Functionalized alkyne bridged dendron based chromophores for dye-sensitized solar cell applications
Received
1st May 2009
, Accepted 3rd June 2009
First published on
1st July 2009
Abstract
We report the results of an investigation on the preparation, spectral and photoelectrochemical properties of alkyne bridged dendron chromophores with carboxylic acid anchoring group, which adhere effectively to nanocrystalline (anatase) TiO2 surfaces yielding incident photon-to-current conversion efficiency values of up to 76% in their photocurrent action spectra, suggesting that the alkyne π-bridge functional moiety is an effective bridging group between chromophore and conduction band of the semiconductor.
Broader context
We report an interesting class of novel sensitizers that contain an alkyne/alkene bridge between the anchoring carboxylic acid group and the donor dendron moiety. These sensitizers anchor effectively onto nanocrystalline (anatase) TiO2 surfaces yielding incident photon-to-current conversion efficiency values of up to 76% in their photocurrent action spectra, suggesting that the alkyne and alkene groups are an effective communicating bridge. Under standard global AM 1.5 solar conditions, the alkyne bridged sensitized cell gave an overall conversion efficiency of 1.55%, while the alkene bridge sensitized cell gave an overall conversion efficiency of 1.20%, demonstrating the influence of electronic coupling between the chromophore and the TiO2 semiconductor on device performance. Density functional calculations interpreted a tunability of band gap by π-conjugation and better electron coupling in the alkyne π-bridged chromophore, which were consistent with the experimental results.
|
Introduction
Ruthenium(II) polypyridine complexes,1 organic sensitizers,2 phthalocyanines3 and porphyrins4 are well established as photosensitizers for use in photovoltaic cells based on nanocrystalline TiO2 films. For high efficiency, several characteristics of the photosensitizer need to be optimized, such as the absorption spectrum which should match as far as possible the solar emission spectrum up to 900 nm (panchromatic response); a high electron-injection efficiency from the chromophore to the conduction band of the semiconductor; good electronic coupling between the chromophore and conduction band of the TiO2 semiconductor; and high affinity for the TiO2 surface. Strong anchoring immobilizes the sensitizer on the nanocrystalline TiO2 surface resulting in efficient electronic communication between the light-harvesting center and the semiconductor substrate, and ascertaining stable cell operation. The immobilization of the sensitizer on the surface of mesoporous TiO2 nanoparticle is achieved by anchoring groups such as carboxylate,5 phosphonate,6 salicylate,7 sulfonate,8 acetyl acetonate9 and catechol.10 Though various anchoring groups have been applied in dye-sensitized solar cells (DSSCs), systematic studies of the influence of the bridging group on electronic coupling between the chromophore and the TiO2 surface are still lacking. This is an important factor to consider in dye design and future organic sensitizer development. The conducting alkyne bridge has been utilized for conjugated donor–acceptor (D–A) systems11 because it is rigid and straight for conducting electrons and at the same time reduces charge recombination.12 In this work, we report the influence of a conducting alkyne bridge on incident photon-to-current and power conversion efficiency of the dye-sensitized solar cell (DSSC).
Experimental
Measurements
1H-NMR spectra were recorded using Varian Oxford 300 MHz spectrometers; chemical shifts were reported in ppm units with tetramethylsilane as an internal standard. Infrared spectra were measured on KBr pellets using a Perkin-Elmer Spectrometer. The mass spectra were taken by a JEOL JMS-AX505WA mass spectrometer. MALDI-TOF mass spectrometry was performed with Voyager-DETM STR Biospectrometry. The elemental analyses were carried out by means of EA1110 (CE Instrument). Steady-state absorption spectra were recorded on a Shimadzu UV-2401PC spectrophotometer and photoluminescence spectra were measured by a steady-state fluorimeter (Edinburgh FS920) with 450W Xe lamp. The excitation light from a 300 mm focal length monochromator was focused on the sample. The fluorescence from the sample was collected and refocused to the emission monochromator with 300 mm focal length. Visible emission spectra were taken by a PMT system (Hamamatsu R955).
