Analysis of 50-y record of surface 137Cs concentrations in the global ocean using the HAM-global database†
Yayoi
Inomata
*,
Michio
Aoyama
and
Katsumi
Hirose
Geochemical Research Department, Meteorological Research Institute, Tsukuba, Ibaraki, 305-0052, Japan. E-mail: yinomata@mri-jma.go.jp; Fax: 29-853-8728; Tel: 29-853-8719
First published on 19th November 2008
Abstract
We investigated spatial and temporal variations in 137Cs concentrations in the surface waters of the global ocean for the period from 1957 to 2005 using the “HAM database – a global version”. Based on the 0.5-y average value of 137Cs concentrations in the surface water in each sea area, we classified the temporal variations into four types. (1) In the North Pacific Ocean where there was high fallout from atmospheric nuclear weapons tests, the rates of decrease in the 137Cs concentrations changed over the five decades: the rate of decrease from the 1950s to the 1970s was much faster than that after the 1970s, and the 137Cs concentrations were almost constant after the 1990s. Latitudinal differences in 137Cs concentrations in the North Pacific Ocean became small with time. (2) In the equatorial Pacific and Indian Oceans, the 137Cs concentrations varied within a constant range in the 1970s and 1980s, suggesting the advection of 137Cs from areas of high global fallout in the mid-latitudes of the North Pacific Ocean. (3) In the eastern South Pacific and Atlantic Oceans (south of 40 °S), the concentrations decreased exponentially over the five decades. (4) In the Arctic and North Atlantic Oceans, including marginal seas, 137Cs concentrations were strongly controlled by discharge from nuclear reprocessing plants after the late 1970s. The apparent half-residence times of 137Cs in the surface waters of the global ocean from 1970 to 2005 ranged from 4.5 to 36.8 years. The apparent half-residence times were longer in the equatorial region and shorter in the higher latitudes. There was no notable difference between the latitudinal distributions of the apparent half-residence times in the Pacific and Indian Oceans. These results suggest that 137Cs in the North Pacific Ocean is transported to the equatorial, South Pacific, and Indian Oceans by the oceanic circulation.
Introduction
Caesium-137 (137Cs), with a half-life of 30.17 y, is the most abundant anthropogenic radionuclide in the marine environment. The main sources of 137Cs are global and local fallout from atmospheric nuclear weapons tests, discharges from nuclear reprocessing plants, and fallout from the Chernobyl accident.1–5 Sixty-nine percent of 137Cs is derived from global fallout, 21% from local fallout, 7% from reprocessing plant discharge, and 3% from the Chernobyl accident.6 Other sources, such as sea-dumping of nuclear waste, routine discharges from nuclear power plants, nuclear reactors, satellite failures, lost nuclear weapons, and the use of radioisotopes in industry and medicine, are minor contributors to the radioactive contamination of the global ocean.6,7The concentrations of 137Cs in surface waters of the global ocean differ from one region to another, depending on the source and injection time, the spatial scale of injection, and currents in the ocean. Higher global fallout of 137Cs occurred mainly in the western part of the North Pacific Ocean (30–45 °N) and in the North Atlantic Ocean (30–50 °N) in the late 1950s and 1960s.8 The peaks 137Cs deposition in the Southern Hemisphere were lower than peaks in the Northern Hemisphere, and it occurred about 1 year later than the Northern Hemisphere peaks as a result of air mass exchange between the Northern and Southern Hemispheres.9,10 The relatively low number of nuclear explosions conducted by France and the United Kingdom also contributed to the small amount of fallout in the eastern Southern Hemisphere. In the Arctic and Atlantic Oceans with marginal seas, discharges from nuclear reprocessing plants are a major source of contamination by 137Cs.6,11–13 Discharges from two nuclear reprocessing plants, the Sellafield plant located on the Irish Sea on the northwest coast of the United Kingdom and La Hague facility located on the northwest coast of France on the English channel, have been the major source of radioactive contamination of the North Atlantic Ocean since 1952.6,14,15 The discharge of 137Cs peaked in the mid- to late 1970s. The fallout from the Chernobyl accident in 1986 contributed notably to 137Cs concentrations in seawater in the Baltic Sea, the Black Sea, and the Mediterranean Sea. In the North Pacific Ocean, this fallout occurred as a single pulse in the region north of latitude 30 °N.16–18 Local underwater nuclear weapons tests, the nuclear weapons accident at Thule, Greenland, and dumping of reactors and radioactive waste in the Kara Sea have all resulted in local peculiarities in the distributions of 137Cs in the Arctic Ocean.19
In the marine environment, 137Cs exists mainly in a dissolved form, and it moves along with water masses. Therefore, 137Cs is considered as one of the useful chemical tracers for the investigation of oceanic processes such as transport of water masses over long distances, and processes occurring in the water column associated with the global ocean water circulation.15,20–23 Furthermore, 137Cs is used in the assessment of anthropogenic radioactive doses to the world population through marine food.24 Concentrations of 137Cs in the global ocean water have been measured over the last five decades by Bundesamt fur Seeschiffapparent und Hydrographie (BSH, Germany), Deutsch Hydrographic Institute German (DHIG, Germany), International Atomic Energy Agency-Marine Environmental Laboratory (IAEA-MEL), Institute of Meteorology and Water Management (IMWM, Poland), Ministry of Agriculture, Fisheries and Food (MAFF, the United Kingdom), Meteorological Research Institute (MRI, Japan), Maritime Safety Agency (MSA, Japan), Riso National Laboratory (RISO, Denmark), and the National Board of Nuclear Safety and Radiation Protection (NBNSRP, Germany) and by projects such as Arctic Monitoring Assessment Programme (AMAP), Geochemical Ocean Sections program (GEOSECS), South Atlantic Ventilation Experiment (SAVE), Transit Tracers in the Ocean (TTO), World Ocean Circulation Experiment (WOCE), and Worldwide Marine Radioactivity Studies (WOMARS).
There are several databases for anthropogenic radionuclide. The “HAM database - a global version” (hereafter, “HAM-global”) was constructed by Aoyama et al.25 and the Global Marine Radioactivity Database (GLOMARD) by IAEA.4,26–29The HAM-global includes 137Cs, 90Sr, and 239,240Pu concentration data in the global ocean measured from 1957 to 2005. HAM-global is available for investigating long-term variation of the 137Cs concentrations in the global ocean over the past five decades. In addition, variations in the 137Cs concentrations can provide important clues for understanding physical oceanographic processes on a decadal scale.
In this paper, we analyze spatial and temporal variations in 137Cs concentrations over five decades in surface waters of the global ocean by using the data in the HAM-global, and we present the temporal variations of 137Cs concentrations in surface waters of each sea area in the global ocean. We introduce 0.5-y average values of 137Cs concentrations for each sea area. Furthermore, we discuss the transport processes affecting surface 137Cs in the global ocean over the past five decades based on the apparent half-residence times.
