The role of anions in surface electrochemistry
D. V.
Tripkovic
,
D.
Strmcnik
,
D.
van der Vliet
,
V.
Stamenkovic
and
N. M.
Markovic
*
Materials Science Division, Argonne National Laboratory, University of Chicago, Argonne, Illinois 60439, USA. E-mail: nmmarkovic@anl.gov
First published on 21st August 2008
Abstract
Some issues of the current state of understanding in the surface electrochemistry are discussed, with emphases on the role of specifically adsorbing anions in hydrogen adsorption and oxide formation, adsorption and ordering of molecular adsorbates and metal ions, metal deposition, restructuring and stability of surface atoms, and kinetics of electrochemical reactions.
1. Introduction
The adsorption of anions on metal electrodes has been one of the major topics in surface electrochemistry. Of particular interest are chemisorbed (also called contact adsorbed or specifically adsorbed) anions, whose adsorption is controlled by both electronic and chemical forces. Halides (F−, Cl−, Br-, I−) and oxyanions (ClO4−, and SO42−) are the most extensively studied specifically adsorbed anions.1,2 As pointed out by Conway,3 the degree of specific adsorption on metal surfaces increases in the following order: F− < ClO4− < SO42− < Cl− < Br− < I−, that reflects the decreasing energy of solvation of these species, with strongly solvated F− and ClO4− being nonspecifically or only weakly adsorbed. In contrast, weakly solvated SO42−, Cl−, Br−, and I− could form direct chemical bonds with the metal surface, resulting in an ionic surface concentration that exceeds one induced by pure electrostatic interaction. It has been realized for a long time that these specifically adsorbing anions have an important, and generally adverse, effect in a number of electrochemical reactions, including adsorption and ordering of adsorbates from a supporting electrolyte, structure of metal substrates, kinetics of electrochemical reactions, metal deposition, and corrosion.The earlier studies, which were carried out on polycrystalline metal electrodes,4 indicated that specific adsorption of anions may affect electrochemical processes in a number of ways: (i) blocking of active sites on which otherwise reactant and/or intermediates could be adsorbed; (ii) modification in adsorption energy for sites adjacent to adsorbed anions; (iii) changes in the potential distribution across the interface; and (iv) surface restructuring. Given that all of these effects may influence electrochemical reactions simultaneously, the role of anions in surface electrochemistry on metal electrodes was considered qualitatively and phenomenologically.
Most of the progress in understanding the role of anions in surface electrochemistry has come from the advent of in situ surface sensitive probes5–11 and the development of the efficient methods to prepare clean single crystal surfaces that are well-characterized with respect to the geometric location and composition of surface atoms.12–15 These well-defined surfaces have offered an ability to find the potential dependence of the surface coverage and structure by anions, the energetics of adsorption, their effects on ordering, reactivity and stability of electrochemical interfaces.
The objective of this Faraday Discussion report is to provide a brief discussion of the importance of specific adsorption of anions in surface electrochemistry. The many investigations concerned with the potential dependence of the phase transitions in ordered anion structures on metal surfaces have been omitted here, in preference of systems for more catalytic interest. We focus on the insight into the physical factors influencing the observed anion effects on adsorption of hydrogen, oxygenated species and CO, restructuring and stability of platinum surface atoms, kinetics of fuel cell reactions, and metal deposition of Cu. The presentation here is restricted to constrained overview; further details (including experimental procedure, etc.) can be found in references cited.
2.1 Adsorption of hydrogen and hydroxyl species
The adsorption of hydrogen (dubbed underpotentially deposited; H+ + e− = Hupd) on Pt single crystal surfaces has been the subject of intensive investigation. As early as 1965, Will recognized that the Hupd formation on platinum single crystals is a function of the crystallographic orientation of the surface atoms, even though the crystals he used were, by today's standards, neither well-characterized nor well-prepared.16 In the late 1970's, structure sensitivity of adsorbed hydrogen was derived from ex-situ ultrahigh-vacuum (UHV) analysis of immersed surfaces4,17–19 and/or from analyzing the results obtained by using very simple flame annealing method, the latter being introduced by Clavilier.12 An examination of the literature from this ex-situ and flame annealing era revealed many conflicting reports about the number, relative size and shape of the Hupd peaks observed on Pt(hkl) in various electrolytes. Interestingly, the major difference was found between the cyclic voltammograms recorded in “non-adsorbing electrolytes, e.g., HF and HClO4.13 For example, while the voltammetry curve for flame annealed Pt(100) in 0.1 M HF (Fig. 1) shows the asymmetry between the positive and negative potential sweep in the Hupd potential region, the Hupd peaks in 0.1 M HClO4 are rather symmetrical. Since F− and ClO4− are not thought to adsorb significantly on Pt, differences in these two electrolytes were proposed to be due residual Cl− impurities in the HF. As seen in Fig. 1 for Pt(100), the addition of HCl to HClO4 in trace amounts has substantial effect on both the Hupd peak position as well as the Hupd peak shape, e.g., the symmetrical voltammetric curve is changed to an asymmetric curve, becoming almost identical to the voltammogram observed in “clean” HF. The sharp peak in the hydrogen region observed in the negative sweep is probably caused by rapid desorption of Cl−, which is accompanied by the concomitant adsorption of Hupd. In contrast to Pt(100), traces of Cl− have much less effect on the hydrogen adsorption on Pt(111), see Fig. 1. It appears that even in the presence of a small concentration of Cl−, the potential of zero charge (pzc) for Pt(111) is still quite positive to the hydrogen adsorption potential region, and thus the specific adsorption of Cl− is negligible in this potential region.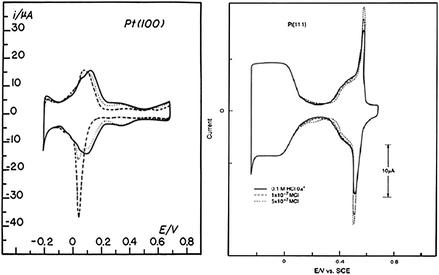 | ||
