Introductory Lecture
Electrocatalysis: theory and experiment at the interface
Marc T. M.
Koper
Leiden Institute of Chemistry Leiden University, PO Box 9502, 2300 RA, Leiden, The Netherlands. E-mail: m.koper@chem.leidenuniv.nl
First published on 22nd August 2008
Introduction
Browsing through the history of Faraday Discussions typically serves as a good indicator of the development of concepts and challenges in physical chemistry. The first Royal Society Discussion Meeting held under the name “Faraday Discussions”, at the University of Manchester in 1947, was on the topic of “Electrode Processes”. Among the list of contributors were illustrious names such as Randles, Levich, Frumkin, Bockris, Butler, Eley, and Heyrovsky†. This was the time that electrochemical measuring techniques, such as electrochemical impedance spectroscopy, were developed, and electrochemists were still very much in the dark about the molecular nature and theoretical description of electrode reactions, in particular hydrogen evolution, which was a prominent discussion topic. One remarkable statement was that made by Eley,1 who seriously questioned whether it would ever be possible to measure on a clean platinum surface in an electrolyte solution.By 1968, at a Faraday Discussion on “Electrode Reactions of Organic Compounds” at the University of Newcastle-upon-Tyne, some remarkable leaps in development had taken place. Most prominently, there was by then a theory of electron transfer reactions, introduced at the meeting by Marcus. The contributions by Parsons and Conway were essentially concerned with the kinetic modeling of electrocatalytic reactions, whereas Hush used semi-empirical quantum-chemical calculations (!) to study bond breaking. Breiter had a contribution on methanol oxidation at platinum, and there was an interesting paper by Brummer and Cahill on the interaction between adsorbates, such as chemisorbed hydrogen and carbon monoxide. The relation with some of the relevant issues still raised at this meeting is remarkable. However, there was the nagging problem of specific adsorption, as put forward by Parsons2: “The greatest uncertainty in the adsorption behaviour arises from the lack of knowledge about the way the relative adsorption of different species depends on the nature of the metal.” Thirsk, in his concluding remarks, also mentioned the “resistance to theoretical attack” of adsorption as one of the main problems of electrochemical science.
Five years later, at a meeting at Oxford University called “Intermediates in Electrochemical Reactions”, one of the founding fathers of electrochemical surface science, Heinz Gerischer, was quite optimistic about the possibilities of new surface-sensitive techniques to probe intermediates in electrode reactions.3 Indeed, this was the meeting where the field witnessed the introduction of new non-electrochemical methods, such as reflectance spectroscopy (Kolb and MacIntyre) and online mass spectrometry (Bruckenstein). However, only few participants of that meeting would have foreseen the explosion of new techniques that was going to transform interfacial electrochemistry in the next 10–20 years. In fact, Randles, in his closing address, seemed rather reserved about the future of electrochemistry.4 He felt that, somehow, “chemistry has lost its glamour”, though “electrochemistry still has vitality and a potential for continuing usefulness”. However, “we should refrain from barren mathematising, we should use the techniques we have where they are most appropriate and think actively about possible new ones”.
The lesson to be learnt? Just browse through the next Faraday Discussion volume 94 on electrochemistry “The Liquid/Solid Interface at High Resolution”, held in Newcastle-upon-Tyne in 1992. There is no lack of glamour in the papers that were contributed to that meeting! Scanning tunneling microscopy and other scanning probe techniques had revolutionized the field of surface science and electrochemists such as Kolb, Itaya, and Bard, amongst many others, were actively studying the unprecedented potential of these new techniques. Also spectroscopic techniques such as infrared spectroscopy, Raman spectroscopy and non-linear methods, such as second harmonic generation, had become part of the modern electrochemist's toolbox. In the flood of STM papers, theory remained somewhat underexposed (apart from one isolated theory paper by Nagy, Heinzinger and Spohr on the modeling of water at platinum5).
The most recent Faraday Discussion meeting on electrochemistry was in 2002 in Berlin, entitled “The Dynamic Electrode Surface”. 2007 Nobel Laureate Gerhard Ertl, Heinz Gerischer's successor at the Fritz-Haber-Institute at Berlin, pointed out in his Introductory Lecture the importance of looking at surface reactions at different time- and length scales, and the different experimental and theoretical tools needed for their proper description.6 Theory and computational chemistry, in particular quantum-chemical calculations based on density functional theory (DFT) and (kinetic) Monte Carlo simulations of surface reactions, were beginning to play an important role in the interpretation of experimental results, as exemplified in the papers by Rikvold,7 Weaver,8 and myself.9 In the electrocatalysis talks, the role of surface diffusion, especially Pt-bonded CO, was a matter of intense debate.
Since 2002, theory and computational chemistry have played an increasingly important role in fundamental electrocatalysis work. Reactions such as hydrogen oxidation and evolution, oxygen reduction, methanol oxidation and carbon monoxide oxidation have all been studied using modern computational techniques, such as first-principles DFT calculations and Monte Carlo simulations, which both complement and give input for the more widespread kinetic modeling approaches. The interaction with experiment has been become very fruitful, and it is this synergy that provided the motivation for the present Faraday Discussion.
My aim in this paper is to illustrate the ideas and concepts on which I believe this interaction or “interface” of theory and experiment should be based, and then to illustrate this on two topics of my own current research focus. Since I am primarily interested in moving electrocatalysis forward as a science, as opposed to “technology”, I will only discuss the general fundamental challenges that we face, as opposed to the specific challenges related to e.g. fuel cell catalysis and the development of better and more efficient low-temperature fuel cells (which is undoubtedly one of the main drivers of the field).
