation of fumaric acid olefinic bond by the [Ru4(η6-C6H6)4H6]Cl2.61](/image/article/2009/GC/b810684c/b810684c-f6.gif) | ||
| Fig. 6 Catalysis cycle of the hydrogenation of fumaric acid olefinic bond by the [Ru4(η6-C6H6)4H6]Cl2.61 | ||
However, even at this high pressure (5.8 MPa), the turnover frequency (TOF) remained relatively low in comparison to the experiments performed with the catalysts described before.
3.2. Solvent catalysis with organometallic complexes
In contrast to aqueous phase catalysis, the use of organometallic catalysts for hydrogenations in organic solvents has been examined in far greater detail and a great variety of complexes have thus far been synthesized. For the hydrogenation of succinic anhydride, and more generally dicarboxylic acid anhydrides, mainly ruthenium and rhodium complexes with phosphine ligands have been reported (cf.Table 4). The hydrogenation product is generally the corresponding lactone—i.e. GBL for succinic anhydride. Apart from this, some studies of unsymmetrical anhydrides enabled a better understanding of the reaction mechanism as well as assessment of the enantioselectivity of the reaction.In contrast to heterogeneous catalysts, the organometallic complexes do not seem to promote over-hydrogenation of GBL and therefore, their use could be quite interesting for the commercial production of this chemical. Furthermore, these organometallic catalysts are generally active at much lower pressures and temperatures.
The first article mentioning this reaction type was published in 1975 by Lyons.63 He studied several complexes with PPh3 ligands and different metal centers. Among [IrCl(CO)(PPh3)2], [RhCl(CO)(PPh)3], [Co2(CO)8] and [RuCl2(PPh3)], the ruthenium complex was the only one active for the production of lactone under the conditions investigated in this article (1 MPa, 100 °C, toluene as solvent).63 It was also found, as would be by other researchers later, that the water produced during the reaction had the unwanted effect of hydrolyzing succinic anhydride to the less active succinic acid.
Five years later, Bianchi et al. tested other ruthenium complexes in dioxane on different carboxylic acids—both aliphatic and aromatic ones and on dicarboxylic acids and their corresponding anhydrides.64 The hydrogenations of succinic acid and anhydride were investigated in the presence of the complex [H4Ru4(CO)8(PBu3)4]. The reaction rate (at 150 °C, 13 MPa) was disappointingly low but a yield and a selectivity of 100% was finally achieved within 48 h.64
More recently, Hara et al. from the Mitsubishi Chemical Corporation presented a more systematic development of an organometallic catalytic system for the hydrogenation of succinic anhydride to GBL.66 Hara et al. described how the ruthenium catalysts reported by Lyons63 and Bianchi et al.,64 despite their ability to produce GBL and no other hydrogenation products, presented some technological drawbacks such as low activities or unfavorable halogen ligands that might corrode the reactor. In the case of the catalyst examined by Lyons,63 Hara et al. also mentioned that the PPh3 ligands were not stable at high temperatures (above 180 °C). Accordingly, there were attempts to develop a catalyst system consisting of a ruthenium salt, alkyl phosphines and an acid promoter in a particular solvent, with the aim of reinforcing the interaction between substrate and catalyst.66
Among the three types of organometallic complexes—anionic, neutral and cationic—a cationic complex was expected to make the carbonyl group of the substrate more accessible to the Ru metal and hence increase the activity. There were attempts to synthesize [RuHX(PPh3)3] complexes, with X being more acidic than Cl. These complexes were formed through the reaction between the corresponding Brønsted acid of the anion and a Ru complex of the type [H2RuP4], P being the phosphine ligand. Although the complex could not be isolated in the pure form, the authors assumed that the catalyst had the following structure:
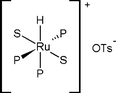
| (S = solvent, P = phosphine ligand)66 |
Through screening of various catalysts, it was found that weakly coordinating anions, like OTs and PF6, yielded higher activities. Additionally, Brønsted acids not only enhance the catalytic activity but also the selectivity toward GBL. A structural change in the Ru complexes was induced, leading to cationic complexes, with an increased stability. Among the acids studied, p-TsOH was found to be the best candidate because of its solubility, resistance to reduction and low price.
After determining the optimal Brønsted acid, the effect of the ligands on the catalytic properties was examined. Linear trialkyl phosphines, like PBu3 or P(octyl)3, gave the best recorded activity. The optimal ratio P(octyl)3/Ru was found to be between 5 and 10 and trialkyl phosphines not only stabilize the ruthenium metal, but also act as a strong base. Hence, without the presence of the Brønsted acid, formation of spiro dilactone was monitored leading to a GBL selectivity of 50%. With the addition of p-TsOH, free P(octyl)3 disappeared and was transformed into a phosphonium salt of P(octyl)3 and p-TsOH.