Data and methods
Database: HAM-a global version
We used the database “Historical Artificial Radionuclides in the global ocean and its marginal seas; HAM database–a global version” (HAM-global) for data analysis,25 which is the updated version of the HAM database.30 The HAM-global includes 31,378 records for 137Cs, 7062 for 90Sr, and 3871 for 239,240Pu concentration data. These data were measured from 1957 to 2005. Of these records, the 22,368 for 137Cs that were obtained from surface waters (0–10 m), were used in this study. The geographical distribution of the sampling locations for these 137Cs data is heterogeneous over the past five decades25 as shown in Fig. S1 (ESI).† The 137Cs concentrations in surface seawater were measured over large areas of the western North Pacific Ocean in the 1960s and 1970s. The number of 137Cs measurements in the Pacific Ocean decreased in the 1980s and 1990s, except in the western North Pacific Ocean and the Sea of Japan. In the Atlantic Ocean, numerous measurements were taken in the northern Atlantic Ocean and its marginal seas in the 1970s and 1980s by several institutes to monitor the fate of discharged radioactivity. In the 1990s and 2000s, the number of 137Cs measurements decreased, and most measurements during this period come from the Sea of Japan and the marginal seas of the Atlantic Ocean.HAM-global includes the measurement errors along with some of the data. Where measurement errors were reported, the standard deviation was within 30%.25 Therefore, we used 30% as the standard deviation for the data entries that did not include the measurement error.
Plutonium-239 (239Pu; half-life [T1/2], 24,000 y), plutonium-240 (240Pu; T1/2, 6000 y), and strontium-90 (90Sr; T1/2, 28.5 y) are also important tracers of water transport and biogeochemical processes. To better understand the transport and distribution of 137Cs in surface seawater, the 239,240Pu/137Cs and 137Cs/90Sr activity ratios in the North Pacific Ocean were also investigated. The data of the 239,240Pu/137Cs and the 137Cs/90Sr activity ratios were 358 and 2238 records, respectively. The cumulative decay corrected radioactivity ratios for 239,240Pu/137Cs since 1945 are also estimated. With respect to the other oceans, the records available for the analysis were only a few. We, therefore, investigated the temporal variations of these ratios in the North Pacific Ocean.
To study the distribution of 137Cs concentrations in surface seawater, we divided the global ocean into 34 latitudinal boxes on the basis of known ocean current systems, latitudinal and longitudinal distributions of 137Cs concentrations, the distribution of global fallout, locations of nuclear reprocessing plants, fallout from the Chernobyl accident, and availability of recent data (Fig. 1). The classification system used for Boxes 1–30 is the same approach as in several previous studies.5,9,27 Boxes 31–34 are novel classifications in this study. Box numbers and the corresponding locations in the world ocean and area abbreviations are listed in Table S1.†
 | ||
| Fig. 1 Latitudinal boxes in the global ocean. (a) the Pacific, Indian, and Atlantic Oceans, (b) Northern European Sea. Box 19 is not assigned. | ||
The 0.5-year average values
The 0.5-y average values of surface 137Cs concentrations are useful for verifying ocean circulation models31,32 and are also used to assess radiation doses to the world population via the marine food supply24,33. Based on the measured data, therefore, we calculated 0.5-y average values of the surface concentrations in each box taking into account advection from the adjacent sea areas (Table S2 in ESI†). The 0.5-y average values for BALT, IRIS, ENGC, BLAS, and MEDS (see Table S1 in ESI† for area abbreviations) cannot be estimated because the surface concentrations were extremely variable owing to the proximity of the discharges from the nuclear reprocessing plants for IRIS and ENGC, and because of heterogeneous fallout from the Chernobyl accident for BALT, BLAS, and MEDS.Estimates of apparent half-residence times
In order to understand global seawater movement by using 137Cs as a chemical tracer, we estimated the apparent half-residence times of surface 137Cs from the corresponding regression lines of their exponential decrease curves, under the assumption that the trend of exponential decrease has continued up to the present. Because global fallout due to atmospheric nuclear tests was a minor contribution after 1970,9 the regression line for each box was determined for the period after 1970. The regression lines in the Arctic and the Atlantic Oceans (Boxes 18, 20–29), except for Box 30, cannot be shown because of the continuing release of large amount of 137Cs from nuclear reprocessing plants and because of fallout from the Chernobyl accident in 1986.The apparent half-residence times of 137Cs in surface waters were estimated by the following equation:
| λCs,ap = λCs,phys + λCs,decay, | (1) |
Results
In this section, we describe the temporal variations of 137Cs concentrations in surface seawater in each box. Note that the maximum observed concentrations and years of occurrence in this study are based on the HAM-global (Table 1). The temporal variations of 137Cs concentrations in surface seawater are displayed in Fig. S2 in the ESI.†| Box | Year | Date | Latitude | Longitude | 137Cs (Bq m−3) | Depth (m) | MRIREFNO b |
|---|---|---|---|---|---|---|---|
| a 30% was used as the standard deviation, because measurement error was not included in the reference. b MRIREFNO is reference of the data and listed in Table S4a and b (ESI). | |||||||
| 1 | 1957 | 26-Sep | 42.00 | 158.00 | 115 ± 11 | 0 | Miyake1961 |
| 2 | 1959 | 11-Sep | 39.57 | 144.13 | 78 ± 15 | 0 | Miyake1961 |
| 3 | 1966 | 21-Jan | 29.88 | 239.97 | 70 ± 8.5 | 0 | Folsom1968 |
| 4 | 1958 | 7-Nov | 24.99 | 134.98 | 122 ± 15 | 0 | Miyake1961 |
| 5 | 1980 | 1-Mar | 16.73 | 190.48 | 47 ± 0.47 | 0 | Noshkin1999 |
| 6 | 1978 | 26-Apr | −1.32 | 145.46 | 10 ± 3.0 | 0 | Domanov1984a |
| 7 | 1978 | 24-Feb | −4.07 | 276.73 | 6.7 ± 2.0 | 0 | Domanov1984a |
| 8 | 1978 | 22-Apr | −7.31 | 158.26 | 8.5 ± 2.6 | 0 | Domanov1984a |
| 9 | 1978 | 2-Apr | −11.51 | 231.89 | 21 ± 6.4 | 0 | Domanov1984a |
| 10 | 1966 | 8-Nov | −38.12 | 178.90 | 12 ± 6.7 | 0 | Folsom1968 |
| 11 | 1966 | 13-Mar | −28.58 | 258.48 | 10 ± 1.5 | 0 | Folsom1968 |
| 12 | 1966 | 28-Aug | −41.32 | 227.73 | 10 ± 3.3 | 0 | Folsom1968 |
| 13 | 1962 | 6-Jan | −67.15 | 67.78 | 6.3 ± 0.4 | 0 | Nagaya1964 |
| 14 | 1964 | 1-May | 41.18 | 137.02 | 26 ± 1.5 | 0 | Nagaya1970 |
| 15 | 1960 | 4-Nov | −9.48 | 113.42 | 9.3 ± 2.8 | 0 | Folsom1968a |
| 16 | 1960 | 27-Oct | −11.97 | 115.43 | 10 ± 3.1 | 0 | Folsom1968a |
| 17 | 1966 | 17-Nov | −55.97 | 175.67 | 17 ± 5.2 | 0 | Folsom1968 |
| 18 | 1981 | 15-Jul | 70.00 | 17.50 | 77 ± 23 | 3 | Casso1984a |
| 19 | 1979 | 31-Aug | 62.00 | 4.00 | 135 ± 41 | 0 | Kautsky1987a |
| 20 | 1986 | 9-May | 54.35 | 11.07 | 2410 ± 506 | 0 | DHIG9997 |
| 21 | 1980 | 15-Jul | 51.73 | 0.88 | 36,304 ± 36 | 0 | MAFF9999 |
| 22 | 1974 | 20-Nov | 54.43 | 356.42 | 204,314 ± 61,294 | 3 | MAFF9999a |
| 23 | 1974 | 13-Dec | 50.50 | 1.00 | 287 ± 86 | 0 | MAFF9999a |
| 24 | 1967 | 24-Jul | 58.57 | 356.22 | 1,409 ± 423 | 0 | MAFF9999a |
| 25 | 1986 | 22-Sep | 41.41 | 29.32 | 360 ± 2.8 | 2 | Busseler1991 |
| 26 | 1986 | 1-May | 43.52 | 16.45 | 245 ± 19.2 | 0 | Franic1993 |
| 27 | 1980 | 10-Aug | 33.12 | 288.56 | 31 ± 0.34 | 2 | Cochran1987 |
| 28 | 1978 | 13-Feb | 13.18 | 296.92 | 21 ± 6.2 | 0 | Domanov1984a |
| 29 | 1961 | 2-Aug | −40.65 | 304.03 | 5.4 ± 0.35 | 0 | Broecker1968 |
| 30 | 1966 | 14-Mar | 48.50 | 153.67 | 18 ± 1.1 | 0 | Folsom1968 |
| 31 | 1968 | 15-Jul | 30.90 | 129.00 | 10 ± 2.2 | 0 | Saruhashi1975 |
| 32 | 1966 | 22-Jun | 20.20 | 115.32 | 16 ± 0.74 | 0 | Folsom1968 |
| 33 | 1966 | 16-Feb | 54.67 | 175.00 | 23 ± 5.6 | 0 | Folsom1968 |
North Pacific Ocean (Boxes 1–3)
In Box 1 (subarctic NPO), the largest global fallout of 137Cs from the nuclear weapons tests occurred in the 1960s. The maximum 137Cs concentration (115 ± 11 Bq m−3) was observed in 1957. The rate of concentration decrease was much faster from the 1950s to the 1970s than rates after the 1970s. Following the 1970s, the concentrations decreased exponentially at a slower rate over the next two decades. The concentrations have remained almost constant at 0.9–3.6 Bq m−3 since the 1990s.Similar temporal variations are found in Box 2 (western NPO). The maximum 137Cs concentration (78 ± 15 Bq m−3) was observed in 1959. The concentrations from 1990 to 2005 were almost constant, ranging between 1.1 and 3.2 Bq m−3.