Theoretical and computational electrochemistry
A famous statement made by Dirac in 192910 claims that “all of chemistry” follows from the laws of quantum mechanics, and the main difficulty lies in the fact that the equations are “too complex to be solved”. However, in his statement Dirac did not foresee (or did not include) the invention of the computer, ultimately made possible by the development of quantum mechanics itself. Much of modern theoretical chemistry is concerned with developing clever ways of using the computing power of computers to solve the complex equations. The success of theoretical chemistry has been immense, as illustrated by the award of the 1999 Nobel Prize to Pople and Kohn, two founding fathers of modern computational quantum chemistry. However, modern quantum chemistry still suffers—to some extent—from Dirac's demon: the number of atoms that can be included in a full-blown “first principles” computation, especially when coupled to dynamics, is limited to at best a few hundred. The impressive work of Neurock and coworkers, as exemplified by their paper in this volume, is a good example of what is currently achievable. The extrapolation to real sizes and realistic time scales is by no means trivial, and involves approximation schemes, the accuracy of which is often unclear and difficult to assess. The accuracy and/or reliability of model calculations (and therefore of the inherent approximations) may be evaluated on the basis on a comparison to experimental data, but this is a practice which, at least in the author's opinion, should be carried out with great care, no matter how successful and gratifying such a comparison may often appear.So what would be an ideal but still practical way to model an electrocatalytic system? In other words: how to go from quantum mechanics to, say, a real voltammogram? First of all, it is imperative that the atomic structure of the electrode surface is known accurately. Experimentally, this implies working with well-defined single crystals, and knowing how the exact structure of the single-crystalline or nanoparticulate surface and possible defects influence reactivity. This aspect becomes less important if one, for instance, compares the activity of a series of metals for a certain reaction, but at the expense of losing quite a bit of detail. Next, one has to set up a hierarchy of calculations that will finally lead to the prediction of the experimental outcome, where typically one level of calculation delivers the input for the next level. For instance, one may carry out a series of first-principles DFT calculations to estimate interaction energies between adsorbates on an electrode surface, and rate constants for reactions between adsorbates (based on transition state theory). These numbers may be input for a lattice-gas kinetic Monte Carlo (KMC) simulation of an extended surface, say 1000 × 1000 lattice points. The KMC simulation may directly give the desired macroscopic variable, such as for instance the electric current, as well as much more, such as the time-dependent distribution of adsorbates on the surface. Essentially, the KMC simulation serves as a way to estimate the system's partition function, i.e. as a simulation method to sample the long-time statistics of the system in a way that would be impossible by quantum chemistry alone. On the other hand, one often approximates the statistics of the system by assuming a perfect mixing, also known as the “mean-field approximation”, which is the basis of most kinetic modeling approaches and of, for instance, the well-known Frumkin isotherm.11 In this case, a KMC simulation should in principle still be carried out in order to confirm the validity of the mean-field approximation. Note, however, that the complexity of the system often forces one to make many more (implicit) assumptions: the lattice-gas approximation, the assumption of the additivity of interaction potentials (sometimes three-particle interactions may be included but even this is an approximation), entropic or other solvent effects are often neglected as first-principles free energy calculations are extremely expensive, assumptions about the exact structure of the surface and role of defects, assumptions about which reactions to include and which not, assumptions about the reaction mechanism (sometimes reactions may be included the rates of which are difficult to calculate and therefore they are estimated or guessed), etc. Although there may be examples where many of these approximations may be (or may appear to be) reasonable for the particular system under consideration, it will still be necessary to compare carefully to experimental data as well as to more accurate simulation data to finally assess the reasonableness of a model.
A key challenge in first-principles simulations of electrode systems is the introduction of the electrode potential. Since all existing quantum chemistry codes work with a fixed number of electrons in the simulation box, rather than with a fixed electrochemical potential of the electrons, applying an electrode potential to the simulation of a half cell is usually approximated by adjusting the charge distribution within the simulation cell. This can be achieved by applying an electric field to the cell,8,12,13 adding or removing electrons to the metal slab representing the electrode,14,15 or adding electron-drawing or withdrawing adsorbates to the metal surface.16 The first two methods should also screen the electrostatic field or charge by an artificial background. The corresponding electrode potential is determined at the end of the fully converged calculation, typically by referring the Fermi level of the metal electrons to a field-free reference somewhere in the simulation cell, in combination with the known relation between the vacuum scale of the metal work function and the normal hydrogen electrode.14–16 Practically all current first-principles calculations including the effect of the electrode potential are carried out in this fashion. The fundamental limitation of these methods is that they are essentially coulostatic methods, and cannot readily be applied to mapping out the reaction path of a charge transfer reaction, as most electrode reactions are. During a charge transfer reaction under coulostatic conditions, the electrode potential will change during the reaction. The preferred way to circumvent this problem is to switch to a grand-canonical simulation method,17 in which the electrochemical potential of the electrons in the half cell is held fixed rather than their number. The computational limitation of this method is that it requires an additional iterative loop in the already very time-consuming simulation, such that in practice the method is still hardly used.