| Fig. 1 Left: Cyclic voltammetry for flame-annealed Pt(100) in 0.1 M HClO4 (solid line); 0.1 M HClO4 + 5 × 10−7 M Cl− (dotted line); 0.1 M HClO4 + 5 × 10−6 M Cl− (dashed line); sweep rate 50 mVs−1; electrode area 0.283 cm2. Right: Cyclic voltammetry for flame-annealed Pt(111) in 0.1 M HClO4 (solid line); 0.1 M HClO4 + 1 × 10−7 M Cl− (dotted line); 0.1 M HClO4 + 5 × 10−7 M Cl− (dashed line); sweep rate 50 mVs−1; electrode area 0.283 cm2. | ||
At more positive potentials, however, the effect of trace level of Cl− on the cyclic voltammetry is observed clearly in the “double layer” potential region and, more significantly, in the so-called “butterfly” potential region where the hydroxyl adsorption (hereafter denoted as OHad) on Pt(111) is accompanied by Cl− desorption and the corresponding sharpening of the butterfly peak.20 Based on this observation alone, it was proposed that Cl− is present as an impurity (at least 10−7 M) even in the most meticulously prepared HClO4. The level of Cl− impurity is significantly smaller in HClO4 than in HF and, therefore, all the differences observed in cyclic voltammetry of Pt(hkl) between these two “non-adsorbing” electrolytes are caused by different amounts of Cl anions present in these solutions.
Further successful “fishing” of trace level of Cl− in HClO4 has been achieved recently from two experimental approaches. One strategy involved utilization of the rotating disk electrode (RDE) technique,21,22 which has allowed Cl− concentration to be enhanced in the vicinity of the electrode surface by increasing the rate of Cl− mass transfer from a bulk of electrolyte to the electrode surface. The second strategy has relied on the fact that the onset of adsorption of anions on metal surfaces with lower values of pzc is shifted towards more negative potentials and, if the geometry of surface atoms is preserved, it would be possible to “catch” a trace level of anions even on a surface with the (111) geometry. A notable published example of these two tactics concerns Cl− adsorption on Pt(111) modified be a pseudomoprhic Pd monolayer.22 As can be seen in Fig. 2, the rotation of the electrode (1600 rpm) has a significant effect on both the shape of the Hupd peaks and on the adsorption of OHad. In particular, the observed Hupd peaks in the voltammogram of the rotated electrode exhibit an asymmetry, in contrast to the relatively symmetrical Hupd peaks observed on a stationary electrode. Interestingly, a very similar asymmetric peak is observed for the adsorption of hydrogen on Pt(100) in HClO4 containing 5 × 10−6 M Cl− (Fig. 1), suggesting that it is possible to detect a trace level of Cl− by increasing mass transport limitations. Notice that the observed voltammetric features in the presence of a small amount of Cl−are qualitatively similar to the effect induced by the rotation of the electrode in pure HClO4, e.g., in the cathodic sweep direction the various peaks merge into a single peak located at 0.2 V. This is a confirmation that an enhanced mass transport of the small amount of Cl− (≈10−7 M) from the bulk of pure HClO4 solution to the electrode surface by forced convection has a similar effect as a small addition of Cl− to the electrolyte. Furthermore, the OH adsorption is largely suppressed by either increasing the rotation rate or by the addition of Cl−, suggesting that trace amounts of Cl− and not the high concentration of perchlorate anions control the adsorption properties of Pt-group metals. Although the binding energy of OHad is stronger on Pd than on Pt, due to strong Pd–Cl− interactions the OHad coverage is higher on Pt(111) than on Pt(111)–1 ML Pd, a fact which will have a consequences for the interpretation of catalytic activity of Pt and Pd surfaces (see section 2.4).
 | ||
| Fig. 2 Cyclic voltammograms of Pt(111)–1 ML Pd (a) in 0.1 M HClO4; (b) 0.1 M HClO4 + 1 × 10−6 M Cl−; (c) same conditions as in (a) but rotation of electrode with 1600 rpm; (d) same conditions as in (b) but rotation of electrode with 1600 rpm; sweep rate 50 mVs−1. | ||
In contrast to trace level of anions, with an increased amount of added Cl− to the HClO4 or in pure HCl electrolyte (Fig. 3) the cyclic voltammograms of Pt single crystals in the Hupd potential region become very sharp and exceptionally symmetrical.13 This is consistent with a coupling between the Hupd and Cl− adsorption–desorption processes, suggesting that a unique synergy of voltammetric features on metallic surfaces in acidic solutions is arising rather through the structure sensitive adsorption of anions than the adsorption of Hupd and OHad. An important consequence of such coupling effects is that even after four decades of comprehensive research on single crystal surfaces in acid solutions we are not able to resolve intrinsic structure sensitivity even for the two elementary steps in surface electrochemistry; e.g., the adsorption of hydrogen and the formation of the hydroxyl layer. Nevertheless, in the various mechanisms for accounting of anion effects on adsorption of hydrogen and hydroxyl species, the authors believe that the coupling of anion adsorption to the hydrogen/hydroxyl adsorption is predominant. Depending on the anion concentration in the bulk of electrolytes, the pseudocapacitive features corresponding to Hupd and OHad may show either an asymmetry or may be sharp and symmetrical.
