Cu2+-induced room temperature hydrogen release from ammonia borane†
Suresh Babu
Kalidindi
,
Jobisha
Joseph
and
Balaji R.
Jagirdar
*
Department of Inorganic & Physical Chemistry, Indian Institute of Science, Bangalore, 560 012, India. E-mail: jagirdar@ipc.iisc.ernet.in; Fax: +91 80 2360 1552; Tel: +91 80 2293 2825
First published on 14th August 2009
Abstract
Mechanical stirring of ammonia borane with CuCl2 in the solid state resulted in the release of hydrogen at room temperature through the intermediacy of [NH4]+[BCl4]−.
Broader contextHydrogen is a very promising candidate that is tipped to meet future energy demands. Developing methodologies to store and release hydrogen around ambient conditions is one of the major hurdles facing the so-called hydrogen economy. Ammonia borane (H3N·BH3) with its striking 19.6 weight% hydrogen is being widely investigated as the material to meet these challenges. The present study reveals a simple but very effective way to release hydrogen from ammonia borane at temperatures as low as room temperature. At a loading of only 5 mol% CuCl2 ∼9 wt% of hydrogen is released in 6.5 h at ∼80 °C, the temperature at which proton exchange membrane (PEM) fuel cells operate. The reaction for hydrogen generation proceeds through a new intermediate [NH4]+[BCl4]− which excludes the formation of borazine byproduct. The simplicity and cost effectiveness of this procedure makes this reaction scheme extremely attractive for real time applications. |
Ammonia borane, AB a compound with 19.6 wt% of hydrogen content is under intense investigation as a chemical hydrogen storage medium in recent times.1 Ammonia borane is a stable white crystalline solid under ambient conditions. Hydrogen can be released from ABviathermolysis or by catalytic dehydrogenation in protic solvents. Metal catalyzed AB hydrolysis or methanolysis of AB has been studied using both precious and first row transition metals.2Regeneration of AB from final products possessing B–O fragments, of hydrolysis or methanolysis reactions, is a highly energy intensive process. Metal complexes such as [Ir(H)2(POCOP)] (POCOP = η3-1,3-(OP-tert-Bu)2C6H3), Ni–NHC (NHC = N-heterocyclic carbene), Cp2Ti derivatives, [Rh(1,5-COD)Cl]2, and Re–nitrosysls have been found to be very effective in dehydrogenation of AB in non-aqueous media.3 The drawback in these cases is that the solvent contributes to the weight of the system leading to an overall lower hydrogen wt% of the system. In this regard, hydrogen release from ABvia solid state reactions is more practicable for mobile applications. The decomposition of neat AB is a multi-step process. Even though AB liberates hydrogen below 100 °C, the kinetics are sluggish with longer induction periods. Rapid release of one equivalent of hydrogen occurs upon melting of AB (around 110 °C). The second equivalent of hydrogen can be liberated from AB above 130 °C.4 Realization of faster hydrogen release rates of two moles of H2 (∼14 wt%) in a proton-exchange membrane fuel cell operating at a temperature below 80 °C is crucial for real time on-board applications.1aIn-situ11B NMR spectral studies on neat AB at 88 °C show that the formation of diammoniate of diborane (DADB) is the critical step in the decomposition of AB.5 Corroborating this, recently it has been shown that addition of DADB to AB decreases the induction time significantly.6 Tom Autery and co-workers used mesoporous silica (SBA-15) for enhanced hydrogen release rates.7 Ionic liquids which can stabilize the ionic DADB have also been found to be effective in dehydrogenation of AB.8LiH and NaH are found to destabilize AB by producing amido boranes.9 But very recently, Fijałkowski and Grochala have shown that thermolysis of sodium amidoborane liberates substantial amount of ammoniavia the [NaNH3]+[BH3(NHNa)BH3]− intermediate.10 In this context, chemical modification of AB for low temperature hydrogen release with high purity is highly desirable.