Material synthesis
Palladium(II) acetate (Pd(OAc)2), tri-o-tolylphosphine (TOP), triethylamine, 2-methyl-3-butyn-2-ol, 4-iodobenzoic acid, and bis(triphenylphosphinepalladium(II) dichloride were obtained from Aldrich Chemical Co. and used without further purification. The third generation aryl–ether dendron ([G3]–Br) was synthesized according to the procedure described in the literature13 and the detailed synthetic procedure for [G3–An]–Br has been described elsewhere.14 The starting material, 4-vinylbenzoic acid methyl ester (1) was prepared by the literature method,15 and the general synthetic strategy for π-extended dendritic anthracene ligands is Heck reaction16 and Sonogashira cross-coupling protocol.17 The vinyl-bridged anthracene derivative ([G3–AnD]–CO2H) was synthesized by the well-known Heck reaction of 4-vinylbenzoic acid methyl ester with [G3–An]–Br and then hydrolyzed. The acetyl-bridged anthracene derivative [G3–AnT]–CO2H was prepared by Sonogashira cross-coupling of 2-methyl-3-butyn-2-ol with [G3–An]–Br. The protecting group was removed with NaOH/toluene, and the resulting alkyne ([G3–AnT]–H) was coupled with 4-iodobenzoic acid to give [G3–AnT]–CO2H as a yellow powder. Chemical structures and the synthesis of π-conjugated anthracene ligands bearing the G3–aryl–ether dendron are shown in Scheme 1.
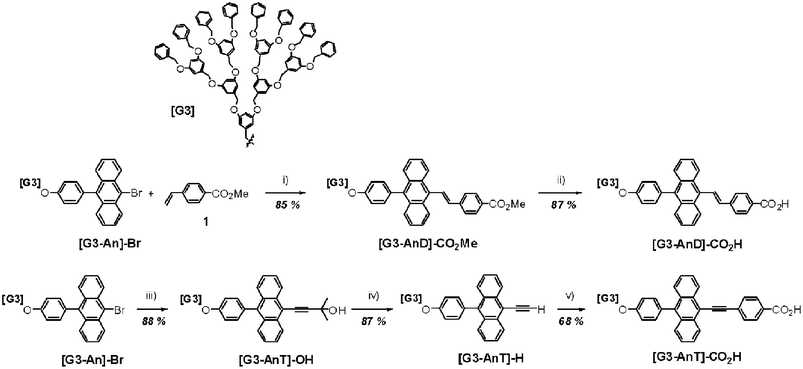 |
| | Scheme 1 Chemical structures and synthesis of π-conjugated anthracene ligands bearing the G3–aryl–ether dendron. | |
Synthesis of [G3–AnD]–CO2H.
KOH (0.5 g, 9.17 mmol) was added to a solution of [G3–AnD]–CO2Me (2.3 g, 1.15 mmol) in 20 mL of ethanol/THF (7 : 3 v/v). The reaction mixture was stirred for 12 h at reflux temperature. The reaction mixture was evaporated to dryness in vacuo. The residual was dissolved with water and then acidified with HCl. This solid was washed sequentially with water, and hexane, giving a yellowish solid (1.98 g, 87%). νmax(KBr pellet)/cm−1 3029, 2867, 1687, 1595, 1449, 1293, 1153, 1049, 831, and 735; δH (300 MHz, CDCl3, Me4Si) 8.37 (d, 2H), 8.22 (d, 2H), 7.79 − 7.70 (m, 4H), 7.40 − 7.26 (m, 46H), 7.18 (d, 2H), 7.05 − 6.99 (m, 2H), 6.78 (d, 2H), 6.69 − 6.66 (m, 12H), 6.61 (t, 1H), 6.55 (m, 6H), 5.11 (s, 2H), 4.99 (d, 20H), 4.96 (s, 8H); MALDI-TOF-MS: m/z calcd for C134H110O17 1990.7743, found (M + 2H)+ 1992.5470.
Synthesis of [G3–AnT]–CO2H.
The mixture of [G3–AnT]–H (2.20 g, 1.180 mmol), CuI (0.02 g, 0.118 mmol), PdCl2(PPh3)2 (0.01 g, 0.118 mmol), PPh3 (0.03 g, 0.118 mmol), and 4-iodobenzoic acid (0.38 g, 1.530 mmol) in 50 mL NEt3/toluene was refluxed for 12 h. After cooling to room temperature, the crude mixture was filtered and purified by column chromatography (1.60 g, 68%). νmax(KBr pellet)/cm−1 3030, 2986, 2192, 1687, 1596, 1450, 1293, 1154, 1050, 831, and 734; δH (300 MHz, CDCl3, Me4Si) 8.71 (d, 2H), 8.22 (d, 2H), 7.88 (d, 2H), 7.75 (d, 2H), 7.62 (m, 2H), 7.41 − 7.25 (m, 44H), 7.17 (d, 2H), 6.77 (d, 2H), 6.69 − 6.66 (m, 12H), 6.61 (t, 1H), 6.55 (m, 6H), 5.10 (s, 2H), 4.99 (s, 20H), 4.95 (s, 8H); MALDI-TOF-MS: m/z calcd for C134H108O17 1988.7587, found (M + H)+ 1989.8315.
