Upgrade of natural gas in rhozeolite-like metal–organic framework and effect of water: a computational study†
Ravichandar
Babarao
and
Jianwen
Jiang
*
Department of Chemical & Biomolecular Engineering, National University of Singapore, Singapore 117576. E-mail: chejj@nus.edu.sg; Fax: +65-6779-1936; Tel: +65-6516-5083
First published on 29th July 2009
Abstract
A computational study is reported for the upgrade of natural gas (CO2/CH4 mixture) in rhozeolite-like metal–organic framework (ZMOF) and the effect of water on upgrade efficiency. CO2 is adsorbed predominantly over CH4, attributed to the strong electrostatic interactions of CO2 with ionic framework and extraframework Na+ ions. With increasing pressure, Na+ ions are coordinated and solvated increasingly by CO2 molecules. The distance between CO2 molecules becomes shorter with increasing pressure, while the distance between Na+ and CO2 remains more or less constant. Compared with other MOFs and nanoporous materials reported to date, rho-ZMOF exhibits exceptionally high selective adsorption for CO2/CH4 mixture. The selectivity is up to 3800 at infinite dilution and 80 at ambient condition. With a trace amount of H2O added into CO2/CH4 mixture, the interaction between CO2 and Na+ is reduced. Consequently, CO2 adsorption drops but CH4 adsorption is not discernibly affected, and the selectivity decreases by one order of magnitude. This work reveals that rho-ZMOF is a promising candidate for the separation of natural gas and H2O has a significant effect.
Broader contextNatural gas is considered as an alternative fuel for the ‘unfriendly’ traditional fossil fuels such as petroleum and coal. To improve the energy efficiency of natural gas, impurity like CO2 needs to be removed. We report a computational study for the adsorptive separation of natural gas (CO2 and CH4 mixture) in rhozeolite-like metal–organic framework. The selectivity is predicted to be up to 3800 at infinite dilution and 80 at ambient condition, substantially higher than in most metal–organic frameworks and nanoporous materials. In the presence of H2O, the selectivity is reduced significantly. This work reveals that a charged metal–organic framework is extremely promising for the upgrade of natural gas and H2O has a very important effect on the selective adsorption. |
I. Introduction
There is considerable interest in using natural gas as an alternative fuel for the ‘unfriendly’ traditional fossil fuels such as petroleum and coal. To improve the energy efficiency of natural gas, impurity like CO2 needs to be removed.1 Techniques proposed to separate CO2 from natural gas include amine absorption, cryogenic distillation, adsorption and enzymatic conversion. Among these, adsorption in porous materials is energetically efficient and economically competitive. The selection of an ideal adsorbent is critical for high-efficacy separation by adsorption. In the past, a large number of experimental and simulation studies have been reported for the adsorptive separation of CO2/CH4 mixture in a variety of nanoporous materials, such as carbons and zeolites, and emerging metal–organic frameworks (MOFs). For example, separation of CO2/CH4 mixture was simulated in MFI with intersecting channels and in CHA and DDR with cages connected by narrow windows,2 and in IRMOF-1 and Cu-BTC.3 Adsorption and separation of CO2 and CH4 were investigated in three types of nanostructures, namely, IRMOF-1, silicalite, and nanoporous carbon. IRMOF-1 was found to have the largest capacity for adsorption of CO2 and CH4, but was unsatisfactory for separation.4 Atomistic simulations were reported for separation of CO2/CH4 and other mixtures in IRMOF-1 and mixture effects were found to play a crucial role in determining the performance.5 CO2/CH4 mixture was studied in carborane-based MOFs and a higher selectivity was reported in MOF possessing exposed metal sites.6 Simulations were performed on the separation of CO2/CH4 in IRMOF-1 and Cu-BTC, and a higher selectivity was observed in Cu-BTC.7,8 Removal of CO2 from binary and ternary mixtures was measured by fixed-bed adsorption in a microporous MOF-508b with one-dimensional pores of 4.0 × 4.0 Å.9 A very large improvement in separation power for CO2 and CH4 was demonstrated in an amine-functionalized MIL-53, with almost infinite selectivity at 1 bar.10Recently, a unique subset of MOFs, zeolite-like MOFs (ZMOFs) have been developed.11–14 They are topologically relevant to inorganic zeolites and exhibit similar structural properties. Nevertheless, the substitution of oxygen atoms in zeolites by organic linkers leads to extra-large cavities and pores in ZMOFs. This edge expansion approach offers a great potential toward the design and synthesis of very open materials. A number of ZMOFs contain ionic frameworks and charge-balancing extraframework ions, e.g., rho-ZMOF synthesized by the assembly of tetrahedral building units with a long ditopic organic linker.11 The presence of extraframework ions in the pores of molecular dimensions increases the interactions with guest molecules; consequently enhances storage, separation or ion-exchange capability.