The temporal variation in Box 3 (eastern NPO) is markedly different from that in Box 2 (western NPO), even though these boxes are located in the same range of latitudes (25–40°N). The concentrations increased from 1959 to 1966, and the maximum concentration of 70 ± 8.5 Bq m−3 was observed in 1966. The concentrations decreased exponentially from 1970 to 1992. Note that the maximum concentration occurred about 3 years after the maximum deposition rate in 1963.9
Subtropical and equatorial Pacific Ocean (Boxes 4–9)
In Box 4 (western NPO), the maximum surface 137Cs concentration of 122 ± 15 Bq m−3 was observed in 1958. After the 1960s, the concentrations decreased exponentially. The rate of the decrease became increasingly lower and concentrations from 1990 to 2005 were almost constant at 1.6–3.7 Bq m−3.In Box 5 (subtropical eastern NPO), the 137Cs concentrations increased after 1960, and the maximum value of 47 ± 0.5 Bq m−3 was observed in 1967. The concentrations in the latter half of the 1960s and the early 1970s showed large spatial variation. Concentrations in the 1980s were similar to those in the 1970s, after which the concentrations decreased. The occurrence of the maximum concentration in the subtropical eastern NPO was delayed 1 year relative to the eastern NPO, located upstream in the general surface circulation.
In Box 6 (equatorial western PO), the 137Cs concentrations were almost constant from the 1960s through the 1980s. After 1990, the concentrations decreased exponentially. The 137Cs concentrations in Box 7 (equatorial eastern PO) showed no temporal changes from 1960 to 1990, although the concentrations in the latter half of the 1960s showed large variation. In Box 8 (subtropical western SPO), the prominent feature is the gradual increase in the temporal variations of the concentrations during the period from the 1960s to the 1980s. After 1980, the surface concentrations decreased exponentially.
In Box 9 (subtropical eastern SPO), there was large variation in the surface 137Cs data obtained before 1980. The maximum concentration (21 ± 6.4 Bq m−3) was observed in 1978. The large variability of the surface 137Cs reflects the spatially heterogeneous distribution in the subtropical eastern SPO. After 1980, the surface concentrations decreased exponentially.
South Pacific and Antarctic Oceans (Boxes 10–13)
In Box 10 (western SPO), the highest surface 137Cs concentration (12 ± 6.7 Bq m−3) was observed in 1966. The concentrations were almost constant in the range from 2.6 to 4.8 Bq m−3 in the 1970s. In the 1990s, the concentrations were lower than those in the 1970s. In Box 11 (eastern SPO) and Box 12 (eastern SO), the temporal variations in the concentrations were higher in the late 1960s than in the early 1960s. The maximum concentrations in Boxes 11 and 12 were 9.6 ± 3.3 Bq m−3 in 1968 and 9.6 ± 1.5 Bq m−3 in 1966, respectively. After the 1970s, the concentrations decreased exponentially. In Box 13 (ANT), the maximum concentration (6.3 ± 0.4 Bq m−3) was observed in 1962, and the concentrations decreased exponentially thereafter.Marginal seas of the Pacific Ocean (Boxes 14, 31–34)
In Box 14 (SOJ), the surface 137Cs concentrations showed a three-step decrease in the rates of decrease, similar to Boxes 1, 2, and 4. The maximum concentration (26 ± 1.5 Bq m−3) was observed in 1964, and the concentrations decreased faster during the 1960s than in later periods. After the 1970s, the concentrations decreased exponentially. The marked increase in concentrations in 1986 is attributable to fallout from the Chernobyl accident. After 1990, the decrease in concentrations was minimal and concentrations remained within the range from 1.9 to 2.2 Bq m−3 into 2005.The surface 137Cs concentrations in Box 31 (SOO) decreased exponentially, and the concentrations in 2004 were in the range of 0.9–2.2 Bq m−3.
In Box 32 (ECS), the highest surface 137Cs concentration (10 ± 2.2 Bq m−3) was observed in 1968. The concentrations decreased exponentially, and values in the 2000s (about 2.0 Bq m−3) were almost the same as those in the western NPO.
In Box 33 (SCS) and Box 34 (BERS), the surface 137Cs concentrations decreased exponentially. The highest concentrations (16 ± 0.7 Bq m−3 for SCS and 22.6 ± 5.6 Bq m−3 for BERS) were observed in 1964.