An interesting approach that allows a rapid assessment of the influence of the electrode potential was suggested by Nørskov et al.,18 and is based on the very simple assumption that the only effect of the electrode potential is to change the energy of the electrons. For the adsorption reaction of hydrogen:
| H+ + e− ⇆ Hads | (1) |
| ΔGreaction(E) = ΔGads(Hads;E) − ΔGads(H+ + e−;E) | (2) |
| dΔGads(Hads)/dE = 0 | (3) |
Electrocatalytic oxidation of carbon monoxide
The oxidation of carbon monoxide is not only one of the favorite model reactions in electrocatalysis, but it is also a hugely important reaction in the development of more efficient low-temperature fuel cells. We have studied the CO electro-oxidation on various stepped single crystals of platinum and rhodium,21–24 and have demonstrated the importance of low-coordination step and defect sites in the oxidation mechanism. However, there remain quite a few fundamental issues that I believe are not yet fully understood. One of them is the importance of COads surface mobility in the oxidative stripping of pre-adsorbed CO, an issue that was quite intensely debated 6 years ago at the Faraday Discussion in Berlin. The other is the nature of the oxygen-donating species. I will discuss these two issues in this and in the next session.From the DFT point-of-view, CO seems to be a somewhat problematic molecule, as DFT has difficulty in correctly predicting the preferred adsorption site on a Pt(111) surface. Whereas experimentally CO prefers atop coordination on Pt(111) (in UHV at low CO coverage),25 most DFT calculations predict the threefold hollow site to be most stable.26 This is typically explained by the tendency of DFT to overestimate bonding interactions, which are stronger for higher coordination. Olsen, Philipsen and Baerends27 have recently shown that using a localized basis set and taking care of achieving full convergence of the DFT calculations, the atop site is found to be the preferred site on Pt(111). Nevertheless, their binding energies seem somewhat low compared to experiment. These observations clearly underpin the statement by Feibelman et al.26 that “DFT calculations cannot be used as black-box simulation tool.” Typically, qualitative or relative predictions are more reliable than quantitative or absolute predictions, having error bars of ca. 0.1 and 0.3–0.5 eV, respectively. At any rate, these calculations suggest the qualitative conclusion that the corrugation potential for CO on Pt(111) should be rather flat, in agreement with the experimental observation that CO mobility on Pt(111) is high.
The oxidation of carbon monoxide, under electrochemical conditions, is believed to follow the Langmuir–Hinshelwood-type mechanism originally suggested by Gilman:28
| H2O + * ⇆ OHads + H+ + e− | (4) |
| COads + OHads → COOHads → CO2 + 2* + H+ + e− | (5) |
The second step in this mechanism, the CO + OH combination reaction, is believed to be rate-determining, primarily because the Tafel slope observed for CO monolayer oxidation is ca. 70–80 mV dec−1,29,30 close to the theoretical value of 60 mV dec−1 expected for an EC mechanism. DFT calculations indeed show that there is sizeable barrier for the CO + OH reaction: Shubina et al.31 report ca. 0.6 eV on Pt(111) in the absence of water, whereas Janik and Neurock32 have obtained values of 0.5–0.3 eV in the presence of water, depending on whether the surface was charged or not. Note that these values apply to T = 0 K, hence these are not free activation energies.
By carrying out extensive chronoamperometry measurements on a series on stepped Pt surfaces in sulfuric acid, we have shown that the rate for CO monolayer stripping is proportional to the step density, and that there is no evidence for CO slowly reaching the active step sites. This strongly suggests that OHads formation takes place preferentially on the step sites, and the CO diffusion on the terrace is rapid, in agreement with the flat corrugation potential predicted by DFT.
More recently, we have performed a similar series of experiments, but in alkaline media.33 Alkaline media typically show higher catalytic activities than acidic media, even if the potential scale is converted to the reversible hydrogen electrode to correct for trivial pH effects.34,35 I believe that these observations are not well understood, and cannot be explained simply by referring to the higher affinity of OH for step sites in alkaline media, as this effect must have been accounted for by the RHE scale.
Fig. 1 shows the CO stripping voltammetry on a number Pt single crystals surfaces in 0.1 M NaOH. The remarkable observation here is that the CO voltammetry exhibits as many as 4 features. We can take the CO stripping voltammetry on Pt(554) as an example. By comparing to Pt(111), Pt(15 15 14), and Pt(553), we can conclude that the high-potential stripping peak between 0.72 and 0.80 V is due to the oxidation of CO on (111) terraces. Inspection of the surfaces with (110) and (100) step sites, we conclude that the stripping peak at around 0.6 V is CO oxidation at (110) sites, and that peak at ca. 0.70 V is due to CO oxidation at (100) sites. Note the small feature at 0.7 V in the stripping curve for Pt(554) (and Pt(553) and Pt(110) as well), which we ascribe to CO oxidation at a small amount of defects of (100) orientation. Finally, a broad low-potential potential feature, which can start at a potential as low as 0.35 V, is observed on all surfaces. This feature was also observed by Spendelow and Wieckowski36 in their studies of CO adlayer oxidation on lightly disordered Pt(111) in alkaline media. By combining voltammetry with scanning tunneling microscopy, they suggested that this feature is due to CO oxidation on small monoatomically high islands on the Pt(111) surface, which present low-coordination sites (essentially kink sites) that are particularly active for CO oxidation. Following their assignment, we suggest that this low-potential feature is CO oxidation on “kink”-type sites, or defects in the steps. This leads to the remarkable observation that a single voltammogram such as that shown for Pt(554) reveals as many as 4 different active oxidation sites for CO: kink sites, (110) step sites, (100) sites, and (111) terrace sites, in decreasing order of activity. Such an observation is only possible if the mobility of CO on the surface is low, as in the case of high CO mobility most if not all CO would oxidize at the most active oxidation sites.