Previous studies on gas separation in MOFs have been conducted exclusively in neutral frameworks. Only recently have we reported the separation of gas mixtures in charged MOFs (both cationic soc-MOF and anionic rho-ZMOF) and unprecedentedly high selective adsorption was predicted for that time being.15–17 In the present study, we simulate the separation of CO2/CH4 mixture in Na+-exchanged rho-ZMOF and the effect of water on separation. Natural gas usually contains a small amount of moisture and it is interesting to examine separation in the presence of water. While experimental measurement on the adsorption of pure gases is relatively straightforward, quantitative measurement of gas mixtures is challenging. In this regard, molecular simulations are useful to predict the behavior of gas mixtures and provide the microscopic insight into underlying mechanisms involved. Indeed, most of the current knowledge for the adsorption and separation of gas mixtures in MOFs has been largely obtained by molecular simulations.
Following from this, molecular models for rho-ZMOF, Na+ and adsorbates are described in Section II. The simulation methods are introduced in Section III for the prediction of adsorption. In Section IV, the simulation snapshot, isotherm and selectivity for CO2/CH4 mixture are presented. The predicted selectivity in rho-ZMOF is compared with reported data in other MOFs. To examine the effect of moisture, adsorption of CO2/CH4/H2O mixture was also shown. Finally, the concluding remarks are summarized in Section V.
II. Molecular models
Rho-ZMOF represents the first example of a 4-connected MOF with a topology of rho-zeolite. It was synthesized by metal–ligand-directed assembly of In atoms and 4,5-imidazoledicarboxylic acid (H3ImDC).11 The space group in rho-ZMOF is Im-3m and the lattice constants are a = b = c = 31.062 Å, as shown in Fig. S1a†. In rho-ZMOF, each In atom is coordinated to four N and four O atoms of four separate doubly deprotonated H3ImDC (HImDC) to form an eight-coordinated dodecahedron. Each independent HImDC is coordinated to two In atoms resulting in two rigid five-membered rings viaN-, O-hetero-chelation. The structure is a truncated cuboctahedra (α-cages) containing 48 In atoms, which link together through double 8-membered rings (D8R). The substitution of oxygens in rho-zeolite with HImDCs generates a very open-framework with extra-large cavity of 18.2 Å in diameter. Unlike rho-zeolite and other rho-aluminosilicate or aluminophosphate, rho-ZMOF contains twice as many positive charges (48 vs. 24) in a unit cell. It is insoluble in water and common organic solvents. Experimental thermogravimetric analysis showed that all residential water molecules can be completely evacuated (calcd 26.9%);11 therefore, anhydrous Na–rho-ZMOF is experimentally feasible.The atomic charges of rho-ZMOF framework atoms were calculated from density functional theory (DFT) based on a fragmental cluster illustrated in Fig. S1b†. It has been widely recognized that quantum mechanically derived charges fluctuate appreciably for small basis sets; however, they tend to converge beyond 6-31G(d) basis set. Consequently, in our DFT calculations, 6-31G(d) basis set was used for all atoms except the transitional metals, for which LANL2DZ basis set was used. The DFT computations used the Lee–Yang–Parr correlation functional (B3LYP) and were carried out with the Gaussian 03 electronic structure package.18 The concept of atomic charges is solely an approximation and no unique straightforward method is currently available to determine atomic charges rigorously. In this study, the atomic charges were estimated by fitting to the ElectroStatic Potential (ESP). The extraframework Na+ ion carried a positive unit charge. In addition to the Coulombic interactions, the dispersion interactions between framework atoms and Na+ ions were represented by Lennard-Jones (LJ) potential with parameters adopted from the Universal Force Field (UFF).19 The cross LJ parameters were estimated by the Lorentz–Berthelot combining rules. A number of simulation studies have shown that UFF can accurately predict adsorption and diffusion of gases in various MOFs.20–23