Indian Ocean (Boxes 15–17)
In Box 15 (ARB), the maximum 137Cs concentration (9.3 ± 2.8 Bq m−3) was observed in 1960. However, for this period there is no data from the ARB. The concentrations decreased gradually during the next four decades, and the values in 1998 ranged between 1.3 and 2.2 Bq m−3. In Box 16 (IO), the maximum concentration was also observed in 1960 at 10.4 ± 3.1 Bq m−3. The surface concentrations from 1962 to the early 1980s were almost constant. After that, the surface concentrations slowly decreased and values in 1998 were in the range of 2–2.5 Bq m−3. In Box 17 (SO), the surface concentrations showed a spatially heterogeneous distribution from the 1960s through the 1980s. The maximum concentration (17.0 ± 5.2 Bq m−3) was observed in 1966. The concentrations from 1966 to 1990 were apparently almost constant. However, the average concentrations after 1990 were lower (1.2 ± 0.5 Bq m−3) than those in previous years.Arctic Ocean and North Atlantic Ocean and marginal seas (Boxes 18, 20, 22–25)
The dominant feature of 137Cs variations in surface seawater in the Arctic Ocean, North Atlantic Ocean, and its marginal seas is that the concentrations were extremely high with large variability. The variation in the surface concentrations corresponds to the variation of discharge from the nuclear reprocessing plants, which had maximum releases in the mid-1970s.15,34 It should be noted that discharges from the nuclear reprocessing plants has been the major sources of 137Cs concentrations after the mid-1970s in comparison with those in the concentrations in the 1960s due to the global fallout from atmospheric weapons tests.The maximum concentration (77 ± 23 Bq m−3) in Box 18 (ARC) was observed in 1981. After that, the concentrations decreased rapidly, and the concentrations in 1995 ranged from 0.4 to 4.9 Bq m−3. There was no indication of marked additional sources by the dumping of radioactive wastes in the ARC.35,36
Surface 137Cs concentration in Box 20 (BARE) decreased exponentially from the maximum value of 135 ± 41 Bq m−3 in 1979 to 2.4 ± 0.2 Bq m−3 in 1995.
In Box 22 (NORS), the surface 137Cs concentrations in the 1960s ranged from 5.6 to 279 Bq m−3. The concentrations rapidly increased after 1976 and reached the maximum value of 36.3 ± 0.04 kBq m−3 in 1980. After that, the concentrations decreased exponentially, and those in the 2000s ranged from 1.9 to 28 Bq m−3. The large spatial variability in the concentrations reflects the heterogeneous distribution of 137Cs in this area. Remobilization of 137Cs from highly contaminated sediments in the IRIS is the dominant source for recently elevated 137Cs in seawater in the NORS.37
In Box 23 (IRIS), the surface 137Cs concentrations in the 1960s ranged between 12 and 20,190 Bq m−3 because of global fallout. The concentrations rapidly increased after the early 1970s in direct response to discharges from the Sellafield nuclear reprocessing plant. The concentrations peaked in the mid- to late 1970s, and the maximum concentration (204 ± 61.2 kBq m−3) was measured in 1974. The concentrations in the IRIS decreased sharply in proportion to the lower rates of radioactive release from the Sellafield plant throughout the 1980s, and between 1995 and 2000,38 they showed no significant change, ranging between 0.3 and 13 Bq m−3. Because the release of 137Cs stopped at the end of 1998, the fluctuation of concentrations in 1995 and 2000 could have been caused by resuspension of sediments and remobilization of 137Cs in the water column.37,39 The impact of the Sellafield discharges in the IRIS was large in comparison with the effects of global fallout due to nuclear weapons testing in the atmosphere prior to the limited test ban treaty of 1963, and fallout from the Chernobyl accident in 1986.40
In Box 24 (ENGC), the maximum surface 137Cs concentration (287 ± 86 Bq m−3) was observed in 1972 owing to discharge from the La Hague nuclear fuel reprocessing facility. After 1980, the concentrations decreased exponentially until 2000. The large fluctuations in this area are related to the variability in the transport of 137Cs from the point of discharge under the current.41
In Box 25 (NNA), trends of the surface 137Cs concentrations were of three types. The first peak with a maximum concentration (1409 ± 423 Bq m−3) measured in the 1960s was attributable to global fallout. Concentrations after 1970 increased and attained a maximum value (1094 ± 4 Bq m−3) in 1980. The concentrations after the 1990s, with a range from 0.9 to 18 Bq m−3, rapidly decreased in proportion to decreases in the release rates from the reprocessing plants.
Baltic Sea (Box 21), Black Sea (Box 26), and Mediterranean Sea (Box 27)
The fallout due to the Chernobyl accident as well as global fallout markedly influenced the surface 137Cs concentrations in the Baltic Sea, Black Sea, and Mediterranean Sea.42,43In Box 21 (BALT), the surface 137Cs concentrations rapidly increased to the maximum value of 2410 ± 506 Bq m−3 in April 1986 as a result of the Chernobyl accident. The range of concentrations just after the accident, measured in May and June 1986, was between 51 and 2410 Bq m−3, which was two orders of magnitude higher than the concentrations in the previous years (12.9–31 Bq m−3). The large variability in the concentrations was due to uneven deposition of the Chernobyl 137Cs in the BALT.44–48 The concentration levels in 2003 (24.9–52.9 Bq m−3) were still high by about a factor of two in comparison with the pre-Chernobyl levels, owing to the closed nature of the BALT and the small exchange of water between the BALT and the NORS. In recent years, BALT has been a main source of 137Cs inflow to the NAO.49
The 137Cs concentrations in Box 26 (BLAS) just after the Chernobyl accident (360 ± 2.8 Bq m−3) were at least 20 times those before the accident (13–22 Bq m−3). The concentrations in the 2000s were similar to those measured in the 1980s. These concentration levels are affected by inflow both from rivers and from Mediterranean waters entering through the Bosphorus.50 Decreases in the 137Cs concentrations in BLAS are caused by radioactive decay, penetration of surface-water137Cs into the deep layers,20 and 137Cs removal through the Bosporus Strait into the MEDS.51
The surface concentrations in Box 27 (MEDS) varied between 0.37 and 43 Bq m−3 before the Chernobyl accident. At the time of the accident in 1986, the concentrations increased to 245 ± 19 Bq m−3. Then, the concentrations rapidly decreased, and the surface concentrations in 1998 ranged between 1.4 and 3.3 Bq m−3. The rapid decrease of the surface 137Cs could be due to the combined effects of radioactive decay and the intrusion of bottom water to the surface.52–55
Atlantic Ocean (Box 28–30)
In Box 28 (NAO) and Box 29 (CAO), the surface 137Cs concentrations in the 1960s and 1970s resulted from global fallout56 and ranged from 3.0 to 23 Bq m−3 in the NAO and from 0.82 to 13 Bq m−3 in the CAO. Increases in the concentrations in the late 1970s and early 1980s are explained by the southward transport of 137Cs discharged from the nuclear reprocessing plants.57The surface 137Cs concentrations in Box 30 (SAO) decreased exponentially after 1960. The highest concentration (5.4 ± 0.4 Bq m−3), caused by global fallout, was observed in 1961.58 The concentrations in 1993 ranged from 0.6 to 1.1 Bq m−3. The 137Cs concentrations in the 1990s (0.8 ± 0.3 Bq m−3) are similar to those in 2002 (0.7 ± 0.2 Bq m−3).59
Discussion
Comparison of 0.5-y average values in the global ocean