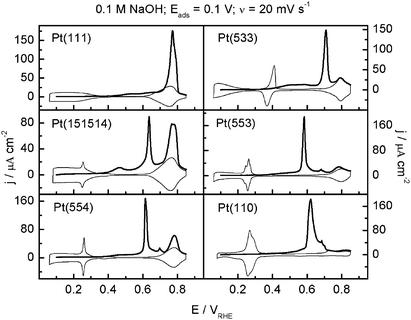 | ||
| Fig. 1 CO stripping (thick solid line) and the subsequent cyclic voltammogram (thin solid line) for Pt(111), Pt(15 15 14), Pt(554), Pt(533), Pt(553) and Pt(110) in 0.1 M NaOH, sweep rate 20 mV s−1, CO adsorbed at a potential of 0.1 V, no CO in solution during stripping. Reproduced with permission from ref. 33. | ||
A simple way to probe the role of low CO mobility is to study the scan rate dependence of the CO stripping voltammetry, as shown in Fig. 2 for Pt(554). It is observed that at high scan rates (500 mV s−1) there is a significant amount of CO oxidizing on the terraces. However, as the scan is lowered, the charge corresponding to CO oxidizing at the terraces decreases until finally at 5 mV s−1 it is almost negligible. This clearly suggests that at low scan rates, CO has more time to diffuse to the step sites and react there. When the amount of CO reacted in the steps or on the terraces, as estimated from the corresponding stripping charges, is plotted as a function of the square root of the scan rate, linear relationships are observed. This “Cottrell-like” behavior is another strong indicator for the important role of slow CO surface diffusion on Pt(111) terraces in alkaline media. The slow diffusion of CO on the (111) terrace is also manifested in the chronoamperometric transients. Kinetic Monte Carlo simulations of a model for CO oxidation on stepped surfaces37 show that in such a case, the transient is composed of two parts: an initial exponential current decay corresponding to a one-dimensional instantaneous nucleation and growth along the step, followed by a peak corresponding to an instantaneous nucleation and growth onto the terrace (see Fig. 3). Experimental transients for CO oxidation on stepped Pt in alkaline media indeed display the same characteristics.38
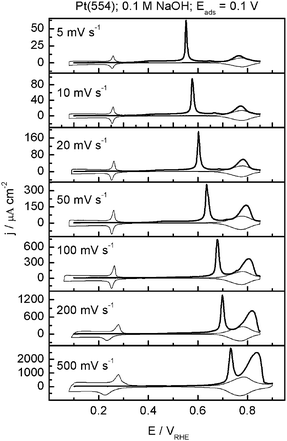 | ||
| Fig. 2 CO stripping (thick solid line) and the subsequent cyclic voltammogram (thin solid line) for Pt(554) in 0.1 M NaOH at different sweep rates, Eads = 0.1 V. Reproduced with permission from ref. 33. | ||
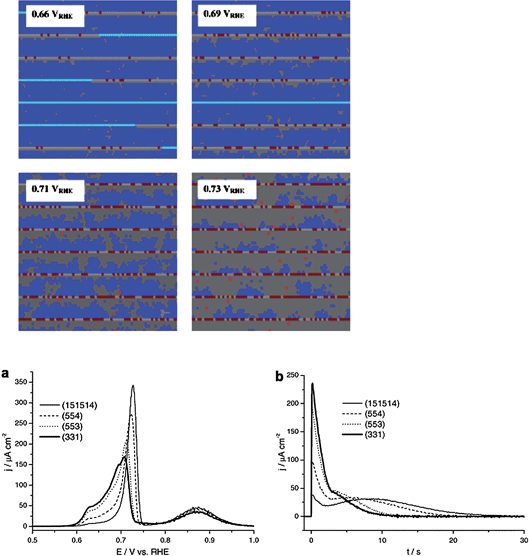 | ||
| Fig. 3 Results of Kinetic Monte Carlo simulations of CO stripping on a stepped surface in the absence of diffusion of CO on the terrace. The stripping voltammetry (a) exhibits three features: CO oxidation at steps (ca. 0.62 V), CO oxidation at terraces (ca. 0.72 V), and OH adsorption on terraces (ca. 0.87 V). The snapshots correspond to a (554) surface, where light (dark) blue is CO on steps (terraces), light (dark) grey is water on steps (terraces) and dark (light) red is OH on steps (terraces). Figure b shows the chronoamperometry at 0.68 V, and clearly displays exponential decay first (oxidation of CO at steps) and then a peak corresponding to CO oxidation on terraces. Reproduced with permission from ref. 37. | ||
The reason for the significantly reduced mobility of terrace-bound CO on stepped Pt electrodes in alkaline media as compared to acidic media has not been fully clarified yet. In acidic media, as well as on Pt(111) in UHV, the high mobility of CO is ascribed to the conclusion made above, namely that CO does not have a strong preference for a specific adsorption site on Pt(111) (as confirmed by DFT, although not in all its details) and therefore it should be able to move over the Pt(111) surface almost barrierless. In alkaline media, the electrode potential is effectively more negative than in acidic media, however, even if the actual potential scale used is the reversible hydrogen electrode (RHE). This implies that the Fermi energy of the Pt in alkaline media (say pH = 13) is about 0.7 eV higher than in acidic media at pH = 1. If the free energy of the adsorbate under consideration is not dependent on pH, such as with CO, this may have a significant influence on the way it binds to the Pt surface. From DFT calculations on both Pt(111) clusters and slabs,39–41 it has been found that CO binds more strongly to the surface at negative potentials, especially to multifold coordination sites such as bridge sites and hollow sites. This preference for multifold coordination at higher Fermi level or more negative potential can be explained qualitatively by the Blyholder model.42 At more negative potential the influence of back donation of metal electrons into the CO 2π* orbital becomes more prominent, and since the interaction between the 2π* orbital and the Pt d band is a bonding interaction, and therefore prefers to interact with as many surface atoms as possible, a stronger preference for multifold coordination may be expected. Recent FTIR experiments in our group43 on CO at Pt(111) in alkaline media confirm the difference in the electronic interaction with acidic media, both through a significant change in C–O stretching frequency (which roughly corresponds to 0.7 V times the Stark tuning slope in cm−1 V−1) as well as a clearly increased band intensity corresponding to bridge-bonded CO. However, a significantly more corrugated binding energy surface for CO on Pt(111) in alkaline media is not straightforwardly implied by these data. On the other hand, the FTIR data seem to suggest that the product of CO oxidation in alkaline media, carbonate, remains adsorbed on the surface until quite positive potentials. Strongly adsorbed carbonate may have a negative effect on the CO surface mobility, similarly to what we concluded for CO oxidation on rhodium single crystals in sulfuric acid,44 where strongly adsorbed sulfate severely hampers CO surface diffusion.