CO2 was represented as a three-site rigid molecule and its intrinsic quadrupole moment was described by a partial-charge model.24 The partial charges on C and O atoms were qC = 0.576e and qO = −0.288e (e = 1.6022 × 10−19 is the elementary charge), respectively. The C–O bond length was 1.18 Å and the bond angle ∠OCO was 180°. CO2–CO2 interaction was modeled as a combination of LJ and Coulombic potentials. CH4 was represented by a united-atom model interacting with the LJ potential. The potential parameters were adopted from the TraPPE force field, which was developed to reproduce the critical parameters and liquid densities of alkanes.25 We also examined the effect of trace amount of H2O on CO2/CH4 separation. H2O was represented by TIP3P model,26 in which O–H bond length was 0.9572 Å and ∠HOH angle was 104.52°. The intermolecular interaction between H2O was modeled by the LJ and Coulombic potentials. Table 1 lists the LJ parameters and atomic charges for extraframework ion and adsorbates.
III. Simulation methods
Grand Canonical Monte Carlo (GCMC) simulations were conducted for the adsorption of CO2/CH4 and CO2/CH4/H2O mixtures in rho-ZMOF at 298 K. The bulk composition was 50:50 (equimolar) for CO2/CH4 mixture, and 50:50:0.1 for CO2/CH4/H2O mixture. The simulation box contained one unit cell of rho-ZMOF and periodic boundary conditions were applied in three dimensions. The framework was considered to be rigid during simulation because adsorption involves low-energy equilibrium configurations and framework flexibility has an insignificant effect. The unit cell was divided into three-dimensional grids with the energy landscape tabulated in advance and then used by interpolation during simulation. In such a way, the simulation was accelerated by two orders of magnitude. In total, 48 extraframework Na+ ions were introduced randomly into the system and allowed to move during simulation. A spherical cutoff of 15.0 Å was used to evaluate the LJ interactions and the usual long-range corrections were used beyond the cutoff. The use of the usual long-range corrections was an appropriate approximation because the error introduced by assuming homogeneity was small compared with the magnitude of long-range corrections. For the Coulombic interactions, a simple spherical truncation could result in unacceptable errors; consequently, Ewald sum with a tin-foil boundary condition was used. The real/reciprocal space partition parameter and the cutoff for reciprocal lattice vectors were chosen to be 0.2 Å−1 and 8, respectively, to ensure the convergence of the Ewald sum. The number of trial moves in a typical simulation was 2 × 107, though additional trial moves were used at high loadings. The first 107 moves were used for equilibration and the subsequent 107 moves for ensemble averages. Six types of trial moves were randomly attempted in the GCMC simulations, namely, displacement, rotation, and partial regrowth at a neighboring position; complete regrowth at a new position; swap with reservoir; exchange of molecular identity. Within statistical uncertainties, the simulation results were found to be independent of the sequence of the trial moves. To examine how the charges on framework and Na+ ions affect separation, additional simulations were performed for CO2/CH4 mixture in a neutral structure, in which the charges on the framework and Na+ ions were switched off. The statistical uncertainties were estimated by block transformation and found to be smaller than the symbol size in the figures presented in Section IV.IV. Results and discussion