We compared the 0.5-y average 137Cs concentration values in each box (Table S2 in the ESI†). Boxes are grouped by geographic region and dominant sources of 137Cs. In the North Pacific Ocean (Boxes 1–3; Fig. 2a), the most prominent features are the three distinct steps in the rate of decrease of the surface concentrations, and the almost constant concentrations after 1990. Before 1970, there is a marked difference between concentrations in the western and eastern NPO, as expected from the geographical patterns of global fallout. This difference reflects complicated processes including west–east differences in the seasonal changes of deposition and in the surface mixed layer in addition to eastward advection. The depth of the surface mixed layer differs between the western and eastern NPO; it is deeper in winter and shallower in summer in the mid-latitudes of the western NPO, whereas it is shallow throughout the year in the mid-latitudes of the eastern NPO.9 The delay between the maximum concentration in the eastern NPO compared with the western NPO suggests that 137Cs deposited on the sea surface in the western NPO was transported eastward and remained in the shallow surface mixed layer in the eastern NPO. These processes have been recognized by processing surface 137Cs concentration histories with the OGCM.31 After 1970, there was no noticeable difference in concentrations between the two areas. The decrease in concentrations after the 1970s may be explained by physical oceanographic processes, such as greater seasonal changes in the surface mixed layer, deep winter convection from cooling, strong seasonal winds, and seasonal changes in radioactive deposition (maximum in spring).9 Following the decrease, there was no notable difference in concentrations in the North Pacific Ocean in the 1990s and 2000s. The circulation patterns in the western North Pacific Ocean are well known; the North Equatorial Current, which flows westward along 10–20 °N to the Philippine Islands, is the dominant current. The western part of the North Equatorial Current connects to the Kuroshio Current.5,60 Recently, subsurface cores with high 137Cs concentrations were discovered in the North Pacific subtropical gyre.9,61 Because there is active vertical mixing of seawater in the mid-latitudes, as compared with the equatorial region, it is possible that 137Cs level in the North Pacific subtropical gyre surface water into the 1990s has been kept constant, which supported by supply due to upward transport from the subsurface 137Cs cores as a result of this active vertical mixing or else from boundary upwelling near the Philippine Islands. This means that recent 137Cs levels measured in the North Pacific subtropical gyre have been augmented by recirculation processes. The temporal variations in surface 137Cs concentrations in the marginal seas of the Pacific Ocean (SOJ, SOO, ECS, SCS, BERS) also show a three-step decreasing trend, as observed in the North Pacific Ocean (subarctic NPO, western NPO, eastern NPO; Fig. 2b). After 1990, there were no obvious differences between concentrations in the North Pacific Ocean and its marginal seas. | ||
| Fig. 2 The 0.5-y average values for 137Cs concentrations in surface seawater in each box. (a) Boxes 1–3 (subarctic NPO, western NPO, eastern NPO); (b) Boxes 14 (SOJ) and 31–34 (SOO, ECS, SCS, BERS); (c) Boxes 4–9 (subtropical western NPO, subtropical eastern NPO, equatorial western PO, equatorial eastern PO, subtropical western SPO, subtropical eastern SPO); (d) Boxes 10–13 (western SPO, eastern SPO, eastern SO, ANT); (e) Boxes 15–17 (ARB, IO, SO); and (f) Boxes 18 (ARC), 20 (BARE), 22 (NORS), 25 (NNA), and 28–30 (NAO, CAO, SAO). | ||
The temporal variations of the surface 137Cs concentrations in the equatorial and South Pacific Oceans (subtropical western and eastern NPO, equatorial western and eastern PO, subtropical western and eastern SPO; Fig. 2c) showed marked latitudinal differences before 1990; concentrations in subtropical western and eastern NPO were higher than those in areas to the south (equatorial western and eastern PO, subtropical western and eastern SPO). In addition, the concentrations in the equatorial and South Pacific Oceans (subtropical western and eastern NPO, equatorial western and eastern PO, subtropical western and eastern SPO) were almost equal to each other from the 1960s to the 1990s. This suggests that concentrations in the equatorial western and eastern PO have gradually increased through southward transport of higher 137Cs water from the mid-latitudes of the North Pacific Ocean through the processes of global ocean circulation, when corrected for the radioactive decay of 137Cs. After 1990, there was no marked latitudinal difference in the concentrations between the subtropical NPO and equatorial PO. This suggests that the surface concentrations were homogeneous in the North Pacific Ocean during the 1990s and the 2000s. To maintain these relatively constant concentrations, there must be a supply of 137Cs to surface waters. Considering that the 137Cs deposition as measured at the MRI, Japan, were in the range of 140 to 350 m Bq m−2 year−1 through the 1999s,62 the contribution of atmospherically deposited 137Cs is negligible compared to the 137Cs concentrations in the surface seawater of the North Pacific Ocean. According to Hamilton et al.,63 most of the 137Cs (95–99%) deposited into the North Pacific Ocean remains in the water column. This suggests that the surface concentrations in the Pacific Ocean could be supported by a supply of water with high 137Cs levels. Results of previous studies show that subsurface cores with high 137Cs concentrations exist in the subtropical North Pacific Ocean.9,61 The high-137Cs cores were formed by subduction of North Pacific Subtropical Mode Water and lighter Central Mode Water in the interior ocean during the past four decades.61 Considering that vertical mixing is active in the mid-latitude region compared with that in the equatorial Pacific Ocean, 137Cs in surface seawater would be supplied from the subducted 137Cs associated with the vertical mixing.
The surface 137Cs concentrations in the South Pacific Ocean (western SPO, eastern SPO, eastern SO) and the Antarctic Ocean (ANT) decreased exponentially (Fig. 2d). Note that the concentrations in the western SPO were relatively higher than those in the eastern SPO, eastern SO, and ANT, suggesting the existence of transport of 137Cs from the equatorial PO. Compared with the western SPO, the surface concentrations in the eastern SPO, eastern SO, and ANT after 1980 were some of the lowest in the Pacific Ocean. In particular, ANT is the area in the global ocean least contaminated by 137Cs, as suggested by Aarkrog.64 These latitudinal differences can be primarily explained by the geographical distribution of radioactive deposition: a small number of nuclear explosions in the eastern Southern Hemisphere,65 limited inter-hemisphere transport of the atmospheric nuclear weapons test-derived 137Cs from the Northern Hemisphere to the Southern Hemisphere,66 and high deposition in the mid-latitude region of both hemispheres because of active stratosphere–troposphere exchange.
In the Indian Ocean (ARB, IO, SO), there is a broad latitudinal variation with higher concentrations in the IO and lower in both ARB and SO (Fig. 2e). Considering that global and local fallout of 137Cs were small in this area, the almost constant levels in the Indian Ocean from the 1960s through the 1980s can be explained by the influx of Pacific Ocean waters through the Indonesian seas, and a system of equatorial currents that recirculates surface water masses in the region.13,27 These processes have been identified by following tritium distributions in seawater.67
In the Arctic and the North Atlantic Oceans, including the marginal seas (Fig. 2f), the most radioactively contaminated sea area was NORS and NNA. The difference in maximum 137Cs concentrations between each area and the time attained these maxima were suggest that 137Cs-labeled seawater due to release from the radioactivity reprocessing plants was transported from IRIS and ENGC through the North Channel to the NORS, NNA, NAO, BARE, and AO by prevailing currents.15,68,69 Compared with the concentrations in the Pacific Ocean and the Indian Ocean, the 137Cs concentrations in the North Atlantic Ocean were notably high. In addition to this, the lower concentrations in these areas (BAR, NORS, IRIS, ENGC, NAO) as shown in Fig. S2 (ESI†) are almost similar levels in the Pacific Ocean and the Indian Ocean. These features suggest that 137Cs concentrations are dominantly controlled by discharges from nuclear reprocessing plants.