Formation of OHads on platinum
All our experiments, as well as those by various other authors, suggest that CO is oxidized by OH that is absorbed in a step or defect on the Pt surface. It is therefore all the more disconcerting that step-bonded OH has remained invisible in both spectroscopic and voltammetric experiments. Its apparent voltammetric invisibility is illustrated in Fig. 4, which compares the voltammetry of Pt(111) and a stepped Pt surface, Pt(15 15 14), in 0.1 M NaOH. Before we will attempt to explain the “anomalous” features of the stepped surface, let us discuss how the Pt(111) voltammogram may be modeled using a combination of DFT and statistical mechanics.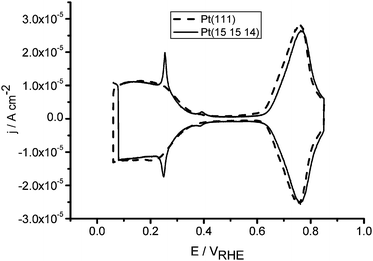 | ||
| Fig. 4 Blank voltammetry of Pt(111) and Pt(15 15 14) in 0.1 M NaOH. For explanation, see text. | ||
The Pt(111) voltammogram displays the well-known reversible features corresponding to H adsorption on the terrace (<0.35 VRHE) and OH adsorption on the terrace (>0.6 VRHE). These regions are reasonably well predicted by DFT calculations. Employing eqn (2) above and the assumptions stipulated there, Rossmeisl et al.45 have calculated from DFT the equilibrium potentials for the reactions:
| H+ + e− ⇆ Hads | (6) |
| H2O ⇆ OHads + H+ + e− | (7) |
At room temperature, it is well known that the coverage θ of H “upd” (underpotential deposition) adsorbates on Pt(111) follow a Frumkin isotherm to a good approximation:
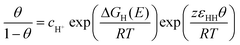 | (8) |
| ΔGH(E)=ΔGH,ads+ e0E | (9) |
From Jerkiewicz's temperature-dependent experiments46 and our Monte Carlo simulations,47 values for the H upd adsorption energy ΔGH(E) and the nearest-neighbor interaction energy εHH may be determined, as summarized in the second column of Table 1. Fig. 5 shows the “hydrogen upd region” predicted by this model, compared to the exact Monte Carlo simulations. It is seen that the Monte Carlo simulation show a bit more structure, due to the relatively strong interactions, ca. 0.047 eV per pair of neighboring H adsorbates. Nevertheless, the mean-field approximation or Frumkin isotherm is a reasonable approximation. In the third column of Table 1, we give the values for the adsorption energy and the nearest-neighbor interaction as estimated from the DFT calculations by Karlberg et al.48 The nearest-neighbor interaction energy is a bit smaller than the experimental estimate, where we note that the DFT calculations were performed without water on the surface. The Monte Carlo isotherm predicted by the DFT values is very close to the mean-field prediction,48 as the lateral interaction is very weak.