 | ||
| Fig. 3 Cyclic voltammetry for flame-annealed Pt single crystals in 0.1 M HCl; sweep rate 50 mVs−1; electrode area 0.283 cm2. | ||
2.2 Adsorption of CO and electrooxidation of CO
Beyond the question of anion effects in determining the pseudocapacitive features in cyclic volammograms, a central issue in electrochemical surface science concerns the role of anions in orchestrating the surface structures (and activity) of co-adsorbed reactants and reaction intermediates. Given its practical importance, a particularly interesting class of electrochemical adlayer systems receiving the widest attention so far is CO adsorption and electrooxidation on Pt(111).23–31 Very recently, our group has focused on studying the role of anions on the electrocehmistry of COad on metal surfaces for two reasons:31 (i) COad is suitable to be characterized by vibrational spectroscopies, e.g., infrared absorption-reflection spectroscopy (IRAS), and to be probed structurally by utilizing in-situ methods of scanning tunneling microscopy (STM) and surface X-ray scattering (SXS); (ii) and in part because the anodic oxidation of COad plays the role of a “test molecule” in electrocatalysis, as it does in gas-phase catalysis. For our purposes here, by summarizing the effects of anions on the potential-dependent vibrational behavior of linear COad band frequencies (vCOl) as well as on corresponding O–C–O vibrational frequency of the dissolved CO2, we shall demonstrate not only that anions are controlling kinetics of CO oxidation reaction through its competition with OHad but, in addition, that they are affecting CO clustering on the surface.Representative cyclic voltammogram of Pt(111) in 0.5 M H2SO4 is shown in Fig. 4a. The presence of hydrogen adsorption region below 0.25 V and bisulfate/hydroxide adsorption region at higher potentials are familiar features of Pt(111) voltammetry in sulfuric acid solution. The CO-stripping curve (recorded simultaneously with IRAS spectra) shows that the onset of CO oxidation commences at ≈0.35 V, forming between 0.35 < E < 0.6 V what we have previously referred to as a pre-ignition potential region.1,25 In the ignition potential region (E < 0.6 V) the stripping voltammetry is characterized by a sharp peak centered at 0.68 V. In both potential regions the CO oxidation reaction takes place in the Langmuir–Hinshelwood type reaction between CO and OH (CO + OH = CO2 + H+ + e−).1,25
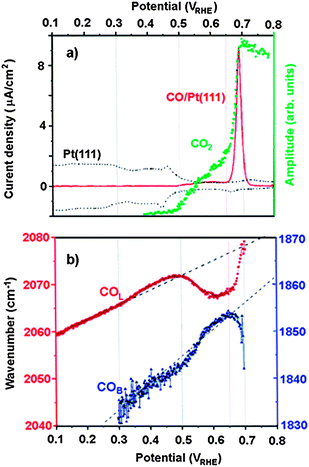 | ||
| Fig. 4 (a) Cyclic voltammogram of Pt(111) in 0.5 M H2SO4 (dashed line) 2 mV s−1; CO stripping current (solid line, 0.5 mV s−1) and corresponding CO2 production (grey dots); (b) vCOvs.E plots recorded simultaneously with CO stripping. | ||
In all previous reports the potential dependence of vCO is seen to be essentially linear, at least to the potentials as high as ≈0.45 V. Fig. 4b shows that the dependence vCOlvs.E is, in fact, nonlinear and that a linear slope is observed only below 0.3 V, e.g., in the potential region where CO adlayer is stable (no CO2 production in Fig. 4a). In contrast, concomitant with the development of CO2 at 0.35 V (grey dots), the vCOl frequencies (solid line) first slightly increase with respect to the expected linear relation (dotted trace in Fig. 4b) then, over the potential range 0.5 < E < 0.62 V, vCOl frequencies downshift substantially and, finally, the marked increase in vCOl frequencies above 0.62 V is mirrored by the rapid CO oxidation (CO2 production in Fig. 4a). Clearly, in addition to the effect of electric field, other factors, such as CO adlayer compression/dissipation should be taken into account to explain the anomalous deviations from a linear dependence. The phenomenon of CO adlayer compression/dissipation is observed in segregated systems where the presence of one species causes the other species to segregate into patches in which the local density is higher/lower than that observed when an equivalent amount of particular species is adsorbed alone. Extending this phenomenon to Fig. 4b, we suggest that the vCOlfrequency deviations arise from different distribution of surface coverage by reactive (CO and OH) and inactive (HSO4) adsorbates and the mutual interaction among them. The most plausible explanation for vCOl blueshift deviation between 0.35 < E < 0.5 V is a relatively simple model, where a small yet clearly discernable CO oxidation (initiated by OH adsorption on defect sites25) is followed by bisulfate anion adsorption on Pt sites, which below the onset of CO oxidation were occupied by COad. Given that the CO–HSO4− interaction is most likely repulsive, the higher vCOl frequency than expected most plausibly reflects mild compression of COad islands (the enhanced dipole–dipole coupling) engendered by HSO4− co-adsorption. On the other hand, marked vCOl redshift observed between 0.4 < E < 0.55 V reflects the reduced dipole–dipole coupling and temporal dissipation of the CO adlayer into less compressed clusters. Such behavior is symptomatic of increased CO oxidation, induced by an enhanced OH adsorption and hindered HSO4− adsorption. In the potential region where intensive CO oxidation occurs (E > 0.65 V), however, the adsorption of bisulfate is more favorable than OH− (surface provides larger ensembles of CO-free Pt sites), acting again as a driving force for COad to cluster into small compressed islands. This, in turn, will enhance the dipole–dipole coupling, e.g., a substational blueshift in the COad vibrational frequency.