Compared with the North Atlantic Ocean, concentrations in CAO and SAO were very low due to low fallout, vertical mixing of the surface water masses, and the influence of less contaminated Antarctic waters.59
Apparent and physical oceanographic half-residence times
We compared the latitudinal distribution of the apparent half-residence times of surface 137Cs concentrations (Fig. 3). The apparent half-residence times in each box ranged from 4.5 to 36.8 years. The most prominent feature is that the apparent half-residence times in the western and eastern Pacific Ocean show a marked latitudinal distribution, with longer residence times near the equator and shorter ones in the mid- to high latitude areas, although the times are also short in the subtropical western SPO. The large uncertainty in the apparent half-residence times in the equatorial region and Southern Hemisphere (between 20 °N and 20 °S; Boxes 5, 7 and 9) was due to the limited data as well as the large sea area included. The latitudinal distribution of the apparent half-residence times in the Indian Ocean was similar to those in the Pacific Ocean. These estimates are similar to those in previous studies.9,27,70 | ||
| Fig. 3 Latitudinal distribution of apparent half-residence times of 137Cs in surface waters of different ocean areas: eastern Pacific Ocean (Boxes 1, 3, 5, 7, 9, 11–13, 34); western Pacific Ocean (Boxes 1, 2, 4, 6, 8, 10, 13, 14, 17, 31–34); Indian Ocean (Boxes 13, 15–17); South Atlantic Ocean (Box 30). The error bars mean 95% confidence level. | ||
The latitudinal variation of the apparent half-residence times may reflect the pathway of basin-scale surface flow: that is, the 137Cs deposited in the subarctic NPO and western NPO was transported eastward (to the eastern NPO) with some penetration under the surface layer. Water with high 137Cs then moved southward following the North Pacific subtropical gyre and was subducted in the subtropical central and eastern North Pacific Ocean. The 137Cs was then transported westward along the equator, where upwelling resulted in increasing the 137Cs concentrations in the equatorial PO9,61,67. The longer apparent half-residence times and the negative values of physical oceanographic half-residence times in the equatorial Pacific Ocean (Fig. S3 in ESI†) suggest a supply of 137Cs from areas of high 137Cs contamination to the equatorial region. In addition, the apparent half-residence times in the IO and ARB were almost the same as in the western South Pacific Ocean. This is a reflection of the inflow of Pacific Ocean water with high 137Cs concentrations via the Indonesian Archipelago. The shorter apparent half-residence times in the subtropical western SPO may be explained by faster transport resulting from outflow of Pacific waters through the Indonesian Seas. The influx of Pacific Ocean water through the Indonesian Seas has been demonstrated by using the distributions of 3H, 90Sr, 137Cs in the Indian Ocean.13,63
Temporal variations of 239,240Pu/137Cs and 137Cs/90Sr radioactivity ratios in the North Pacific Ocean
Fig. 4 shows the temporal variations of the 239,240Pu/137Cs and 137Cs/90Sr activity ratios in the surface water of the North Pacific Ocean and its marginal seas (Boxes 1–7, 14, 31–34). Variations in the 239,240Pu/137Cs activity ratios in the surface waters of the North Pacific Oceans were in the range of (0.15–18) × 103. After the 1990s, the ratio was slightly higher with large variations. These ratios are lower than the cumulative decay corrected deposition ratios of 137Cs and 239,240Pu. Plutonium is effectively removed from surface layers by adsorption onto sinking particles.71 A typical vertical profile shows a surface minimum and a gradual decrease with increasing depth.72 The lower 239, 240Pu/137Cs activity ratios in surface waters compared with the integrated ratios reflect the removal of 239,240Pu associated with biogeochemical processes in the surface water.71 The higher 239,240Pu/137Cs activity ratios with large variability of the ratios suggest that 239,240Pu and 137Cs subducted into the subsurface ocean is upward transported in the surface seawater. In other word, this implies the recirculation of 239,240Pu and 137Cs in the North Pacific Ocean. The variations in the 239,240Pu/137Cs activity ratios, therefore, support the interpretation that 137Cs in the subsurface waters, which is recently measured, comes from upward transport associated with seawater advection.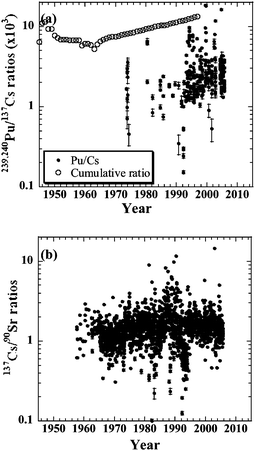 | ||
| Fig. 4 Temporal variations of (a) 239,240Pu/137Cs activity ratios, and (b) 137Cs/90Sr activity ratios in surface waters in the North Pacific Ocean and its marginal seas (Boxes 1–7, 14, 31–34). The cumulative decay corrected 239,240Pu/137Cs deposition ratios since 1945 are also plotted in (a). | ||
The average 137Cs/90Sr activity ratio in the surface waters of the North Pacific Oceans was 1.52 ± 0.82, excluding several data points with unusually high ratios. This value is consistent with the traditionally expected activity ratio in global fallout of 1.45.56 It was reported that an average activity ratio was 1.7 with essentially the same value throughout the Pacific, Atlantic, and Indian Oceans.56 The 137Cs/90Sr radioactivity ratios in the north Pacific Ocean are almost equal to those in the surface seawater in the global ocean.
Conclusions
137Cs is one of several extremely useful chemical tracers of ocean water circulation. In this study, we used the HAM-global database to follow spatial and temporal variations of 137Cs concentrations in the surface waters of the global ocean during the past five decades.The 0.5-y average value of 137Cs concentrations in the surface water in each sea area had been estimated. Based on the 0.5-y average value, we classified the temporal trends of surface 137Cs concentrations during the past five decades into four patterns:
(1) In the western North Pacific Ocean, which includes the area of the highest global fallout because of atmospheric nuclear weapons tests, the rate of decrease in the 137Cs concentrations from the 1950s to the 1970s was much faster than after 1970. After 1990, the surface concentrations were almost constant in the North Pacific Ocean.
(2) In the eastern North Pacific, equatorial Pacific, and Indian Oceans, the surface concentrations in the 1970s and 1980s were almost equal to each other. This suggests that the surface concentrations in these areas increased gradually as a result of the advection of high 137Cs water from the North Pacific Ocean over these two decades. After 1990, the surface concentrations decreased exponentially.
(3) In the eastern South Pacific and Antarctic Oceans, the surface concentrations decreased exponentially after the 1960s. The Antarctic Ocean is still the most uncontaminated area in the global ocean.
(4) The temporal variations of surface 137Cs concentrations in the Arctic Ocean, North Atlantic Ocean and its marginal seas were dominated by discharges from nuclear reprocessing plants. The temporal variations in each box suggest that 137Cs discharged from the nuclear reprocessing plants is transported to the North Atlantic Ocean by prevailing currents.
The apparent-half residence times of 137Cs in surface water in each box of the global ocean, except for north of 40 °S in the Atlantic Ocean, ranged from 4.5 to 36.8 y. The apparent half-residence times showed a clear latitudinal variation, with longer times in the equatorial and subequatorial regions and shorter times in the mid- to high latitudes. There were no noticeable latitudinal differences between the Pacific and Indian Oceans. Latitudinal differences can be explained by the transport of 137Cs in seawater according to global thermohaline circulation. 137Cs from global fallout injected into the mid-latitude region of the western North Pacific Ocean was transported into the eastern Pacific Ocean, where it penetrated under the surface layer of the subtropical gyre. The 137Cs was then upwelled in the equatorial Pacific and transported into the western South Pacific Ocean, as well as into the Indian Ocean through the Indonesian Seas. The temporal variations of the 239, 240Pu/137Cs support our interpretations of the surface water circulation using 137Cs.