The locations and dynamics of extraframework Na+ ions in rho-ZMOF have been reported in our previous study,17 and are briefly described here. As shown in Fig. 1, two types of binding sites for Na+ ions exist in rho-ZMOF. Site I is in the single 8-membered ring (S8R) and at the entrance to the α-cage. Two S8Rs in neighboring unit cells form a double 8-membered ring (D8R). The distance from site I to the nearest In atoms in S8R is 5.0–5.3 Å and is approximately 7.8 Å to the next-to-nearest In atoms in the D8R. Site II is in the α-cage and proximal to the moiety of organic link. In a unit cell, 26 Na+ ions are located at site I and the remaining at site II. Site I has a larger coordination number than site II with the neighboring atoms and thus interacts more strongly with framework. The two types of binding sites in rho-ZMOF are similar to those in inorganic rho-zeolite.27 In the latter, however, an additional type of site is located at the center of the D8R and equally distanced from both S8Rs. | ||
Fig. 1
Binding sites of Na+ ions in rho-ZMOF. Site I ( ) is in the single 8-membered ring and site II ( ) is in the single 8-membered ring and site II ( ) is in the α-cage. Color code: In, ) is in the α-cage. Color code: In,  ; N, ; N,  ; C, ; C,  ; O, ; O,  ; and H, ○. ; and H, ○. | ||
The mobility of Na+ ions was quantified by mean-squared displacements (MSDs). MSD at site I is nearly flat with a negligible value of 0.15 Å2. In contrast, MSD at site II initially increases and then approaches a constant about 1.3 Å2. The mobility at site II is greater than at site I due to the relatively weaker interaction with framework and the larger void space around site II. The mobility of extraframework ions in rho-ZMOF is generally small. This is attributed to the degenerated favorable sites away from each other, which largely prohibits ion hopping from one site to the other. In addition, the steric hindrance of metal atoms and organic linkers also reduces ion mobility. In a separate study, we have found that adsorption of H2O in rho-ZMOF leads to a redistribution of Na+ ions and a slight enhancement of Na+ ion mobility.
For adsorption of equimolar CO2/CH4 mixture in Na-rho-ZMOF, Fig. 2 illustrates the typical locations of CO2 molecules in the S8R. At 10 kPa, CO2 molecules adsorb preferentially near Na+ ions. This is because CO2 has a quadrapolar moment and can strongly interact with Na+ ions, which act as additional adsorption sites for CO2. With increasing pressure to 500 and 3000 kPa, Na+ ions are coordinated by more CO2 molecules, i.e., increasingly solvated by surrounding CO2 molecules. The distance between CO2 molecules (CCO2–CCO2) becomes shorter with increasing pressure, while the distance between Na+ and CO2 (Na+–OCO2) remains more or less constant. This implies an enhancement in the cooperative interactions between CO2 molecules. As pressure rises, however, more CO2 molecules are adsorbed in the α-cages and the electrostatic interactions between CO2 and Na+ ions are largely reduced. Upon comparison, the enhanced cooperative interactions between CO2 molecules are negligible.
 | ||
| Fig. 2 Locations of CO2 molecules in the S8R for CO2/CH4 mixture at 10, 500 and 3000 kPa. Na+ ions and CO2 molecules are represented by balls and sticks, respectively. The distances of CCO2–CCO2 (orange) and Na+–OCO2 (green) are in angstroms. | ||
To explore the structural information of CO2 and CH4 in rho-ZMOF, the radial distribution functions g(r) between Na+ ions and adsorbates are shown in Fig. 3 at 10, 500, and 3000 kPa. A pronounced peak in g(r) for Na+–CO2 is observed at r = 3.6 Å at all the three pressures, nevertheless, no such peak exists for Na+–CH4. This confirms that CO2 interacts with Na+ ions more strongly than CH4. With increasing pressure, the peak height in g(r) for Na+–CO2 drops, whereas the coordination number of CO2 molecules surrounding Na+ ions increases (data not shown). As a consequence, Na+ ions are solvated by more CO2 molecules. For Na+–CH4, g(r) does not change much with pressure because the adsorption of CH4 is vanishingly small.