To test further the anion-induced island compression model, CO oxidation experiments were carried out in CO saturated H2SO4 (Fig. 5a), HClO4 (Fig. 5b) and KOH (Fig. 5c) solutions. We chose to show the data for vCOvs.E relationships in CO saturated solution to demonstrate that the results summarized in Fig. 4 are not unique to the CO stripping experiments. In particular, above 0.55 V in H2SO4 solution the anomalous vCOl deviations are also observed in the presence of CO in solution. The comparison between the anion-dependent vCOl deviations in the solution containing strongly (HSO4−) and weakly (ClO4−) adsorbing anions shows that weakly adsorbing perchloric acid anions allow formation of reactive OH (which may aid the dissipation of CO islands) without compressing the remaining CO into smaller islands. Because OH− adsorption rather than weak anion adsorption is preferred at higher potentials, then the vCOl redshift at E > 0.65 V follows from the fast oxidative removal of COad and, thus, the reduced dipole–dipole coupling. Consequently, instead of a “U-shaped” vCOlvs.E dependence, characteristic for H2SO4, in HClO4 only moderate non-linear deviations are observed in the same potential range. Not surprisingly, in the solution containing only OH− anions, the effect of facile oxidation of CO adlayer (vCOl redshift in Fig. 4c), induced by the adsorption of reactive OH−, is even more pronounced. Consequently, the anomalous “Stark-tuning” behavior is completely missing in KOH solution (Fig. 5c).
 | ||
| Fig. 5 v CO vs. E plots for COad (light grey: linearly bonded, vCOl; grey: multicoordenated bonded, vCOm; dark grey: bridge bonded vCOb) in the different electrolytes. Notice the existence of multicoordinated CO (COm) in the CO saturated solution; for details see ref. 31. | ||
A related, yet separate, issue concerns the possible role of anions in influencing the kinetics of CO oxidation reaction. Although the role of anions in electrocatalysis of a fuel cell reaction will be discussed in section 2.4 below, for our purposes here, we continue with our discussion by analyzing the role of anions in the electrooxidation of COad on Pt(111). Fig. 5 shows that the kinetics of CO oxidation are strongly pH dependent; a notable feature is that the kinetics in alkaline solution is faster than in acid media. If the L–H mechanism is operative the higher catalytic activity in the alkaline solution (evidenced by the lower CO oxidation potential, CO2 production) implies that the surface coverage by OH− is larger in alkaline than in acid solution. There is a strong competition between OH− and anions for the Pt sites in acid solution, and therefore the surface coverage by OH− is significantly reduced with respect to alkaline solution and thus the kinetics is strongly inhibited at low pH. The explanation, for the remarkable effect of pH on the rate of CO oxidation is the pH-dependent adsorption of OH− which, in turn, is determined by competitive adsorption of anions. Therefore, as in the case of Hupd and OHad, it appears that the ordering of COad in an adlayer (see ref. 25), the morphology of the adlayer during CO oxidation (Fig. 4), the structure sensitive kinetics of COad electrooxidation (see ref. 1 and 25), and the rate of CO oxidation on the same electrode but in various electrolytes (Fig. 5) is completely determined by the structure sensitive adsorption of anions. In what follows, we demonstrate that the same applies for metallic adsorbates as well.
2.3 Underpotential deposition of Cu
Another fundamental question concerns the possible role of anions in both structural ordering of underpotentially deposited (upd) metals as well as in the kinetics of metal deposition. An essentially well studied example, which also illustrates the importance of SXS in analyzing metal–anion adsorbing structures is the formation of Cu adlayer(s) on Pt(111) in acidic media. The interpretation of processes associated with the formation of the Cu monolayer on Pt(111), and the nature of the Pt(111)–Cu structure, have been the subject of considerable controversy. Overviews of several different perspectives can be found in references.32–34 The rotating ring disk (RRDE) method33,34 was successfully applied to investigate the kinetics of Cu2+ deposition and to determine the potential dependent surface coverage by Cuupd (Θcu(upd)).33,34Fig. 6 shows that Cu deposition in 0.1 M HClO4 is independent of electrode rotation, manifesting that Cu deposition is a kinetically limited reaction in the absence of specifically adsorbing anions. The effect of addition of Cl− becomes apparent in the voltammetry of Fig. 6b, with the two major distinguished features: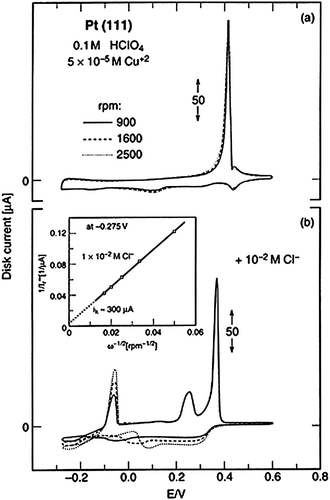 | ||
| Fig. 6 Cyclic voltammogram for Cu UPD on the Pt(111) disk electrode in 0.1 M HClO4; (b) Cyclic voltammogram for Cu UPD on the Pt(111) disk electrode in 0.1 M HClO4 + 1 × 10−2 M Cl−; inset; Levich plot for unshielded ring currents at −0.257 V. | ||
1. The kinetics of Cu UPD is dramatically enhanced in the presence of Cl−, resulting in the diffusion-limiting transport of Cu2+ in the presence of Cl−; e.g. in both the UPD and overpotential deposition (OPD) potential regions the current is proportional to ω1/2. The promotive effect of Cl− can be rationalized based on appreciative polarizability and highly deformable hydration shells of Cl−. One might anticipate that such a Cl− anion may perturb the solvation shell of strongly hydrated Cu2+, thereby promoting electron transfer kinetics in the discharge reaction of Cu2+ to Cu0.