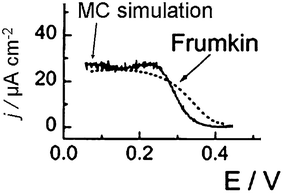 | ||
| Fig. 5 Monte Carlo simulation and mean-field “Frumkin” approximation of the hydrogen region on Pt(111) using the parameters in the second column of Table 1. | ||
The “OH adsorption region” on Pt(111) is more difficult to model. A simple model suggested by Rossmeisl et al.49 elsewhere in this volume models the OH adsorption on Pt(111) by a Langmuir isotherm with a maximum OH coverage of 1/3 ML. Using the DFT value for ΔGOH,ads mentioned above (0.81 eV), a reasonable agreement with experiment is obtained although a few important details are not reproduced or explained. The final coverage of OH on Pt(111) is ca. 0.4 ML in acidic media but in fact depends on pH, being slightly higher in alkaline media. The voltammogram (i.e. the derivative of the isotherm) displays a sharp peak in acidic media (but not in alkaline media), which has been explained as an order–disorder phase transition in the OH adlayer,47 or as caused by the adsorption of two different kinds of OH.50
Fig. 4 compares the voltammetry of Pt(111) in 0.1 M NaOH with that of a Pt(15 15 14) surface, which has 30-atom wide (111) terraces separated by steps of (110) orientation. Whereas on the Pt(111) terrace H and OH adsorption lead to two separate features, introduction of step sites leads to only one additional feature in the voltammetry at ca. 0.25 V (note a small feature at ca. 0.4 V in Fig. 4 which corresponds to step sites of (100) orientation). Furthermore, the feature is sharp instead of broad, implying attractive lateral interactions, which is at least unexpected. The charge corresponding to this peak is ca. 1 electron per step atom in acidic media,51 the reason why traditionally it has been attributed to hydrogen adsorption on the step site. However, if that were true, where is the feature corresponding to OH adsorption on the step site? Finally, whereas the features corresponding to H and OH adsorption on the (111) terrace show no significant pH dependence on the RHE scale, the step-related feature is observed at a more positive potential in alkaline media, shifting by ca. 10 mV pH−1 on an RHE scale.52
Table 1, fourth column, gives the values for ΔGH,ads and εHH estimated from the peak corresponding to the (110) step site in perchloric acid solution,53 if it is assumed that only H adsorbs on the step with a maximum step coverage of 1. Note that these numbers would imply that adsorbed H not only experiences attractive interactions on step sites, but also that H has a lower affinity to step sites than to terrace sites. This is in disagreement with ultra-high vacuum results, where it has been found that H has a higher affinity for step sites.54 By studying the co-adsorption of H and water on a stepped Pt surface in UHV, we have recently shown that these results can also not be explained by the interaction with water. In fact, a Pt surface covered with H, be it on terraces or in steps, tends to be hydrophobic.55
A more general theory for sharp voltammetric peaks was formulated recently in relation to a model for the voltammetry of Pt(100) in bromide containing solution.56 The adsorption of bromide on Pt(100) is accompanied by a sharp peak in which adsorbed H is quickly replaced by adsorbed bromide (see Fig. 6). We have modeled this voltammetric peak using a simple lateral interaction model that was solved using Monte Carlo simulations. First, we estimated the adsorption energy and interaction of upd H on Pt(100) by fitting the blank voltammetry of Pt(100) in perchloric acid solution, the results of which are also given in Table 1. Note that, as expected, H adsorbs more strongly on Pt(100) than on Pt(111), but that the interactions between the adsorbed H are weaker than on Pt(111). These results are in good qualitative agreement with DFT calculations,48 as given in the last column of Table 1.
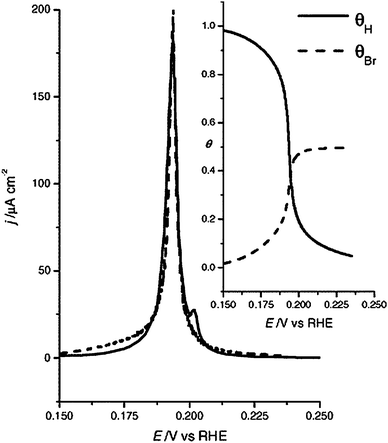 | ||
| Fig. 6 Modeling hydrogen and bromine competitive adsorption from the voltammogram of Pt(100) in HClO4 0.1 M + KBr 10−2 M (solid line, positive scan), by using Monte Carlo simulations using the interaction energies mentioned in the text (dashed line). The inset shows the corresponding coverages of hydrogen (solid line) and bromine (dashed line) as a function of the potential. Reproduced with permission from ref. 56. | ||
Introducing the potential dependent co-adsorption of Br into the model, a sharp peak is obtained, giving a good fit of the experiment (dashed line in Fig. 5), if we assume ΔGBr,ads = −0.27 eV and εHBr = 0.055 for a pair of H and Br sitting on neighboring sites, and an infinite repulsion between two Br on neighboring sites. The latter assumption leads to a maximum coverage of 0.5 ML of Br, in a c(2 × 2) adlayer, in agreement with experiment.57 Because the interaction between H and Br is stronger than between H, and the interaction between two adsorbed Br at next-nearest neighbor sites is small, the interparticle repulsion exceeds the sum of the intraparticle repulsion and the effective interaction for competing adsorbates may be negative (i.e. attractive). For a mean-field based derivation of this condition, we refer the reader to the original paper.56 This condition is typically satisfied if a small and a large adsorbate compete for surface sites.
A similar explanation may be applied to the sharp peak observed in the “hydrogen region” of stepped Pt surfaces. If we assume that the actual reaction corresponding to that peak is:
| Hads + xH2O ⇆ xOHads + (1 + x)H+ + (1 + x)e− | (10) |
Since it is very difficult to see adsorbed OH on Pt in a spectroscopic experiment, we have recently tried to adsorb OH in a step site of a stepped Pt(533) surface in UHV. Our tactic was similar to a method employed previously for Pt(111): by pre-adsorbing atomic oxygen and subsequently dosing water, O will react with H2O to form chemisorbed OH on the Pt(111) surface.58 In a temperature-programmed desorption experiment, this manifests as a water desorption peak that appears at higher temperature than without pre-adsorbed O. This apparent hydrophilicity is due to the stabilization of water by the exothermic reaction of water with O to OH. A similar experiment with atomic oxygen pre-adsorbed in the steps of a Pt(533) surface, without O on the terraces, does not yield a clear apparent stabilization of a monolayer of water when subsequently dosed on the Pt(533)–Ostep surface.59 This would suggest that the reaction to OH does not take place to a significant extent in the step sites, presumably because the relative stability of atomic oxygen is higher in the step than on the surface. This obviously raises the question what the product of water dissociation is under electrochemical conditions. I believe that this is a crucial question for which at this moment we do not have a consistent answer.