Dye-sensitized solar cells.
The detailed fabrication procedure for dye-sensitized solar cells was described in a previous publication.18 A paste composed of ellipsoidal TiO2 particles for the transparent nanocrystalline layer was coated on the TiCl4 treated FTO glass plates (Nippon Sheet Glass, 4 mm thickness) by repetitive screen printing to obtain the required thickness, 12 µm. The TiO2 electrodes were immersed into the [G3–AnD]–CO2H and [G3–AnT]–CO2H solutions composed of 0.1 mM in ethanol and kept at room temperature for 15 h. The electrolyte (A6986) composed of 0.6 M BMII (butylmethylimidazolium iodide), 0.1 M LiI, 0.05 M I2, 0.05 M tert-butylpyridine, in valeronitrile and acetonitrile (15 : 85) ratio used. The current–voltage characteristics were measured using a 450 W Xenon light source that was focused to give 100 mW cm−2, the equivalent of one sun at AM 1.5, at the surface of the test cell. In order to reduce scattered light from the edge of the glass electrodes of the dyed TiO2 layer, a light-shading mask was placed onto the DSCs, so the active area of DSCs was fixed at 0.2 cm2.
Results and discussion
The chemical structures of all dendritic ligands were identified by FT-IR, 1H-NMR, MALDI-TOF mass spectrometry, elemental analysis, absorption and emission spectroscopy. In particular, the observed δ values for methyl protons in [G3–AnT]–OH and for terminal alkyne proton (–C≡CH) in [G3–AnT]–H are 1.85 and 4.08 ppm, respectively. Moreover, in the FT-IR spectrum, a vibrational band at 3278 cm−1, corresponding to the –C≡C–H stretch vibration of the terminal alkyne, was clearly visible, together with a band at 1595 cm−1 which may be attributed to the aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch vibration (see Fig. 1).
C stretch vibration (see Fig. 1).
![FT-IR spectra of a series of [G3-AnX]-CO2H (X = D and T).](/image/article/2009/EE/b908670f/b908670f-f1.gif) |
| | Fig. 1 FT-IR spectra of a series of [G3-AnX]-CO2H (X = D and T). | |
 |
| | Fig. 2 1H-NMR spectra of ethynyl-brigded anthracene derivatives. | |
![The anthracene ligands ([CH3O–AnX]–CO2H, X = D and T), derived from a semi-empirical method (AM1).](/image/article/2009/EE/b908670f/b908670f-f3.gif) |
| | Fig. 3 The anthracene ligands ([CH3O–AnX]–CO2H, X = D and T), derived from a semi-empirical method (AM1). | |
UV-vis absorption and photoluminescence (PL) spectra of π-extended anthracene ligand bearing G3–aryl–ether dendron ([G3–AnX]–CO2H, X = D and T) in solution and as solid state are shown in Fig. 4 and Fig. 5, respectively. The absorbance of the aryl–ether typed dendrons in dendritic π-extended anthracene ligands appeared at 285 nm and the anthracene units absorbed in the range from 350 nm to 480 nm. The introduction of the aryl–ether dendron into π-extended anthracene ligands does not influence the rotation barrier between adjacent π-extended phenyl ring and anthracene ring hydrogens (see Fig. 3). This restrictive rotation barrier leads to π-extended anthracene ligands with the aryl–ether dendron which have well-resolved structures, similar to that of the 9,10-DPA derivative.19 Also, similar UV-vis absorption behavior of π-extended anthracene ligand ([G3–AnX]–CO2H, X = D and T) was observed in THF solutions. Recently, Bhaskar et al. reported that the absorption maxima of alkyne π-bridged chromophores are blue-shifted relative to their alkene counterparts in donor–π–acceptor molecules.20 By contrast, the alkyne π-bridging centered anthracene ligand shows a red-shifted absorption, since the alkyne π-bridge has a more effective and directional π-conjugation than an alkene π-bridge, as illustrated in Fig. 3.