2. Cl− produces a splitting of the voltammetric Cu UPD peak.32–34 This finding encouraged a careful evaluation of this system with SXS and RRDE methods. In combination, these studies have demonstrated that Cuad coverage and thus structure in between the two peaks (Fig. 7) is completely determined by the co-adsorbing anions (Cl− or Br−). In nearly halide free supporting electrolytes, Cu appears to be deposited as metallic islands (or “patches”) having the Pt lattice constant, i.e., pseudomorphic adlayer.32–34 In the presence of halides, however, a multi-step deposition occurs with the formation of ordered anion adlattices. A representation of a possible Pt(111)–Cuupd-anion structural model as a function of Cuupd coverage is shown in Fig. 7 (for more details see ref. 34). Clearly, Cuupd is either sandwiched between the Pt surface and anions or is in contact with the anions adsorbed on the adjacent Pt sites, in Fig. 7 states 1 to 4. At higher surface coverages by Cuupd (0.5 < ΘCu < 1), anions are entirely displaced from the surface by Cuupd (state 3). Note that the state 3 is representative of an ordered (4 × 4) Cuupd–Clad (Brad) bilayer structure, which is formed in between the Cu UPD peaks in the cyclic voltammetry (Fig. 7), each with coverage of 0.585 ML. The final step in the Cu UPD is the filling-in of the Cuupd monolayer to form a bilayer phase: i.e., a pseudomorphic (1 × 1) Cuupd monolayer.
 | ||
| Fig. 7 The representation of the proposed Cu-halide structure on Pt(111) in which the Pt atoms are shown by open circles and halide overlayer atoms by filled circles. Bottom: proposed mechanism for Cu UPD in the presence of halide anions. For details see text. | ||
The observation that anions are affecting the kinetics of metal deposition as well as ordering of metal adlayers has far-reaching implications in surface electrochemistry. While both Cu and halides are known to transfer most (or all) of their charge upon chemisorption, the Coulombic stabilization afforded by an ionic-solid-style adlattice may well trigger their common formation at electrochemical interfaces. Understanding the factors controlling the nature and occurrence of such double layer coadsorbate adlattices is clearly a topic worthy of theoretical as well as greater experimental attention in the near future.
2.4 Electocatalysis of fuel cell reactions
The term electrocatalysis is commonly employed to describe the study of electrode processes where charge-transfer reactions have a strong dependence on the nature of the electrode material.35–37 Not surprisingly, virtually every electrochemical reaction where chemical bonds are broken or formed is electrocatalytic, and the kinetics varies by many orders of magnitude for different electrode materials. This is true even for the simplest electrochemical reaction where chemical bonds are broken, the hydrogen evolution/oxidation reaction (HER/HOR), and for the more complex reactions such as the oxygen reduction reaction (ORR), and certainly for small organic molecules such as the oxidation formic acid and methanol. Although largely different in nature, the kinetics of electrochemical reactions involving either inorganic or organic compounds is governed by the same electrocatalytic law: while the reaction rate passes through a maximum for metals adsorbing reaction intermediates moderately, the reaction rate is very slow on metals which adsorb intermediates either strongly or weakly, i.e., the Sabatier Principle. The establishment of more quantitative relationships between the energetics of intermediates and the rate of electrochemical reaction is somewhat difficult owing in part to the absence of directly measured values for the surface–intermediate bond energy, and in part to the fact that an electrocatalytic reaction occurs on electrode surfaces which are always modified by reaction intermediates and “spectator” species. Therefore, for the future development of electrocatalysis as science, it is essential that these difficulties/complexities be overcome in order to progressively link the atomic/molecular-level properties of the electrochemical interface to the macroscopic kinetic process. The aim of this section is to summarize some of these relationships and to give selected examples of each, with particular emphasis on the systems that have been studied because of their inherent interest in the development of efficient energy converting systems.
While numerous details remain uncertain, this reaction scheme involves the adsorption of CH3OH (HCOOH) (kad), followed by the (non-faradic) dehydration of CH3OH (HCOOH), and the formation of chemisorbed “poison” (reaction 2) in competition with the direct dehydrogenation path via one or more reactive intermediates. As we shall see below, the rate of this step is determined by the surface coverage of Hupd, anions (Aad), OHad, and “poisoning” species. The major “poisoning” species was identified clearly as adsorbed CO. Besides being a “poison”, COad may also act as an intermediate, where some fraction of the COad can be further oxidized to produce CO2 (reaction 4). The active surface oxidant is most likely adsorbed OHad, as proposed in the section above for COadoxidation. Following the reaction scheme for oxidative removal of COad, the adsorption of oxygenated species is in a strong competition with anion adsorption (kA), and consequently the rate of reaction 4 (kox) is determined by the delicate balance between the rate constants kp, kOH, and kA. Of the various proposed rate determining steps for accounting for oxidative removal of CO,1 the authors believe that the adsorption of OH (kOH) and, thus, competition between OH− and anion adsorption is controlling the rate of CO electrooxidation.