 | ||
| Fig. 3 Radial distribution functions between Na+ ions and adsorbates for CO2/CH4 mixture at 10, 500 and 3000 kPa. | ||
Fig. 4 shows the adsorption isotherm and selectivity for CO2/CH4 mixture in rho-ZMOF. There is a sharp increase in CO2 adsorption at low pressures. With increasing pressure, CO2 adsorption tends to approach saturation. The isotherm belongs to type I, the signature of adsorption in microporous adsorbents. Over the entire range of pressure, CO2 is predominantly more adsorbed than CH4 due to two reasons. First, CO2 is a three-site molecule and has a much stronger interaction with the framework than CH4. Second, the charged framework and the presence of Na+ ions induce strong electrostatic interactions with CO2 molecules. The separation of gas mixture is quantified by selectivity Si/j = (xi/xj)(yj/yi), where xi and yi are the mole fractions of component i in adsorbed and bulk phases, respectively. The selectivity at infinite dilution is equal to the ratio of Henry's constants KH(CO2)/KH(CH4). From separate simulations, we found KH(CO2)/KH(CH4) ≈ 3800 for CO2/CH4 mixture at 298 K. With increasing pressure, the selectivity drops and is approximately 80 at 1 atm. This is a consequence of the significant reduction in the electrostatic interactions between CO2 and rho-ZMOF with increased loading. The predicted selectivity for CO2/CH4 mixture in rho-ZMOF is significantly higher than reported data in IRMOF-1 (2–3),4,7,8carborane-based MOFs (∼17),6 mixed-ligand MOFs (∼30),28Cu–BTC (6–9),7,8 and MOF-508b (3–4).9 However, it is lower than the selectivity in amine-functionalized MIL-53.10
 | ||
| Fig. 4 (a) Isotherms and (b) selectivities for CO2/CH4 and CO2/CH4/H2O mixtures. The bulk composition is 50:50 for CO2/CH4, and 50:50:0.1 for CO2/CH4/H2O. | ||
To quantitatively examine the effect of charges, Fig. S2† shows the isotherm and selectivity for CO2/CH4 mixture in a neutral structure by switching off the charges on framework and ions. Compared with Fig. 4, CO2 adsorption decreases, whereas CH4 adsorption increases. The selectivity decreases by 2 to 3 orders of magnitude and exhibits qualitatively different behavior. With increasing pressure, the selectivity initially decreases and then increases. The initial decrease is attributed to the heterogeneous distribution of adsorption sites and the later increase is due to the cooperative interactions between CO2 molecules. This reveals that the charges on framework and extraframework ions play a very important role in the selective adsorption of CO2/CH4 mixture.
As we can see in Fig. 4, adsorption of CO2/CH4 mixture in rho-ZMOF is significantly affected by H2O. With even 0.1% of H2O, CO2 adsorption drops significantly, while CH4 adsorption is not discernibly affected. Despite its negligible composition, the extent of H2O adsorption is much greater than CO2 (not shown). This is because H2O is highly polar, interacts with charged framework and ions substantially more strongly than CO2, and therefore has a significant effect on CO2 adsorption. Upon addition of H2O, the selectivity of CO2/CH4 mixture is reduced approximately by one order of magnitude. To provide a more detailed insight, Fig. 5 shows the locations of CO2 and H2O molecules in the S8R for CO2/CH4/H2O mixture at 500 kPa. There are a large number of H2O molecules around Na+ ions, which are considered as being hydrated. Compared with Fig. 2 (also at 500 kPa), the number of CO2 molecules in Fig. 5 is fewer and the distance from CO2 to Na+ is longer, changing from 2.5–2.6 Å to 4.0–4.4 Å. This implies that the interaction between CO2 and Na+ is reduced in the presence of H2O.