We recently reported Co2+-, Ni2+- and Cu2+- assisted AB hydrolysis for hydrogen generation.2e It was demonstrated that metal atoms/clusters formed in-situ during the hydrolysis were the real catalysts. Herein, we report our studies on the Co2+-, Ni2+- and Cu2+-induced H2 release from AB in the solid state.11
Mechanical stirring of CoCl2, NiCl2 and CuCl2 with AB in the solid state resulted in significant hydrogen evolution at 60 °C. Fig. 1a shows a comparison of Co2+-, Ni2+-and Cu2+- (M2+/AB = 0.15) induced H2 evolution from AB at 60 °C. As evident from the figure, Cu2+-induced H2 evolution was found to be more efficient than that of Co2+ and Ni2+. Moreover, in the case of Cu2+, liberation of H2 started almost instantaneously, whereas in the case of Co2+ and Ni2+, H2 release was slower during the initial periods of thermolysis. For a 4 h reaction time, the Co2+-, Ni2+- and Cu2+-induced reaction generated ∼1.1, 0.5, and 2.0 equivalents of hydrogen respectively. Gislon and co-workers reported ball milling of AB with 1 wt% of H2PtCl6 that resulted in the liberation of about 1 mole of hydrogen at 138 °C in 1.25 h.12 Ammonia borane loaded with 5 wt% Li/CMK-3 framework released ∼7 wt% (excluding Li/CMK) of hydrogen around 60 °C.13 Even though lithium and sodium amido boranes were shown to liberate two moles of hydrogen with good kinetics, recent studies have shown that these materials release a substantial amount of ammonia along with H2. Hydrogen released from AB induced by Cu2+ (Cu2+/AB = 0.15) was found to be more than that of Co2+ and Ni2+; 2 moles of H2 were liberated in 4 h at 60 °C, which is well below the fuel cell operating temperature. Considerable amounts of H2 release was noted even at room temperature. The CuCl2–AB mixture was stirred at room temperature for 24 h. The room temperature hydrogen release for various Cu2+ concentrations is shown in Fig. 1b. To the best of our knowledge, this is the best system realized to date for the release of hydrogen from AB in the solid state. In this context the results obtained here are significant; around 8 wt% of hydrogen was obtained in the Cu2+-induced reaction at 60 °C, in addition to our findings that the H2 release can take place at room temperature.‡
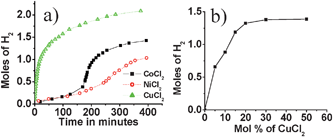 | ||
| Fig. 1 (a) Hydrogen evolution from M2+–AB mixtures at 60 °C (M2+ = Co2+, Ni2+, and Cu2+; M2+/AB = 0.15); (b) Cu2+-induced hydrogen evolution from AB at room temperature for various Cu2+ concentrations. | ||
Volumetric H2 measurements for H2 release at 60 °C were carried out by varying Cu2+ concentration (Fig. 2a) and also by varying the temperature (Fig. 2b) for a fixed Cu2+ concentration (Cu2+/AB = 0.05). Increase in Cu2+ concentration or temperature resulted in faster H2 release from AB.
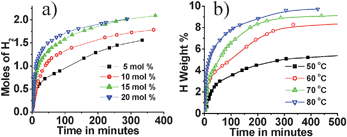 | ||
| Fig. 2 (a) Hydrogen generation from Cu2+–AB composite at 60 °C for various Cu2+ concentrations; (b) Hydrogen generation from Cu2+–AB composite (Cu2+/AB = 0.05) at different temperatures. | ||
Fig. 3a shows a comparison between the thermal behavior of neat AB and the Cu2+/AB combination. Neat ammonia borane showed two weight losses as expected around 100 °C and 150 °C.4 The Cu2+/AB system showed an initial weight loss of around 10 wt% below 60 °C, which was assigned to the loss of water from CuCl2 and also H2 release from AB. Evidence for this assignment was obtained from mass profiles of m/z = 2 (Fig. 3b) and m/z = 17,18 (see ESI†). Exposure of anhydrous CuCl2 to the atmosphere although brief, (1–2 min) during sample preparation and loading, in the TGA-MS instrument could not be avoided thereby enabling the sample to pick up some water from the atmosphere. The 10 wt% loss suggests the presence of approximately one water molecule per CuCl2. For Cu2+/AB = 0.05, a maximum of 0.016 equivalent of AB can undergo hydrolysis reaction, which is almost negligible. No detectable borate species were observed by IR and NMR spectroscopies, suggesting that the H2 evolution was solely due to thermolysis of AB and ruled out the hydrolysis pathway. Attempts to detect ammonia which has a m/z value very close to that of water were unsuccessful. No change in the pH of water in a bubbler through which the evolved gas was continuously purged, was noted. Borazine which is a major impurity in the decomposition of neat AB was not observed in the mass profile of m/z = 80. These observations rule out NH3 and borazine evolution and show that the H2 evolved is highly pure.
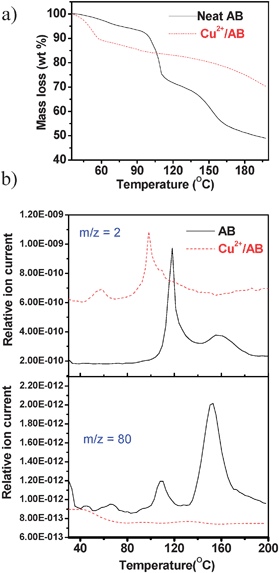 | ||
| Fig. 3 (a) TG profiles of neat AB (solid line) and Cu2+/AB composite (dashed line); (b) Synchronous MS profiles of m/z = 2 (H2), m/z = 80 (borazine). The ramp rate is 2 °C min−1. | ||
In order to probe the reaction further, we recorded 11B MAS-NMR spectrum of Cu2+/AB combination after thermolysis at 60 °C for 1.5 h. The NMR spectrum (Fig. 4) showed four broad peaks centered around δ −28, 3, 12, and 23.