Important examples of the role of anions in electrooxidation of small organic molecules includes the effects of Cl− on oxidation of methanol (Fig. 8). As anticipated, increasing the Cl− concentration inhibits significantly the methanol electrooxidation on Pt(111) and even more on Pt(100). On Pt(100) the effect of Cl− is generally more complicated than on Pt(111), showing a strong dependence of the pre-history of the electrode; with the inhibition becoming stronger with each successful sweep. This “ageing” effect appeared to be related to both the stronger adsorption of Cl− on Pt(100) than on Pt(111), requiring a lower potentials to desorb Clad from Pt(100) than Pt(111) and, as we discuss in the last section, due to anion structure sensitive halide-induced restructuring of surface atoms. Nevertheless, on both surfaces the inhibition of methanol oxidation is caused by blocking of surface sites by Clad, e.g., the anion adsorption is predominant over adsorption of methanol (deactivating step 1) and effectively blocks the formation of hydroxyl adlayer and thus inhibiting oxidative removal of COad from the surface (step 4). The same is valid for the oxidation of formic acid, see ref. 1 and references cited therein.
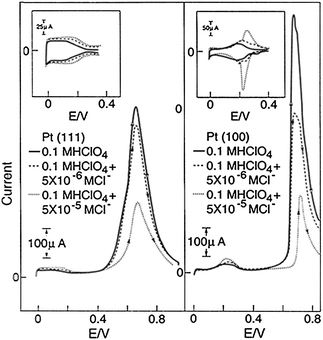 | ||
| Fig. 8 Cyclic voltammetry for flame-annealed Pt(100) in 0.1 M HClO4 with addition of HCl. Inset: the effect of Cl−on hydrogen adsorption-desorption pseudocapacitance. | ||
 | (1) |
In deriving eqn 1 it is assumed that while the (1 − Θad) term is determined by the surface coverage by blocking adsorbates (Hupd, OHad, anions), the reactive intermediates (O2−)ad and (OH−)ad are adsorbed only at a low coverage, i.e. they are not a significant part of Θad. All these factors are uniquely related to the electronic properties of electrode materials and the nature of solution used, either through the adsorption energy of reaction intermediates, which is controlled by the position of the d-band center,39,40 or through the potential of zero charge (Epzc),41 which defines the onset potential of adsorption of spectator species. In what follows, we use this simple rate equation to discuss the role of anions in the ORR on IB group metals in acidic media. For comparison, the corresponding results for the ORR on Pt(111) are also included in the same figure.
Comparison between the cyclic voltammograms and the ORR on the IB group metals reveals that the order of activity is closely related to the fractional surface coverage by spectator anion/OHad species at the constant potential, which for the IB metals increases from Cu to Ag to Au. This, in turn, is related to the values of Epzc of coinage metals. Evaluating Epzs (i.e., the potential at which the surface charge Q equals 0) of metal–aqueous electrochemical interfaces has long been recognized as a key requirement for understanding the double-layer properties of these important systems. However, the experimental evaluation and even the meaning of Epzc for metal interfaces are complicated by the occurrence of potential-dependent chemisorption. As a consequence, it is very difficult, if not impossible, to evaluate the exact values for Epzc. Because of that, a useful consideration will be to find the trend in Epzc on the IB group metals and to explore how this trend correlates with the adsorption of spectator species, and thus with the surface activity. Following the relationship between the Epzc and the work function (Φ) measured in vacuum42 it is clear that Epzc on IB group metals should increase in the order Cu(111) < Ag(111) < Au(111). Considering that the onset of adsorption of anions from supporting electrolytes should follow the same trend as Epzc, it is not surprising that the anion adsorption from electrolyte is observed first on Cu than on Ag and finally on Au (see Fig. 9). Therefore, the present results indicate that the major effect of Epzc is in controlling the adsorption isotherm of spectator species, i.e., the (1 − Θad) term in eqn (1), and thus the availability of active metal sites at the constant overpotential for adsorption of O2 and intermediates. Despite our conclusion about the importance of Epzc in surface electrochemistry of the ORR, there are number of challenges that need to be met in the future in order to understand electrocatalysis on atomic and molecular levels. Of those, a fundamental issue concerns the developments of theoretical and/or computational framework that can at least rationalize, and ultimately understand, the importance of Epzc in surface electrochemistry.