![Effect of solvent polarity on absorption and emission spectra of [G3–AnX]–CO2H (X = D (top) and T (bottom)).](/image/article/2009/EE/b908670f/b908670f-f4.gif) |
| | Fig. 4 Effect of solvent polarity on absorption and emission spectra of [G3–AnX]–CO2H (X = D (top) and T (bottom)). | |
The fluorescence of the G3–aryl–ether dendron, which appeared at the maximum wavelength of 345 nm, was totally quenched when it was excited at 285 nm of the dendron absorption band, but this significant decrease in fluorescence of the aryl–ether dendron was accompanied by a strong increase in the fluorescence intensity of the π-extended anthracene moiety. These results show that a highly efficient energy transfer from the aryl–ether dendron to the π-extended anthracene moiety takes place.
![Photocurrent density–voltage characteristics (top) and incident monochromatic photon-to-current conversion efficiency (bottom) of a 12 µm TiO2 film derivatized with [G3–AnD]–CO2H and [G3–AnT]–CO2H.](/image/article/2009/EE/b908670f/b908670f-f6.gif) |
| | Fig. 6 Photocurrent density–voltage characteristics (top) and incident monochromatic photon-to-current conversion efficiency (bottom) of a 12 µm TiO2 film derivatized with [G3–AnD]–CO2H and [G3–AnT]–CO2H. | |
The photovoltaic performance characteristics of [G3–AnD]–CO2H and [G3–AnT]–CO2H sensitized solar cells are shown in Fig. 6. The incident monochromatic photon-to-current conversion efficiency (IPCE) of the [G3–AnT]–CO2H sensitized DSSC plotted as a function of excitation wavelength exhibit a high value, 76% at 460 nm. The IPCE of the [G3–AnD]–CO2H sensitized solar cell shows 73% at 400 nm and a blue-shifted response when compared to [G3–AnT]–CO2H, which is consistent with the absorption spectra shown in Fig. 3. Under standard global AM 1.5 solar conditions, the [G3–AnT]–CO2H sensitized cell gave a short circuit photocurrent density (JSC) of 3.70 mA cm−2, an open circuit voltage (VOC) of 631 mV and a fill factor (FF) of 0.67, corresponding to an overall conversion efficiency η21 of 1.55%. The JSC from the integrated IPCE of 3.65 mA cm−2 shows that the spectral mistmatch is less than 2%. Under the same conditions, the [G3–AnD]–CO2H sensitized cell yields a lower JSC of 2.90 mA cm−2 due to a blue-shifted light harvesting which leads to a lower η of 1.20%.
Further density functional B3LYP/3-21G* calculations were also performed for these three compounds. Corresponding orbital energy levels from HOMO-4 to LUMO+4 are shown in Fig. 7. It can be found that the HOMO levels of these three compounds are similar, while the LUMO levels decrease from [G3–AnS]–CO2H (S: single bond), [G3–AnD]–CO2H to [G3–AnT]–CO2H, which causes the HOMO–LUMO gaps also decrease in the same order. This order corresponds well with the red-shift from [G3–AnD]–CO2H to [G3–AnT]–CO2H in the UV-vis spectra shown in Fig. 4, Fig. 5 and the IPCE of the corresponding solar cells shown in Fig. 6.
![Orbital energy levels from HOMO-4 to LUMO+4 of [G3–AnS]–CO2H, [G3–AnD]–CO2H to [G3–AnT]–CO2H.](/image/article/2009/EE/b908670f/b908670f-f7.gif) |
| | Fig. 7 Orbital energy levels from HOMO-4 to LUMO+4 of [G3–AnS]–CO2H, [G3–AnD]–CO2H to [G3–AnT]–CO2H. | |
It is also noted that the HOMO, LUMO and LUMO+1 are separated from the other orbital, while the other orbitals are very close to each other. Molecular orbital distribution from HOMO-2 to LUMO+2 of [G3AnT]–CO2H are shown in Fig. 8. It is obvious from this that LUMO and LUMO+1 are localized on the moiety from the anthracene to carboxylic acid group, which is appropriate to inject photo-excited electrons into the conduction band of the TiO2 semiconductor. HOMO-1 and HOMO-2 are mainly localized on the dendron moiety. Electrons excited from these orbitals to LUMO and LUMO+1 indicate effective oriented electron transfer from the dendron moiety to the anchoring group, which to some extent demonstrates the validity of the strategy to connect the dendron to the anthracene. Molecular orbital distribution from HOMO-2 to LUMO+2 of [G3AnD]–CO2H is shown in Fig. 9. LUMO and LUMO+1 are also localized on the moiety from the anthracene to the carboxylic acid group, like [G3AnT]–CO2H. The HOMO-1 and HOMO-2 are mainly localized on the dendron moiety, which are similar to those of [G3AnT]–CO2H. However, the dihedral angle between the anthracene plane and the plane of the alkene group as well as the carboxylphenyl group, about 57° according to the DFT calculations, decreases the conjugation between these two moieties compared with those of [G3AnT]–CO2H, which are almost in the same plane. This may be one of the reason that [G3AnT]–CO2H performs better than [G3AnD]–CO2H as described above.