Acknowledgements
We thank the scientists and researchers who published the results compiled in HAM database – a global version. The authors thank the IAEA for providing numerous data. We thank Ms. T. Kudo for her work on creating the database. We also thank S. Shimada for her work in preparing the figures. This study is part of the research program “Study of long-term behavior of radionuclides in the ocean” funded by MEXT (the Ministry of Education, Culture Sports, Science and Technology). This study is also supported by a Grant-in-Aid for Science Research (KAKENHI 18310017) of the MEXT.References
- B. G. Bennett, Proceedings of an international symposium on environmental impact of radioactive releaseastern SOrganized, the international atomic energy agency, IAEA-SM-339/131, Vienna, 1995, pp. 3–12 Search PubMed.
- United Nations, Sources and effects of ionizing radiation, United Nations, New York, 2000 Search PubMed.
- H. D. Livingston and P. P. Povinec, A millennium perspective on the contribution of bomb fallout radionuclides to ocean science, Fallout from atmospheric nuclear tests –impact on science and Society, NCRP Annual meeting, IAEA-MEL-03/2001, 2001 Search PubMed.
- P. P. Povinec, K. Hirose, T. Honda, T. Ito, E. Scott and O. Togawa, J. Environ., Radioact., 2004, 76, 113–137 CrossRef CAS.
- IAEA, Worldwide marine radioactivity studies (WOMERS) Radionuclide levels in oceans and seas, IAEA-TECDOC-1429, Vienna, 2005 Search PubMed.
- A. Aarkrog, Deep-Sea Research II, 2003, 50, 2596–2606 Search PubMed.
- H. D. Livingston and P. P. Povinec, Open & Coastal Management, 2000, 43, 689–712 Search PubMed.
- M. Aoyama, K. Hirose and Y. Igarashi, J. Environ. Monit., 2006, 8, 431–438 RSC.
- K. Hirose and M. Aoyama, Deep-Sea Res.II, 2003a, 50, 2675–2700 Search PubMed.
- K. Hirose, M. Aoyama, K. Katsuragi and Y. Sugimura, J. Meteor. Soc. Jn., 1987, 65, 259–277 Search PubMed.
- F. Nyffeler, A. A. Cigna, H. Dahlgaard and H. D. Livingston, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique 1996, pp. 1–22 Search PubMed.
- A. Aarkrog, J. Environ. Radioact., 1994a, 25, 21–35 Search PubMed.
- P. P. Povinec, R. Delfanti, J. Gastaud, J. L. Rosa, U. Morgenstern, B. Oregioni, M. K. Pham, S. Salvi and Z. Top, Deep-Sea Res. partII, 2003a, 50, 2751–2760 Search PubMed.
- P. J. Kershaw, R. J. Pentreath, D. S. Woodhead and G. J. Hunt, A review of radioactivity in the Irish Sea, MAFF Aquatic Environment Monitoring Reastern POrt, 1992, 32, Lowestoft Search PubMed.
- P. P. Povinec, Bailly du Bois, P. Kershaw, P. J. Nies and P. Scotto, Deep-Sea Res. partII, 2003c, 50, 2785–2801 Search PubMed.
- M. Aoyama, K. Hirose and Y. Sugimura, J. Radioanal. Nucl. Chem., 1987, 115, 291–306 CrossRef.
- M. Aoyama, K. Hirose and Y. Sugimura, J. Environ. Radioact., 1991, 13, 103–115 CrossRef.
- M. Aoyama, Geophys. Res. Lett., 1988, 15, 327–330 CrossRef.
- P. Strand, M. Sickel, A. ArkrogJ. M. Bewers, Y. Tsaturov and S. Magnusson, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique 1996, pp. 95–119 Search PubMed.
- J. V. Staneva, K. O. Buesseler, E. V. Stanev and H. D. Livingston, J. Geophys. Res., 1999, 104, 11099–11114 CrossRef CAS.
- T. Miyao, K. Hirose, M. Aoyama and Y. Igarashi, Geophys. Res. Lett., 2000, 27, 3731–3734 CrossRef CAS.
- IAEA, Worldwide marine radioactivity studies (WOMERS), IAEA-MEL, Monaco, 2001a Search PubMed.
- IAEA, Marine Radioactivity Studies in the World Ocean (MARS), IAEA-MEL, Monaco, 2001b Search PubMed.
- A. Aarkrog, M. S. Baxter, A. O. Berttencourt, R. Bojanowski, A. Bologa, S. Vharmasson, I. Cunha, R. Delfanti, E. Duran, E. Holm, R. Jeffree, H. D. Livingston, S. Mahapanyawong, H. Nies, I. Osvath, Li. Pingyu, P. P. Povinec, A. Sanchez, J. N. Smith and D. Swift, J. Environ. Radioact., 1997, 34, 69–90 CrossRef CAS.
- M. Aoyama, K. Hirose, Y. Inomata and J. A. Sanchez-Cabeza, submitted to Progress. Oceanogr.
- P. P. Povinec, H. D. Livingstone, S. Shima, M. Aoyama, J. Gastaud, I. Goroncy, K. Hirose, L. Huynh-Ngoc, Y. Ikeuchi, T. Ito, J. La Rosa, W. K. Liong, L. Lee, H. Moriya, S. Mulslow, B. Oregioni, H. Pettersson and O. Togawa, Deep-Sea Res. partII, 2003b, 50, 2607–2637 Search PubMed.
- P. P. Povinec, A. Aarkrog, K. O. Buesseler, R. Delfanti, K. Hirose, G. Hong, G. T. Ito, H. D. Livingston, H. Nies, V. E. Noshkin, S. Shima and O. Togawa, J. Environ. Radioact., 2005, 81, 63–87 CAS.
- IAEA, World marine radioactivity database (GLOMARD), IAEA-TECDOC-1146, IAEA, Vienna, 2000 Search PubMed.
- P. P. Povinec, J. Gayol and O. Togawa, World marine radioactivity database (GLOMARD), IAEA-SM-354/143P, Vienna, 2001 Search PubMed.
- M. Aoyama and K. Hirose, Sci. World J., 2004, 4, 200–215 Search PubMed.
- D. Tsumune, M. Aoyama and K. Hirose, J. Geophys. Res., 2003, 108 DOI:10.1029/2002JC001434.
- M. Nakano and P. P. Povinec, Deep-Sea Res. partII, 2003, 50, 2803–2816 CrossRef CAS.
- IAEA, sourceastern SOf radioactivity in the marine environment and their relative contributions to overall dose assessment from marine radioactivity (MARDOS), IAEA-TECDOC-838, IAEA, Vienna, 1995 Search PubMed.
- G. T. Cook and A. B. MacKenzie, J. Environ. Radioact., 1997, 35, 227–241 CrossRef CAS.
- A. Aarkrog, H. Dahlgaard and S. P. Nielsen, Sci. Total Environ., 1999, 237/238, 143–151 CrossRef.
- M. S. Baxter, S. Ballestra, J. Gastaud, T. F. Hamilton, I. Harms, L. Huynh-NGOC, L. L. W. Kwong, I. Osvath, P. Parsi, H. Pettersson, P. P. Povinec and A. Sanchez, Proceedings of an international symposium on environmental impact of radioactive releaseastern SOrganized, the international atomic energy agency, IAEA-SM-339/185, Vienna, 1995, pp. 125–141 Search PubMed.
- P. I. Mitchell, P. J. Kershaw, and L. Leon Vintro, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 155–175 Search PubMed.