Conclusions
The papers in this volume amply illustrate the glamour and vitality of modern electrocatalysis and interfacial electrochemistry, and the immense impact that modern spectroscopic techniques and modern computational chemistry, and especially their combination, are having on our understanding of the electrochemical interface at the molecular level. Heinz Gerischer, in his Introductory Lecture to the Faraday Discussion 56 in 1973,3 quoted Julius Tafel from his 1904 paper in which he introduced the famous Tafel equation:60 “The problem of electrode polarisation in electrolysis has been studied scientifically for about one hundred years. It is, therefore, scarcely possible to find some new aspects which previously have not been touched already, either in experiment or speculations.” Now, another one hundred years later, I would conclude almost the opposite: there are many aspects that we have not yet explored and many observations that we still do not fully understand. However, the potential of the modern tools that we have at our disposal to tackle these issues is formidable and there is no question in my mind that this will lead to major advances in both the understanding and the applications of electrochemistry.These are indeed exciting times to be an electrochemist. Not only is the unprecedented power of modern experimental and computational tools an excellent enabler for innovative fundamental research work, with the ever louder cry for alternative and more sustainable energy sources and devices, electrochemistry has every reason to put itself at the center of attention. Even prominent non-electrochemists admit that our future energy technology will have electrochemistry as one of its cornerstones. In a 2007 Science paper, Whitesides and Crabtree61 have identified long-term research areas that should not be forgotten, and many of them are of a partial or even complete electrochemical nature. In the largely personal translation of this electrochemist:
1. The oxygen electrode (both oxygen reduction and oxygen evolution),
2. (Electro-)catalysis by design (in essence the theme of this meeting),
3. Various aspects of photoelectrocatalysis,
4. (Electro-)chemistry of carbon dioxide,
5. (Electro-)chemistry of complex systems (“emergent behavior”, nonlinearity, innovative (electro-)chemical engineering),
6. Efficiency of energy use,
7. (Electro-)chemistry of small molecules (H2O, CO, small inorganic nitrogen compounds),
8. New (but sensible) ideas.
These long-term research areas provide us with plenty of challenges to explore the potential of the interface between theory and experiment in electrocatalysis, and will continue to be discussion topics at Faraday Discussions in the decades to come.
References
- D. D. Eley, Discuss. Faraday Soc., 1947, 1, 129 Search PubMed.
- R. Parsons, Discuss. Faraday Soc., 1968, 45, 40 RSC.
- H. Gerischer, Faraday Discuss. Chem. Soc., 1973, 56, 1 RSC.
- J. E. B. Randles, Faraday Discuss. Chem. Soc., 1973, 56, 379 RSC.
- G. Nagy, K. Heinzinger and E. Spohr, Faraday Discuss., 1992, 94, 307 RSC.
- G. Ertl, Faraday Discuss., 2002, 121, 1 RSC.
- S. J. Mitchell, S. Wang and P. A. Rikvold, Faraday Discuss., 2002, 121, 53 RSC.
- S. A. Wasileski and M. J. Weaver, Faraday Discuss., 2002, 121, 285 RSC.
- M. T. M. Koper, N. P. Lebedeva and C. G. M. Hermse, Faraday Discuss., 2002, 121, 301 RSC.
- P. A. M. Dirac, Proc. R. Soc. London, Ser. A, 1929, A123, 714 CrossRef.
- A. N. Frumkin, Z. Phys. Chem., 1926, 35, 792 Search PubMed.
- P. S. Bagus, G. Pacchioni and M. R. Philpott, J. Chem. Phys., 1989, 90, 4287 CrossRef CAS.
- M. T. M. Koper, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, B. E. Conway and R. E. White, Kluwer Academic/Plenum Press, New York, 2003, vol. 36, p. 51–130 Search PubMed.
- C. D. Taylor, S. A. Wasileski, J.-S. Filhol and M. Neurock, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 165402 CrossRef.
- M. Otani and O. Sugino, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 115407 CrossRef.
- E. Skulason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Phys. Chem. Chem. Phys., 2007, 9, 3241 RSC.
- A. Y. Lozozvoi, A. Alavi, J. Kohanoff and R. Lynden-Bell, J. Chem. Phys., 2001, 115, 1661 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lundqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886 CrossRef CAS.
- K. J. Vetter and J. W. Schultze, Ber. Bunsen-Ges. Phys. Chem., 1972, 76, 920 CAS.
- W. Schmickler and R. Guidelli, J. Electroanal. Chem., 1987, 235, 387 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, E. Herrero, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2000, 487, 37 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Phys. Chem. B, 2002, 106, 12938 CrossRef CAS.
- T. H. M. Housmans, J. M. Feliu and M. T. M. Koper, J. Electroanal. Chem., 2004, 572, 79 CrossRef CAS.
- T. H. M. Housmans and M. T. M. Koper, J. Electroanal. Chem., 2005, 575, 39 CrossRef CAS.
- (a) D. F. Ogletree, M. A. Van Hove and G. A. Somorjai, Surf. Sci., 1986, 173, 351 CrossRef CAS; (b) B. E. Hayden, K. Kretzschmar, A. M. Bradshaw and R. G. Greenler, Surf. Sci., 1985, 149, 394 CrossRef CAS.