![Molecular orbital distribution from HOMO-2 to LUMO+2 of [G3AnT]–CO2H.](/image/article/2009/EE/b908670f/b908670f-f8.gif) |
| | Fig. 8 Molecular orbital distribution from HOMO-2 to LUMO+2 of [G3AnT]–CO2H. | |
![Molecular orbital distribution from HOMO-2 to LUMO+2 of [G3AnD]–CO2H.](/image/article/2009/EE/b908670f/b908670f-f9.gif) |
| | Fig. 9 Molecular orbital distribution from HOMO-2 to LUMO+2 of [G3AnD]–CO2H. | |
To the best of our knowledge, these results represent a breakthrough in the design and development of alkyne bridged dendron chromophores, and therefore we believe that the data and findings of this study should spark broad interest in the field of dendron-based sensitized solar cells, useful for photovoltaic windows that transmit part of the visible light and harvest in the blue part of the spectrum.
Conclusions
We have established that alkyne π-bridged chromophores exhibit superior incident monochromatic photon-to-current conversion efficiency and enhanced red response compared to alkene π-bridged chromophores. Under standard global AM 1.5 solar conditions the former (alkyne π-bridged) sensitized cell gave an overall conversion efficiency of 1.55%. On the other hand, the alkene π-bridge sensitized cell gave an overall conversion efficiency of 1.20% demonstrating the influence of electronic coupling between the chromophore and the TiO2 semiconductor on device performance. Density functional calculations interpreted a tunability of band gap by π-conjugation and better electron coupling in the alkyne π-bridged chromophore which was consistent with the experimental results.
Acknowledgements
This research was supported by the Korea Foundation for International Cooperation of Science & Technology through the Global Research Lab (GRL) Program funded by the Ministry of Science and Technology, and MKE (The Ministry of Knowledge Economy), Korea, under the ITRC support program supervised by IITA (Institute for Information Technology Advancement) (IITA-2008-C1090-0804-0013). JHY, MKN and MG thank the Swiss National Science Foundation for financial support.
References
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphrybaker, E. Muller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS; M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613 CrossRef CAS; M. K. Nazeeruddin, Q. Wang, L. Cevey, V. Aranyos, P. Liska, E. Figgemeier, C. Klein, N. Hirata, S. Koops, S. A. Haque, J. R. Durrant, A. Hagfeldt, A. B. P. Lever and M. Grätzel, Inorg. Chem., 2006, 45, 787 CrossRef CAS; C. S. Karthikeyan, H. Wietasch and M. Thelakkat, Adv. Mater., 2007, 19, 1091 CrossRef CAS; F. Matar, T. H. Ghaddar, K. Walley, T. DosSantos, J. R. Durrant and B. O'Regan, J. Mater. Chem., 2008, 18, 4246 RSC.
- D. P. Hagberg, T. Edvinsson, T. Marinado, G. Boschloo, A. Hagfeldt and L. C. Sun, Chem. Commun., 2006, 2245 RSC; S. Ito, H. Miura, S. Uchida, M. Takata, K. Sumioka, P. Liska, P. Comte, P. Pechy and M. Grätzel, Chem. Commun., 2008, 5194 RSC; D. Kuang, S. Uchida, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 1923 CrossRef CAS; J.-H. Yum, D. P. Hagberg, S.-J. Moon, K. M. Karlsson, T. Marinado, L. Sun, A. Hagfeldt, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2009, 48, 1576 CrossRef CAS.