- P. J. Kershaw, D. C. Denoon and D. S. Woodhead, J. Environ. Radioact., 1997, 44, 191–221.
- N. T. Mitchell and A. K. Steele, J. Environ. Radioact., 1988, 6, 163–175 CrossRef CAS.
- P. Guegueniat, J. Hermann, P. Kershaw, P. Baillu du Bois and Y. Baron, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 121–154 Search PubMed.
- H. Nies, Plutonium and 137Cs in the water column of the Northwest Atlantic: Interim Oceanographic description of the North-East Atlantic site for the disposal of low level radioactive waste, OECD, Paris, 1989a, pp. 77–81 Search PubMed.
- S. Ballestra, R. Bojanowski, R. Fukai and D. Vas, Proc. Int. Symposium on the Behavior of Long-lived Radionuclides in the Marine Environment, Luxembourg, EUR-Reastern POrt 9214: 215–232, 1984 Search PubMed.
- C. Papucci, S. Charmasson, R. Delfanti, C. Gasco, P. Mitchell and J. A. Sanchez-Cabeza, in Radionuclides in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 177–197 Search PubMed.
- A. Aarkrog, J. Environ. Radioact., 1988, 6, 151–162 CrossRef CAS.
- H. Nies, The distribution of the Chernobyl fallout over the Baltic sea and its change during 1987 and 1988 in seawater, Baltic Sea Environment Proceedings, No.31, 1989b, pp. 31–41 Search PubMed.
- H. Nies, Monitoring of radioactive substances in the Baltic Sea, HELCOM seminar for experts from Estonia, Latvia, Lithuania, and Russia on the implementation of HELCOM Arrangements, other international instruments and related matters. Riga, Sept, 1993. Baltic Sea Environment Proc. No 59, 1994, pp. 61–70 Search PubMed.
- H. Nies and S. P. Nielsen, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 219–229 Search PubMed.
- G. Lujanine, K. Joksas, B. Silobritiene and R. Morkuniene, in Radionuclides in the environment, 2006, pp. 165–180 Search PubMed.
- K. O. Buesseler and H. D. Livingston, in Radionuclides in the Oceans Inputs and Inventories ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 200–217 Search PubMed.
- V. V. Kanivets, O. V. Voitsekhovitch, V. G. Simov and Z. A. Golubeva, J. Environ. Radioact., 1999, 43, 121–135 CrossRef CAS.
- V. N. Egolov, P. P. Povinec, G. G. Polikarpov, N. A. Stokozov, S. B. Gulin, L. G. Kulebakina and I. Osvath, J. Environ. Radioacit., 1999, 43, 137–155 Search PubMed.
- S.-H. Lee, J. J. La Rosa, I. Levy-Palomo, B. Oregioni, M. Pham, P. P. Khanh, P. P. Povinec and E. Wyse, Deep-Sea Res. partII, 2003, 50, 2817–2834 CrossRef CAS.
- R. Delfanti, B. Klein and C. Papucci, J. Geophys. Res, 2003, 108 DOI:10.1029/2002JC001371.
- H. Florou, P. Kritidis, F. Vosniakos, J. Trindafyllis, R. Delfanti, C. Papucci, A. Cigna, G. G. Polikarpov, V. N. Egoorov, A. S. Bologa and V. Patrascu, Fresenius Environ. Bull., 2003, 12, 3–9 CAS.
- S.-H. Lee, F. R. Mantoura, P. P. Povinec, J. A. Sanchez-Cabeza, J.-L. Pontis, A. Mahjoub, M. A.NoyreddineBoulahdid, L. Chouba, M. Samaali and N. Reguigui, Radioact. Environ., 2006, 8, 137–147 Search PubMed.
- Y. Bourlat, J.-C. Millies-Lacroix, G. Le Petit and J. Bourguignon, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 75–93 Search PubMed.
- J. K. Cochran, H. D. Livingston, D. J. Hirschberg and L. D. Surprenant, Earth and Planet Sci. Lett., 1987, 84, 135–152 CrossRef.
- W. S. Broecker and H. J. Simpson, A Summary of Lamont Sr90 and Cs137 Measurements on Ocean Water Samples, Health and Safety laboratory Reastern POrt HASL-197, 1968, I-9–I-104 Search PubMed.
- S. B. Gulin and N. A. Stokozov, J. Environ. Radioact., 2005, 83, 1–7 CrossRef CAS.
- S. E. Wijffels, M. M. Hall, T. Joyce, D. J. Torres, P. Hacker and F. Firing, J. Geophys. Res., 1998, 103, 12985–13009 CrossRef.
- M. Aoyama, M. Hirose, K. Nemoto, Y. Takatsuki and D. Tsumune, Geophys. Res. Lett., 2008, 35, L01604, DOI:10.1029/2007GL031964.
- Y. Igarashi, M. Aoyama, K. Hirose, M. Takashi, K. Nemoto, M. Tomita and T. Fujiwara, J. Radiat. Res., 2003, 44, 319–328 Search PubMed.
- T. F. Hamilton, J.-C. Millies-Lacroix and G. H. Hong, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 29–58 Search PubMed.
- A. Aarkrog, Q. J. Chen, J. Clausen, G. C. Christensen, H. Dahlgaard, K. Ellis, H. Hansen, H. Holm, H. P. JoensenS. P. Nielsen and M. Strandberg, Environmental radioactivity in the North Atlantic Region. The Faroe Islands and Greenland included 1990–1991, RISO-R-622, Riso National Laboratory, Roskilde, Denmark, 1994b Search PubMed.
- Y. Bourlat, J. C. Millies-Lacroix, G. L. Petit and R. Nazal, J. Radioanal. Nucl. Chem., 1995, 197, 387–408 CAS.
- M. Yamada and Z.-L. Wang, Sci. Total Environ., 2007, 382, 342–350 CrossRef CAS.
- R. A. Fine, R. Lukas, F. M. Bingham, M. J. Warner and R. H. Gammon, J. Geophys. Res., 1994, 99, 25063–25080 CrossRef.
- A. Aarkrog, H. Dahlgaard, I. Hallstadius, H. Hansen and M. Holm, Nature, 1983, 304, 49–51 CrossRef CAS.
- E. Holm, P. Roos, D. Josefsson and B. Persson, in Radionuclide in the Oceans Inputs and Inventories, ed. P. Gúegúeniat, P. Germain and H. Metivier, Les Editions de Physique, 1996, pp. 59–74 Search PubMed.
- M. Aoyama, K. Hirose, K. Komura, R. F. C. Mantoura, P. P. Povinec, J.-A. Sanchez-Cabeza and D. Tsumune, in Environmental modeling and radioecology, Proceedings of the international symposium on environmental modeling and radioecology, ed. S. Hisamatsu, H. Kakiuchi and N. Akata, Rokkasyo, Japan, 2006b, pp. 95–100 Search PubMed.
- K. Hirose, M. Aoyama, M. Fukazawa, C. S. Kim, K. Komura, P. P. Povinec and J. A. Sanchez-Cabeza, Sci. Tol. Environ., 2007, 381, 243–255 Search PubMed.
- V. T. Bowen, V. E. Noshkin, H. D. Livingston and H. L. Volchok, Earth Planet. Sci. Lett., 1980, 49, 411–434 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1 to S4; Fig. S1 to S3. See DOI: 10.1039/b811421h |
| This journal is © The Royal Society of Chemistry 2009 |