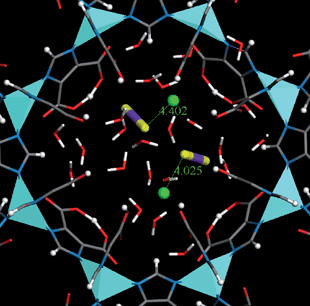 | ||
| Fig. 5 Locations of CO2 and H2O molecules in the S8R for CO2/CH4/H2O mixture at 500 kPa. Na+ ions are represented by balls, CO2 and H2O molecules are represented by sticks. The distances of Na+–OCO2 are in angstroms. | ||
Fig. 6 shows the radial distribution functions g(r) between Na+ and adsorbates for CO2/CH4/H2O mixture at 500 kPa. A very high peak is observed in g(r) of Na+–H2O, indicating a strong affinity between Na+ and H2O. However, no peak is observed here for Na+–CO2, which is in contrast to Fig. 3. These further suggest that H2O competes the adsorption sites with CO2 and exerts a significant effect on CO2 adsorption. Therefore, it is important to remove moisture before the separation of CO2/CH4 mixture.
 | ||
| Fig. 6 Radial distribution functions between Na+ ions and adsorbates for CO2/CH4/H2O mixture at 500 kPa. | ||
H2O significantly reduces the selectivity of CO2/CH4 mixture in rho-ZMOF over the whole range of pressure under study. This is remarkably different from the effect of H2O on separation of CO2/H2 mixture in soc-MOF, wherein the selectivity increases slightly at low pressures and decreases at high pressures upon addition of H2O.16 For CO2/H2 mixture in soc-MOF, H2O strongly binds onto the exposed In atoms of trimer [In3O(CO2)6]. On average, a trimer is surrounded by three H2O molecules and the distance is approximately 3.2–3.4 Å from the O atom of H2O to the indium atom of trimer. The bound H2O molecules around trimer act as additional sites for CO2 adsorption. Only at high pressures, H2O enters the carcerand-like capsules and starts to interact with the extraframework NO3− ions located in the capsules. However, CO2 cannot enter because H2O has a much stronger interaction with NO3− ions. As a consequence, the selectivity for CO2/H2 in soc-MOF slightly rises at low pressures because of the promoted adsorption of CO2 by H2O. In contrast, competitive adsorption occurs at high pressures, H2O substitutes pre-adsorbed CO2 and consequently the selectivity drops.16
V. Conclusions
We have simulated the separation of CO2/CH4 mixture in novel rho-ZMOF and the effect of water on the separation. CO2 is preferentially more adsorbed over CH4 because both the ionic framework and the large density of extraframework ions exert strong electrostatic interactions with CO2. Furthermore, Na+ ions act as additional adsorption sites and augment the interactions with CO2. At low pressures, CO2 is adsorbed proximally to Na+ ions. With increasing pressure, Na+ ions are coordinated and solvated by CO2 molecules. The selectivity of CO2 over CH4 is approximately 3800 at infinite dilution and 80 at 298 K and 1 atm, the typical condition for pressure swing adsorption in practice. The selectivity is reduced by one order of magnitude in the presence of trace mount of H2O, and by 2 to 3 orders of magnitude in a neutral structure with the charges on framework and ions switched off. The predicted selectivity for CO2/CH4 mixture in rho-ZMOF is exceptionally higher than in most MOFs reported to date. H2O has a pronounced effect on the selectivity; therefore, a pre-treatment is required for the upgrade of natural gas.Acknowledgements
This work was supported by the National University of Singapore and the Singapore National Research Foundation.References
- D. Singh, E. Croiset, P. L. Douglas and M. A. Douglas, Energy Convers. Manage., 2003, 44, 3073 CrossRef CAS.
- R. Krishna, J. M. van Baten, E. Garcia-Perez and S. Calero, Chem. Phys. Lett., 2006, 429, 219 CrossRef CAS.
- R. Krishna and J. M. van Baten, Chem. Eng. Sci., 2008, 63, 3120 CrossRef CAS.
- R. Babarao, Z. Q. Hu, J. W. Jiang, S. Chempath and S. I. Sandler, Langmuir, 2007, 23, 659 CrossRef CAS.