- J. J. Cid, J. H. Yum, S. R. Jang, M. K. Nazeeruddin, E. M. Ferrero, E. Palomares, J. Ko, M. Grätzel and T. Torres, Angew. Chem., Int. Ed., 2007, 46, 8358 CrossRef CAS; P. Y. Reddy, L. Giribabu, C. Lyness, H. J. Snaith, C. Vijaykumar, M. Chandrasekharam, M. Lakshmikantam, J. H. Yum, K. Kalyanasundaram, M. Grätzel and M. K. Nazeeruddin, Angew. Chem., Int. Ed., 2007, 46, 373 CrossRef CAS.
- Q. Wang, W. M. Carnpbell, E. E. Bonfantani, K. W. Jolley, D. L. Officer, P. J. Walsh, K. Gordon, R. Humphry-Baker, M. K. Nazeeruddin and M. Grätzel, J. Phys. Chem. B, 2005, 109, 15397 CrossRef CAS.
-
M. K. Nazeeruddin and M. Grätzel, Encyclopedia of Electrochemistry: Semiconductor Electrodes and Photoelectrochemistry, Wiley-VCH, Weinheim, 2002 Search PubMed.
- P. Péchy, F. P. Rotzinger, M. K. Nazeeruddin, O. Kohle, S. M. Zakeeruddin, R. Humphrybaker and M. Grätzel, J. Chem. Soc., Chem. Commun., 1995, 65 RSC.
- F. Campus, P. Bonhote, M. Grätzel, S. Heinen and L. Walder, Sol. Energy Mater. Sol. Cells, 1999, 56, 281 CrossRef CAS.
- Z. S. Wang, F. Y. Li and C. H. Huang, J.
Phys. Chem. B, 2001, 105, 9210 CrossRef CAS.
- T. A. Heimer, S. T. Darcangelis, F. Farzad, J. M. Stipkala and G. J. Meyer, Inorg. Chem., 1996, 35, 5319 CrossRef CAS.
- C. R. Rice, M. D. Ward, M. K. Nazeeruddin and M. Grätzel, New J. Chem., 2000, 24, 651 RSC.
- J. M. Tour, Adv. Mater., 1994, 6, 190 CrossRef CAS; R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350 CrossRef.
- S. A. Vail, P. J. Krawczuk, D. M. Guldi, A. Palkar, L. Echegoyen, J. P. C. Tome, M. A. Fazio and D. I. Schuster, Chem.–Eur. J., 2005, 11, 3375 CrossRef CAS.
- C. J. Hawker and J. M. J. Frechet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- N. S. Baek, Y. H. Kim, S. G. Roh, B. K. Kwak and H. K. Kim, Adv. Funct. Mater., 2006, 16, 1873 CrossRef CAS.
- Y. J. Miao and G. C. Bazan, Macromolecules, 1997, 30, 7414 CrossRef CAS.
- N. S. Baek, H. K. Kim, E. H. Chae, B. H. Kim and J. H. Lee, Macromolecules, 2002, 35, 9282 CrossRef CAS; K. L. Paik, N. S. Baek, H. K. Kim, J. H. Lee and Y. Lee, Macromolecules, 2002, 35, 6782 CrossRef CAS.
- E. E. Nesterov, Z. G. Zhu and T. M. Swager, J. Am. Chem. Soc., 2005, 127, 10083 CrossRef CAS.
- S. Ito, P. Chen, P. Comte, M. K. Nazeeruddin, P. Liska, P. Péchy and M. Grätzel, Prog. Photovoltaics: Res. Appl., 2007, 15, 603 Search PubMed.
- E. P. Kirby and R. F. Steiner, J. Phys. Chem. B, 1970, 74, 4480 Search PubMed;
B. M. Krasovitskii and B. M. Bolotin, Organic Luminescent Materials, VCH, Weinheim, 1988 Search PubMed;
J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum, New York, 1999 Search PubMed.
- A. Bhaskar, G. Ramakrishna, Z. K. Lu, R. Twieg, J. M. Hales, D. J. Hagan, E. Van Stryland and T. Goodson, J. Am. Chem. Soc., 2006, 128, 11840 CrossRef CAS.
- The overall conversion efficiency derived from the equation: η = (JSC × VOC × FF)/light intensity.
|
| This journal is © The Royal Society of Chemistry 2009 |
Click here to see how this site uses Cookies. View our privacy policy here. 