Among the solvents studied, TGM (tetraglyme), dodecyl benzene, and sulfolane allowed high GBL yield. As succinic anhydride is not very soluble in the last two solvents, TGM was regarded as the optimal solvent.
The technology utilizing this novel homogeneous catalytic system to produce GBL was commercialized in 1997 by the Mitsubishi Chemical Corporation. A plant with a GBL capacity of 10,000 t/a was constructed initially but underwent subsequent enlargement to 15,000 t/a in 2002. This process is the first one to be used for the full-scale commercial production of GBL which employs a homogeneous catalyst. It represents also the first use of Ru complexes for the production of a chemical at a rate of over 10,000 t/a.67 Hara et al. have also patented a similar homogeneous catalytic system for the production of BDO in 1991.68
A lot of effort has been put into betterment of the understanding of the reaction mechanism of the hydrogenation of dicarboxylic acid anhydrides to lactones. To that end, the hydrogenation of unsymmetrical anhydrides into unsymmetrical lactones has been studied. Two reaction pathways are possible (cf.Fig. 7). Morand and Kayser tried to achieve the regioselective reduction of the less hindered carbonyl group of the anhydride to yield the corresponding unsymmetrical lactones (4).69 They found that the catalyst proposed by Lyons63 was able to catalyze such a reaction in toluene at 100 °C and 2.1 MPa, and that this was in contrast to LiAlH4 or Na-EtOH, which preferentially catalyzed the reduction of the more hindered carbonyl group (5).
 | ||
| Fig. 7 Reaction mechanisms of the hydrogenation of asymmetrical anhydrides using ruthenium complexes, proposed by Ikariya et al.64 | ||
Ikariya et al. also investigated the catalytic reaction of unsymmetrical anhydrides with the four Ru catalysts RuCl2(PPh3)3, Ru3Cl4(DIOP)3, RuCl2(TTP) and Ru2Cl4(DPPB)3.70 Like Morand and Kayer,69 they obtained the hydrogenation product of the less hindered carbonyl group (4) as the major product.70
The first three catalysts gave maximum yields of 50–60% at 120 °C and 1 MPa. As shown in Fig. 7, they also assumed that the reaction occurred by an initial attack of ruthenium to the carbonyl group and successive C-O bond cleavage of one of the C–O bonds.70
Ikariya et al.75 and then Osakada et al.71 tried to explain more precisely the reaction mechanism of the transformation of succinic anhydride into GBL using [RuH2(PPh3)4].
Osakada et al. isolated the intermediate complexes formed by the initial catalyst complex with the anhydride by C–O bond cleavage.71 Then, upon contact with hydrogen at elevated temperatures (180 °C, 1.2 MPa), or with hydrogen chloride, or carbon monoxide at atmospheric pressure, these intermediate complexes release the lactones through reduction of formyl or acyl groups in the carboxylate ligands followed by intramolecular condensations. The two proposed mechanisms are shown in Fig. 8.
![(a) Reaction mechanism of the hydrogenation of succinic anhydride using [RuH2(PPh3)4] as catalyst; (b) structure of the reaction intermediate.71](/image/article/2009/GC/b810684c/b810684c-f8.gif) | ||
| Fig. 8 (a) Reaction mechanism of the hydrogenation of succinic anhydride using [RuH2(PPh3)4] as catalyst; (b) structure of the reaction intermediate.71 | ||
In the research field of water-stable organometallics, new catalysts have to be developed for the downstream transformations of bio-derived chemicals. For succinic acid, complexes for reactions in organic solvents have been reported. For those types of reactions, the reaction conditions mentioned in the literature are much less severe than those reported for heterogeneous metallic catalysts and could therefore lead to a reduction of the operating and equipment costs. However, systematic catalyst design and optimization of the reaction conditions must be performed in order to improve the catalyst activities that so far are relatively low.