 | ||
| Fig. 9 (a) Cyclic voltammograms of (111) single crystal orientation of Au, Ag, Cu and Pt in 0.1 M HClO4 at 298 K, and 50 mV s−1. CVs are shifted along the i-axis from the origin for clarity. (b) ORR polarization curves in positive going sweep at 50 mV s−1 and 1600 rpm; (b″) corresponding currents detected on the ring electrode at 1.2 VRHE. | ||
2.5 Surface restructuring
Finally, we now turn the discussion on the role of anions in restructuring of Pt surface atoms, where only few selected systems have been studied so far. In general, modern surface crystallographic studies have shown that on the atomic scale even the most clean metal surfaces tend to minimize their surface energy by two kinds of surface atom rearrangements, relaxation and reconstruction, which collectively may be called restructuring. Relaxation of metal surfaces is usually defined as a small interlayer spacing change relative to the ideal bulk lattice.43–47 The displacements should be small compared to the near-neighbor distance, such that no bond-breaking/bond-making events take place within the substrate. Adsorbate induced relaxations occur in many varieties: as interlayer spacing changes, where in the near-surface region the top layer of metal atoms relaxes either inward or outward from the bulk atoms; lateral relaxation, in which adsorbates shift near-surface atoms parallel to the surface (frequently inducing collective rotation of substrate atoms around the adsorbate sites); and layer buckling, whereby a coplanar atomic layer loses its coplanaraty because of an adsorbate pulls or pushes some substrate atoms out of the plain relative to the lattice atoms. Reconstruction, on the other hand, involves large atomic displacements both perpendicular to and parallel to the surface plane, so that causes re-bonding and change in the periodicity within the substrate. In our laboratory, the examination of surface restructuring has been persuaded by means of both SXS and STM, especially for the effect of anions on lifting the surface reconstruction of Au(hkl),11halide effects on relaxation of Pt surface atoms and very recently on changing the surface morphology of Pt surface atoms by fast mass transport of adatoms from the terrace to the step sites. For the latter system, Fig. 10 shows such an example of STM images for the effect of Br− on restructuring the “perfect” Pt(111) surface in 0.1 M HClO4. The protocol in these experiments was the same as one described recently in ref. 48: (1st step) in an electrochemical cell twenty electrochemical oxidation–reduction cycles in Ar saturated solution were applied from 0.005 V to 0.95 V; (2nd step) following this so-called “oxide annealing” preparation method, the electrode was held at 0.05 V and covered with COad; (3rd step) the CO-protected electrode was transferred into the STM cell, and (4th step) after the STM images were obtained, the electrode was returned to the experimental cell to record cyclic voltammetry. As shown in Fig. 10, STM images for the oxide-annealed surfaces are dominated by large clusters with diatomic (0.5 nm) and some triatomic (0.75 nm) heights which are randomly distributed over the (111) surface terraces. In the presence of Br−, not only is a reduction in the number and size of the islands observed after Br-annealing up to 0.95 V but, in addition, adislands completely disappear and are replaced by a series of smooth terrace-step structures. Since this transformation only occurs after Br-annealing up to 0.95 V, we conclude that it is Br− coadsorption with OH−, as opposed to simple Br− adsorption, that leads to step formation. It is plausible that Br− acts as a surfactant that transports adatoms from terraces to steps. Step formation therefore appears to involve a delicate balance between the tendency of adsorbed oxygenated species to induce adisland formation at high potentials and the contrasting tendency of Brad to facilitate diffusion of adatoms/adislands across the surface with concomitant incorporation into step edges. The above example shows unique ability of anions to trigger stepwise alternations in surface stability and morphology viaelectrode potential and much more can be learned regarding the anion role in controlling mobility and ordering of Pt surface atoms that is utterly inaccessible in UHV-based systems. | ||
| Fig. 10 STM images (100 × 100 nm size) of Pt(111) covered by CO for: (left) oxide-annealed surface (20 potential cyles) up to 0.95 V and (right) Br-annealed surface. The images (Utip = 0.15 V;Itip = 1 nA) illustrate the presence of islands on oxide-annealed surface and steps on Br-annealed surface. | ||
In summary, this article presented some facets of our current state of physicochemical understanding that anions have in surface electrochemistry. By utilizing both powerful in-situ techniques for structural determination and electrochemical methods for adsorption and kinetic analysis of electrochemical processes, we illustrate the remarkable insight into the effects of specifically adsorbed anions on the adsorption of hydrogen, oxygen, oxygenated species, CO, surface restructuring, stability of platinum surface atoms, kinetics of fuel cell reactions, and metal deposition of Cu.
Acknowledgements
This work was supported by the contract (DE-AC02-06CH11357) between the University of Chicago and Argonne, LLC, and the US Department of Energy.References
- N. M. Markovic and P. N. Ross, Surf. Sci. Rep., 2002, 45, 117 CrossRef CAS.
- O. M. Magnussen, Chem. Rev., 2002, 102, 679 CrossRef CAS.
- B. E. Conway, in Progress in Surface Science, ed. S. Davison, Pergamon Press, Fairview Park, NY, 1984, p. 1 Search PubMed.
- J. C. Huang, W. E. O'Grady and E. Yeager, J. Electrochem. Soc., 1977, 124, 1732 CrossRef CAS.
- K. Itaya, Prog. Surf. Sci., 1998, 58, 121 CrossRef CAS.
- M. G. Samant, M. F. Toney, G. L. Borges, K. F. Blurton and O. R. Melroy, J. Phys. Chem., 1988, 92, 220 CrossRef CAS.
- B. M. Ocko, J. Wang, A. Davenport and H. Isaacs, Phys. Rev. Lett., 1990, 65, 1466 CrossRef CAS.
- M. F. Toney and B. M. Ocko, Synchrotron Radiat. News, 1993, 6, 28 Search PubMed.
- I. M. Tidswell, N. M. Markovic and P. N. Ross, Phys. Rev. Lett., 1993, 71, 1601 CrossRef CAS.