- P. J. Feibelman, B. Hammer, J. K. Nørskov, F. Wagner, M. Scheffler, R. Stumpf, R. Watwe and J. Dumesic, J. Phys. Chem. B, 2001, 105, 4801 CrossRef CAS.
- R. A. Olsen, P. H. T. Philipsen and E. J. Baerends, J. Chem. Phys., 2003, 119, 4522 CrossRef CAS.
- S. Gilman, J. Phys. Chem., 1964, 68, 70 CAS.
- L. Palaikis, D. Zurawski, M. Hourani and A. Wieckowski, Surf. Sci., 1988, 199, 183 CrossRef CAS.
- N. P. Lebedeva, M. T. M. Koper, J. M. Feliu and R. A. van Santen, J. Electroanal. Chem., 2002, 524–525, 242 CrossRef CAS.
- T. E. Shubina, C. Hartnig and M. T. M. Koper, Phys. Chem. Chem. Phys., 2004, 6, 4125 Search PubMed.
- M. Janik and M. Neurock, Electrochim. Acta, 2007, 52, 5517 CrossRef CAS.
- G. García and M. T. M. Koper, Phys. Chem. Chem. Phys., 2008, 10, 3802 RSC.
- N. M. Markovic and P. N. Ross Jr., Surf. Sci. Rep., 2002, 45, 117 CrossRef CAS.
- J. S. Spendelow, J. D. Goodpaster, P. J. A. Kenis and A. Wieckowski, J. Phys. Chem. B, 2006, 110, 9545 CrossRef CAS.
- J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2007, 9, 2654 RSC.
- T. H. M. Housmans, C. G. M. Hermse and M. T. M. Koper, J. Electroanal. Chem., 2007, 607, 67.
- G. García and M. T. M. Koper, in preparation.
- M. T. M. Koper and R. A. van Santen, J. Electroanal. Chem., 1999, 476, 64 CrossRef CAS.
- M. T. M. Koper, R. A. van Santen, S. A. Wasileski and M. J. Weaver, J. Chem. Phys., 2000, 113, 4392 CrossRef CAS.
- D. Curulla Ferré and J. W. Niemantsverdriet, Electrochim. Acta, 2008, 53, 2897 CrossRef CAS.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772 CrossRef CAS.
- G. García, P. Rodriguez and M. T. M. Koper, in preparation.
- T. H. M. Housmans and M. T. M. Koper, Electrochem. Commun., 2005, 7, 581 CrossRef CAS.
- J. Rossmeisl, J. K. Nørskov, C. D. Taylor, M. J. Janik and M. Neurock, J. Phys. Chem. B, 2006, 110, 21883.
- G. Jerkiewicz, Prog. Surf. Sci., 1998, 57, 137 CrossRef CAS.
- M. T. M. Koper and J. J. Lukkien, J. Electroanal. Chem., 2000, 485, 161 CrossRef CAS.
- G. S. Karlberg, T. F. Jaramillo, E. Skulason, J. Rossmeisl, T. Bligaard and J. K. Nørskov, Phys. Rev. Lett., 2007, 99, 126101 CrossRef CAS.
- J. Rossmeisl, G. S. Karlberg, T. Jaramillo and J. K. Nørskov, Faraday Discuss., 2008, 140 10.1039/b802129e , paper 16.
- A. Berná, V. Climent and J. M. Feliu, Electrochem. Commun., 2007, 9, 2789 CrossRef CAS.
- J. Clavilier, K. El Achi and A. Rodes, J. Electroanal. Chem., 1989, 272, 253 CrossRef CAS.
- M. J. T. C. van der Niet, N. García-Araez, J. M. Feliu and M. T. M. Koper, in preparation.
- M. T. M. Koper, J. J. Lukkien, N. P. Lebedeva, J. M. Feliu and R. A. van Santen, Surf. Sci., 2001, 478, L339 CrossRef CAS.
- A. T. Gee, B. E. Hayden, C. Mormiche, T. S. Nunney, J. Chem. Phys.112, p. 7660 Search PubMed.
- M. J. T. C. van der Niet, I. Dominicus, M. T. M. Koper, L. B. F. Juurlink, Phys. Chem. Chem. Phys Search PubMed, submitted.
- N. Garcia-Araez, J. J. Lukkien, M. T. M. Koper and J. M. Feliu, J. Electroanal. Chem., 2006, 588, 1 CrossRef CAS.
- N. Garcia-Araez, V. Climent, E. Herrero and J. M. Feliu, Surf. Sci., 2004, 560, 269 CrossRef CAS.
- C. Clay, S. Haq and A. Hodgson, Phys. Rev. Lett., 2004, 92, 046102 CrossRef CAS.
- M. J. T. C. van der Niet, I. Dominicus, O. T. Berg, L. B. F. Juurlink and M. T. M. Koper, in preparation.
- J. Tafel, Z. Phys. Chem., 1904, 50, 641 Search PubMed.
- G. M. Whitesides and G. W. Crabtree, Science, 2007, 315, 796 CrossRef CAS.
Footnote |
| † At the meeting it was pointed out by Professor Roger Parsons, who was also present at the Faraday Discussion 1 in 1947 as a graduate student of Professor Bockris, that the contributors from the Soviet Union and Eastern Europe (Frumkin, Levich, Ershler, Heyrovsky) were not able to be present at the meeting at Manchester. This severely limited discussion of their important work and significantly delayed the impact of their contributions. This clearly illustrates the importance of discussion in person which characterises the Faraday Discussions. |
| This journal is © The Royal Society of Chemistry 2009 |