4. Hydrogenation transfer
For hydrogenation reactions, a hydride donor is required. Hydrogen gas has the advantage of being a relatively cheap starting material that is available in large quantities. However, one major problem of using hydrogen gas is the requirement that the reaction be performed under pressure. If high pressure is necessary, special equipment is required and the operating and equipment costs increase. As mentioned before, Osakada et al.71 tried, for example, to reduce the carbonyl group of an anhydride using HCl or CO. Similarly, various other articles discuss the possible reduction of dicarboxylic acids or anhydrides using hydrogen donors other than hydrogen gas.Taking the work of Takahashi et al.76 as an example, a vapor phase reduction of dicarboxylic anhydride or esters on hydrous zirconium oxide granules with 2-propanol as hydrid donor and dioxane as solvent was outlined. This reaction was executed at atmospheric pressure in a temperature range of 280–330 °C. The reaction of succinic anhydride gives rise to THF and GBL with selectivities of 62% and 20%, respectively. It was noted in this context that the reducibility of secondary alcohols is higher than that of primary alcohols. Depending on the anhydride to alcohol ratio, the main product can be either the lactone (ratio 1:130) or THF (ratio 1:260).76 As for other reported heterogeneous reactions, the temperature seems to have a considerable impact on the selectivity: for temperatures between 280 and 300 °C, the main product is reported to be GBL whereas at temperature higher than 305 °C THF is produced as the major product.76
Another reduction method without hydrogen gas has been described by Ohta et al.72 In this work diphenylsilane was used as a hydride donor for the reduction of carboxylic acid to alcohol, involving the use of an organometallic catalyst: [RhCl(COD)]2/4PPh3 in THF. The reaction is performed at low temperatures between 25 °C and 50 °C, but has the drawback of requiring diphenylsilane, a high cost, non-water-soluble hydride donor.
5. Conclusion
The downstream transformation of bio-derived chemicals is a fast growing sector. The production of bio-based organic acids as a (partial) replacement for oil-derived building-block chemicals is of particular interest and certainly requires further attention.77 The currently limited market for succinic acid (16,000 t/a) is for instance predicted to increase to as much as 270,000 t/a within a couple of years.77 Efforts must therefore be focused on the development of water-stable and highly active catalysts (metal containing ones or organometallic complexes) for the direct aqueous treatment of this chemical with particular emphasis on environmental considerations.List of symbols
2-pyrr, 2-pyrrolidone; BDO, 1,4-butanediol; COD, cyclo-octadiene; DAM, diammonium maleate; DAS, diammonium succinate; DIOP, 2S,3S-O-isopropylidene-2,3-dihydroxy-1,4-bis(diphenylphosphino)butane; DME, ethylene glycoldimethylether; DPPB, 1,4-bis(diphenylphosphino)butane; F.Ac., fumaric acid; GBL, γ-butyrolactone; M.Ac., maleic acid; M.Anh., maleic anhydride; MTPA+I−, 1-methyl-1-azonia-3,5-diaza-7-phosphaadamantane iodide; NMP, N-methyl-2-pyrrolidone; NMS, N-methylsuccinimide; PTA, 1,3,5-triaza-7-phosphaadamantane; PTMEG, polytetramethylene ether glycol; S.Ac., succinic acid; S.Anh., succinic anhydride; TGM, tetraglyme; THF, tetrahydrofurane; TPPMS, sodium triphenylphosphine-3-monosulfonate; TPPTS, trisodium triphenylphosphine-3,3′,3″-trisulfonate; TTP, tetra-p-tolylporphyrinato dianion.Acknowledgements
This work was generously supported by the International Graduate School of Science and Engineering (IGSSE) at the Technische Universität München, particularly by an IGSSE Ph.D. grant for Clara Delhomme.Notes and references
- M. Paster, J. L. Pellegrino, and T. M. Carole, Prospects for a Bio-based Industry, U.S. Department, of Energy Report, 2003 Search PubMed.
- B. Kamm and M. Kamm, in Advances in Biochemical Engineering/Biotechnology, White Biotechnology, ed. R. Ulber and D. Sell, Springer, Heidelberg, 2007, vol. 105, ch. 4, pp. 175–203 Search PubMed.
- J. B. McKinlay, C. Vieille and J. G. Zeikus, Appl. Microbiol. Biotechnol., 2007, 76(4), 727 CrossRef CAS.
- Y. S. Huh, Y. S. Jun, Y. K. Hong, H. Song, S. Y. Lee and W. H. Hong, Proc. Biochem., 2006, 41, 1461 CrossRef CAS.
- A. Lubineau, J. Augé and M. C. Scherrmann, in Aqueous Phase Organometallic Catalysis, 2nd edition, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, 2004, pp. 27–43 Search PubMed.
- W. A. Herrmann and F. E. Kühn, in Aqueous Phase Organometallic Catalysis, 2nd edition, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, 2004, pp. 44–56 Search PubMed.
- H. C. Hails, Org. Proc. Res. Dev., 2007, 11, 114 Search PubMed.