- S. Keskin and D. S. Sholl, Ind. Eng. Chem. Res., 2009, 48, 914 CrossRef CAS.
- Y. S. Bae, O. K. Farha, A. M. Spokoyny, C. A. Mirkin, J. T. Hupp and R. Q. Snurr, Chem. Commun., 2008, 4135 RSC.
- Q. Y. Yang and C. L. Zhong, J. Phys. Chem. B, 2006, 110, 17776 CrossRef CAS.
- A. Martin-Calvo, E. Garcia-Perez, J. M. Castillo and S. Calero, Phys. Chem. Chem. Phys., 2008, 10, 7085 RSC.
- L. Bastin, P. S. Barcia, E. J. Hurtado, J. A. C. Silva, A. E. Rodrigues and B. Chen, J. Phys. Chem. C, 2008, 112, 1575 CrossRef CAS.
- S. Couck, J. F. M. Denayer, G. V. Baron, T. Remy, J. Gascon and F. Kapteijn, J. Am. Chem. Soc., 2009, 131, 6326 CrossRef CAS.
- Y. L. Liu, V. C. Kravtsov, R. Larsen and M. Eddaoudi, Chem. Commun., 2006, 14, 1488 Search PubMed.
- Y. Q. Tian, Y. M. Zhao, Z. X. Chen, G. N. Zhang, L. H. Weng and D. Y. Zhao, Chem.–Eur. J., 2007, 13, 4146 CrossRef CAS.
- M. Dinca, W. S. Han, Y. Liu, A. Dailly, C. M. Brown and J. R. Long, Angew. Chem., Int. Ed., 2007, 46, 1419 CrossRef CAS.
- D. F. Sava, V. C. Kravtsov, F. Nouar, L. Wojtas, J. F. Eubank and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 3768 CrossRef CAS.
- R. Babarao, J. W. Jiang and S. I. Sandler, Langmuir, 2009, 25, 5239 CrossRef CAS.
- J. W. Jiang, AIChE J., 2009, 55, 2422 CrossRef.
- R. Babarao and J. W. Jiang, J. Am. Chem. Soc., 2009, 131, 11417 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 03, Revision D.01 ed.Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef CAS.
- A. Vishnyakov, P. I. Ravikovitch, A. V. Neimark, M. Bulow and Q. M. Wang, Nano Lett., 2003, 3, 713 CrossRef CAS.
- G. Garberoglio, A. I. Skoulidas and J. K. Johnson, J. Phys. Chem. B, 2005, 109, 13094 CrossRef CAS.
- A. I. Skoulidas and D. S. Sholl, J. Phys. Chem. B, 2005, 109, 15760 CrossRef CAS.
- R. Babarao and J. W. Jiang, Langmuir, 2008, 24, 6270 CrossRef CAS.
- A. Hirotani, K. Mizukami, R. Miura, H. Takaba, T. Miya, A. Fahmi, A. Stirling, M. Kubo and A. Miyamoto, Appl. Surf. Sci., 1997, 120, 81 CrossRef CAS.
- M. G. Martin and J. I. Siepmann, J. Phys. Chem. B, 1998, 102, 2569 CrossRef CAS.
- P. Mark and L. Nilsson, J. Comput. Chem., 2002, 23, 1211 CrossRef CAS.
- Y. Lee, B. A. Reisner, J. C. Hanson, G. A. Jones, J. B. Parise, D. R. Corbin, B. H. Toby, A. Freitag and J. Z. Larese, J. Phys. Chem. B, 2001, 105, 7188 CrossRef CAS.
- Y. S. Bae, L. K. Mulfort, H. Frost, P. Ryan, S. Punnathanam, L. J. Broadbelt, J. T. Hupp and R. Q. Snurr, Langmuir, 2008, 24, 8592 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic charges of rho-ZMOF framework. Adsorption isotherm and selectivity of CO2/CH4 mixture in a neutral framework by switching off the charges on framework and Na+ ions. See DOI: 10.1039/b909861e |
| This journal is © The Royal Society of Chemistry 2009 |