- C. Lucas, N. M. Markovic and P. N. Ross, Surf. Sci., 1996, 340, L949.
- D. M. Kolb, Prog. Surf. Sci., 1996, 51, 109 CrossRef CAS.
- J. Clavilier, J. Electroanal. Chem., 1980, 107, 211 CrossRef CAS.
- N. Markovic, M. Hanson, G. McDougall and E. Yeager, J. Electroanal. Chem., 1986, 214, 555 CrossRef CAS.
- M. Wasberg, L. Palaikis, S. Wallen, M. Kamrath and A. Wieckowski, J. Electroanal. Chem., 1988, 256, 51 CrossRef CAS.
- L. A. Kibler, M. Cuesta, M. Kleinert and D. M. Kolb, J. Electroanal. Chem., 200, 484, 73 Search PubMed.
- F. Will, J. Electrochem. Soc., 1965, 112, 451 CAS.
- A. T. Hubbard, R. M. Ishikawa and J. Katekaru, J. Electroanal. Chem., 1978, 86, 271 CrossRef CAS.
- A. S. Homa, E. Yeager and B. D. Cahan, J. Electroanal. Chem., 1983, 150, 181 CrossRef CAS.
- F. T. Wagner and P. N. Ross Jr., J. Electroanal. Chem., 1983, 150, 141 CrossRef CAS.
- N. Markovic and P. N. Ross, J. Electroanal. Chem., 1992, 330, 499 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, J. Phys. Chem., 1995, 99, 3411 CrossRef CAS.
- M. Arenz, V. Stamenkovic, T. J. Schmidt, K. Wandelt, P. N. Ross and N. M. Markovic, Surf. Sci., 2003, 523, 199 CrossRef CAS.
- I. Villegas and M. J. Weaver, J. Chem. Phys., 1994, 101, 1648 CrossRef CAS.
- I. Villegas, X. Gao and M. J. Weaver, Electrochim. Acta, 1995, 40, 1267 CrossRef CAS.
- N. M. Markovic, B. N. Grgur, C. A. Lucas and P. N. Ross, J. Phys. Chem. B, 1999, 103, 487 CrossRef CAS.
- A. Wieckowski, M. Rubel and C. Gutiérrez, J. Electroanal. Chem., 1995, 382, 97 CrossRef CAS.
- H. Kita, H. Naohara, T. Nakato, S. Taguchi and A. Aramata, J. Electroanal. Chem., 1995, 386, 197 CrossRef CAS.
- A. Rodes, R. Gomez, J. M. Feliu and M. J. Weaver, Langmuir, 2001, 16, 811.
- N. M. Markovic, C. Lucas, A. Rodes, V. Stamenkovic and P. N. Ross, Surf. Sci., 2002, 499, 149.
- N. M. Markovic, C. Lucas, B. N. Grgur and P. N. Ross, J. Phys. Chem., 1999, 103, 9616 Search PubMed.
- V. Stamenkovic, K. C. Chou, G. A. Somorjai, P. N. Ross and N. M. Markovic, J. Phys. Chem. B, 2005, 1, 1.
- N. Markovic and P. N. Ross, Langmuir, 1993, 9, 580 CrossRef CAS.
- N. M. Markovic, H. A. Gasteiger and P. N. Ross, Langmuir, 1995, 11, 4098 CrossRef CAS.
- N. M. Markovic, C. Lucas, H. A. Gasteiger and P. N. Ross, Jr., Surf. Sci., 1997, 372, 239 CrossRef CAS.
- N. Kobosev and W. Monblanova, Acta Physiochem. URSS, 1934, 1, 611 Search PubMed.
- W. T. Grubb, Nature, 1963, 198, 883 CAS.
- J. O. M. Bockris and A. K. N. Reddy, Modern Electrochemistry, Plenum Press, New York, 1970 Search PubMed.
- T. D. Jarvi and E. M. Stuve, in Electrocatalysis, ed. J. Lipkowski and P. N. Ross, p. 75, Wiley-VCH, Inc., New-York, 1998 Search PubMed.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Nørskov, Angew. Chem., Int. Ed., 2006, 45, 2897 CrossRef CAS.
- B. B. Blizanac, V. Stamenkovic and N. M. Markovic, Z. Phys. Chem., 2007, 221, 1379 CrossRef CAS.
- S. Trasati, J. Electroanal. Chem., 1972, 39, 163 CrossRef CAS.
- G. A. Somorjai and M. A. Van Hove, Prog. Surf. Sci., 1989, 30, 201 CrossRef CAS.
- G. A. Somorjai, Introduction to Surface Chemistry and Catalysis, John Wiley & Sons, New York, 1993 Search PubMed.
- P. A. Thiel and P. J. Estrup, in The Hanbook of Surface Imiging and Visualization, ed. A. T. Hubbard, CRC Press, Boca Raton, 1995 Search PubMed.
- R. I. Masel, Principles of Adsorption and Reaction on Solid Surfaces, John Wiley & Sons, Inc., 1996 Search PubMed.
- M. A. Van Hove, in Physics of Covered Solid Surfaces, ed. Landolt-Boernstein, 1999 Search PubMed.
- D. Srtmcnik, P. Rebec, M. Gaberscek, D. Tripkovic, V. Stamenkovic, C. Lucas and N. M. Markovic, J. Phys. Chem. C, 2007, 111, 18672 CrossRef.
| This journal is © The Royal Society of Chemistry 2009 |