- E. M. Miller, US Pat., 4 001 282, 1977.
- J. Kanetaka, T. Asano and S. Masamune, Ind. Eng. Chem., 1970, 62(4), 24 CrossRef CAS.
- Y.-L. Zhu, G.-W. Zhao, J. Chang, J. Yang, H.-Y. Zheng, H.-W. Xiang and Y.-W. Li, Catal. Lett., 2004, 96(3–4), 123 CrossRef CAS.
- R. M. Deshpande, V. V. Buwa, C. V. Rode, R. V. Chaudhari and P. L. Mills, Catal. Commun., 2002, 3, 269 CrossRef CAS.
- S. Varadarajan and D. J. Miller, Biotechnol. Prog., 1999, 15, 8451.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107(6), 2411 CrossRef CAS.
- T. Werpy, J. G. Frye, Jr., Y. Wang, and A. H. Zacher, US Pat., 6 670 300, 2002.
- M. A. Mabry, W. W. Prichard, and S. B. Ziemecki, US Pat., 4 550 185, 1985.
- M. A. Mabry, W. W. Prichard, and S. B. Ziemecki, US Pat., 4 609 636, 1986.
- B. W. Griffiths, and J. B. Michel, US Pat., 4 659 686, 1987.
- V. N. M. Rao, US Pat., 4 782 197, 1988.
- J.-A. T. Schwartz, Int Pat., WO 96/27436, 1996.
- J.-A. T. Schwartz, US Pat., 5 478 952, 1995.
- P. A. Tooley, and J. R. Black, US Pat., 5 985 789, 1999.
- R. E. Bockrath, D. Campos, J.-A. T. Schwartz, and R. T. Stimek, US Pat., 6 008 384, 1999.
- R. V. Chaudhari, C. V. Rode, R. M. Deshpande, R. Jaganathan, T. M. Leib and P. L. Mills, Chem. Eng. Sci., 2003, 58, 627 CrossRef CAS.
- N. N. Thakar, R. Jaganathan and R. V. Chaudhari, AIChE J., 2003, 42(12), 3199 CrossRef.
- D. Campos, and G. M. Sisler, US Pat., 6 670 490, 2003.
- D. Campos, and F. D. Strickland, Int Pat., WO 04/058397, 2004.
- J. R. Budge, T. G. Attig, and S. E. Pedersen, US Pat., 5 473 086, 1995.
- S. E. Pedersen, J. G. Jr,. Frye, T. G. Attig, and J. R. Budge, US Pat., 5 698 749, 1997.
- J. R. Budge, T. G. Attig, and R. A. Dubbert, US Pat., 5 969 164, 1999.
- J. R. Budge, T. G. Attig, and R. A. Dubbert, US Pat., 6 486 367, 2002.
- R. P. Hepfer, C. T. Miller, G. A. Norenberg, T. G. Attig, and J. R. Budge, US Pat., 6 989 455, 2006.
- M. Kitson, and P. S. Williams, US Pat., 4 985 572, 1991.
- M. Kitson, and P. S. Williams, US Pat., 5 149 680, 1992.
- A. Bhattacharyya, and M. D. Maynard, US Pat., 2006/0004212, 2006.
- A. Bhattacharyya, and M. D. Maynard, Int Pat., WO 06/007523, 2006.
- A. Bhattacharyya, and M. D. Maynard, Eur Pat., EP 1773746, 2007.
- T. Werpy, J. G. Frye, Jr., Y. Wang and A. H. Zacher, Int Pat., WO 02/102511, 2002.
- H. P. Liao, US Pat., 3 080 377, 1963.
- W. H. Walldorf and N. von Kutepow, US Pat., 3 198 808, 1965.
- E. J. Hollstein and W. A. Butte, US Pat., 3 812 148, 1975.
- F. A. Pesa and A. M. Graham, US Pat., 4 263 175, 1981.
- M. S. Matson, US Pat., 4 904 804, 1990.
- H. zur Hausen and W. Otte, US Pat., 4 780 547, 1988.
- U. Koehler, and H. Siegel, US Pat., 5 101 045, 1992.
- H.-J. Weyer, R. Fischer, and W. Harder, US Pat., 5 157 127, 1992.
- H.-J. Weyer, and R. Fischer, US Pat., 5 434 273, 1995.
- T. Werpy, J. G. Frye, Jr., Y. Wang, and A. H. Zacher, US Pat., 6 632 951, 2003.
- T. Werpy, J. G. Frye, Jr., Y. Wang and A. H. Zacher, US Pat., 6 670 483, 2003.
- T. Werpy, J. G. Frye, Jr., Y. Wang and A. H. Zacher, Int Pat., WO 02/102772 A1, 2002.
- T. Werpy, J. G. Frye, Jr., Y. Wang and A. H. Zacher, US Pat., 6 706 893, 2004.
- T. Werpy, J. G. Frye, Jr., J. F. White, J. E. Holladay, and A. H. Zacher, US Pat., 2007/0173643, 2007.
- G. Chichery, P. Benite, and P. Perras, US Pat., 3 448 118, 1969.
- Guide to Cleaner Technologies–Alternatives to Chlorinated Solvents for Cleaning and Degreasing, US Environmental Protection Agency Report, EPA/625/R-93/019, 1994.
- Routes to Pyrrolidones, Nexan's ChemSystems PERP Report 94/95 S1, 1997.
- F. Joo, L. Somsak and T. Beck, J. Mol. Catal., 1984, 24, 71 CAS.
- F. Joo, P. Csiba and A. Benyei, J. Chem. Soc. Chem. Commun., 1993, 1602 RSC.
- F. Joo, J. Kovacs, A. Benyei, L. Nadasdi and G. Laurenczy, Chem. Eur. J., 2001, 7(1), 193 CrossRef CAS.
- J. Kovacs, T. J. Decuir, J. H. Reibenspies, F. Joo and D. J. Darensbourg, Organometallics, 2000, 19, 3963 CrossRef CAS.
- F. Joo, L. Nadasdi, A. Benyei and D. J. Darensbourg, J. Organomet. Chem., 1993, 512, 45.
- F. P. Pruchnik, P. Smolenski, E. Galdecka and Z. Galdecki, New J. Chem., 1998, 22(12), 1395 RSC.
- G. Meister, G. Rheinwald, H. Stoeckli-Evans and G. Süss-Fink, J. Chem. Soc. Dalton Trans., 1994, 3215 RSC.
- L. Pu, L. Ye and Y. Yuanqi, J. Mol. Catal. A: Chem., 1999, 138, 129 CrossRef CAS.
- J. E. Lyons, J. Chem. Soc. Chem. Commun., 1975, 412 RSC.
- M. Bianchi, G. Menchi, F. Francalaci and F. Piacenti, J. Organomet. Chem., 1980, 188, 109 CrossRef CAS.
- P. Frediani, L. Rosi, M. Frediani, G. Bartolucci and M. Bambagiotti-Alberti, J. Agri,. Food Chem., 5 101 045, 1992 Search PubMed.
- Y. Hara, H. Kusaka, H. Inagaki, K. Takahashi and K. Wada, J. Catal., 2000, 194, 188 CrossRef CAS.
- Y. Hara and K. Takaha, Catal. Surv. Jpn., 2002, 6(1/2), 73 CrossRef CAS.
- Y. Hara, and H. Inagaki, US Pat., 5 077 442, 1991.
- P. Morand and M. Kayser, J. Chem. Soc. Chem. Commun., 1976, 314 RSC.
- T. Ikariya, K. Osakada, Y. Ishii, S. Osawa, M. Saburi and S. Yoshikawa, Bull. Chem. Soc. Jpn., 1984, 57, 897 CrossRef CAS.
- K. Osakada, T. Ikariya and S. Yoshikawa, J. Organomet. Chem., 1982, 231, 79 CrossRef CAS.
- T. Ohta, M. Kamiya, M. Nobutomo, K. Kusui and I. Furukawa, Bull. Chem. Soc. Jpn., 2005, 78, 1856 CrossRef CAS.
- J. Kovacs, L. Nadasdi, G. Laurenczy and F. Joo, Green Chem., 2003, 5, 213 RSC.
- B. J. Frost, C. M. Mebi and P. W. Gingrich, Eur. J. Inorg. Chem., 2006, 6, 1182 CrossRef.
- T. Ikariya, K. Osakada and S. Yoshikawa, Tetrahedron Lett., 1978, 39, 3749 CrossRef.
- K. Takahashi, M. Shibagaki and H. Matsushita, Bull. Chem. Soc. Jpn., 1992, 65, 262 CrossRef CAS.
- M. Sauer, D. Porro, D. Mattanovich and P. Branduardi, Trends Biotechnol., 2008, 26, 100 CrossRef CAS.
Footnote |
| † Sources: lit,1 Goliath 2005 (http://goliath.ecnext.com/), SRI Consulting 2006 (http://www.sriconsulting.com/), ICIS 2006 (http://www.icis.com/Home/Default.aspx). |
| This journal is © The Royal Society of Chemistry 2009 |