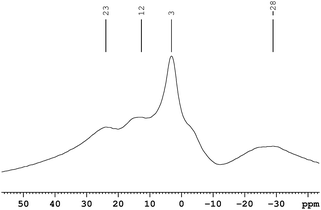 | ||
| Fig. 4 11B MAS NMR spectrum of Cu2+/AB composite after 1.5 h of thermolysis at 60 °C. | ||
The latter two peaks can be assigned to B(H)N2 and BN(H)2 of polymeric amino borane (PAB).14 We tentatively assign the peak centered around δ 3 to [NH4]+[BCl4]− based on the following: (a) the 11B NMR resonance for H3N·BCl3 was observed at δ 4.1 by Hu and Geanangel15 and (b) Landesman and Williams found that the chemical shifts of BCl3.NR3 and BCl4− to be very similar.16
The broad peak around δ −28 can be assigned to µ-aminodiborane [B2H5(µ-NH2)].15,17 Also a shoulder was observed around δ −2 which can be assigned to the Cl2BHNH3 species.15 The powder XRD pattern (see ESI†) of the final product showed reflections corresponding to Cu(0) and very weak reflections assignable to NH4Cl. All of these observations are consistent with eqn (1).
| H3N·BH3 + 2CuCl2 → [NH4]+[[BCl4]− + H2 + 2Cu(0) | (1) |
We believe that the formation of [NH4]+[BCl4]− is the nucleation step in the Cu2+-induced decomposition of AB. [NH4]+[BCl4]− can propagate the reaction by reacting further with AB and can form intermediates of the type ClBH2NH3 and Cl2BHNH3. The latter species was observed in the 11B MAS-NMR spectrum. Also, the observation of µ-aminodiborane corroborates the formation of ClBH2NH3 as an intermediate. Similar type of intermediates were observed when BCl3 was treated with AB in ether solution.15 To test the catalytic activity of in-situ generated Cu(0) nanoparticles, Cu nanoparticles (∼20 nm) were added to neat AB and thermolysis was carried out at 60 °C. Only 0.47 moles of H2 were liberated in 17 h from this reaction. Solid state decomposition of neat AB involves formation of the diammoniate of diborane (DADB) in the nucleation step. Addition of CuCl2 to AB provides an alternative path for the decomposition of ABvia the intermediate [NH4]+[BCl4]−. This eliminates the route to borazine formation and changes the overall feature of hydrogen release.
In conclusion, addition of CoCl2, NiCl2 or CuCl2 salts to neat AB resulted in significant destabilization of AB. Cu2+-induced dehydrogenation of AB released a considerable amount of hydrogen even at room temperature. For Cu2+/AB = 0.05, 2 moles of H2 were liberated in 6.5 h, which is equal to 9 wt% of the system. Intermediacy of [NH4]+[BCl4]− eliminates the formation of borazine impurity thereby affording pure H2. These features and the additional cost effectiveness of the first row transition metal salts used for AB dehydrogenation make this reaction scheme extremely attractive.
Acknowledgements
We are grateful to the Board of Research in Nuclear Sciences, Department of Atomic Energy, India for the financial support. SBK thanks the Council of Scientific & Industrial Research, India for a fellowship.Notes and references
- (a) T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116 CrossRef CAS; (b) F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613 RSC; (c) B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479 RSC; (d) C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279 RSC.
- (a) F. Cheng, H. Ma, Y. Li and J. Chen, Inorg. Chem., 2007, 46, 788 CrossRef CAS; (b) T. J. Clark, G. R. Whittell and I. Manners, Inorg. Chem., 2007, 46, 7522 CrossRef CAS; (c) M. Chandra and Q. Xu, J. Power Sources, 2007, 168, 135 CrossRef CAS; (d) S. B. Kalidindi, U. Sanyal and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2008, 10, 5870 RSC; (e) S. B. Kalidindi, M. Indirani and B. R. Jagirdar, Inorg. Chem., 2008, 47, 7424 CrossRef CAS; (f) S. B. Kalidindi, A. A. Verneker and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2009, 11, 770 RSC; (g) J.-M. Yan, X.-B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2289.
- (a) M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048 CrossRef CAS; (b) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS; (c) Y. Luo and K. Ohno, Organometallics, 2007, 26, 3597 CrossRef CAS; (d) C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424 CrossRef CAS; (e) Y. Jiang and H. Berke, Chem. Commun., 2007, 3571 RSC.
- (a) F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Rößler and G. Leitner, Thermochim. Acta, 2002, 391, 159 CrossRef CAS; (b) G. Wolf, J. Baumann, F. Baitalow and F. P. Hoffmann, Thermochim. Acta, 2000, 343, 19 CrossRef CAS.
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 9, 1831 RSC.
- D. J. Heldebrant, A. Karkamkar, N. J. Hess, M. Bowden, S. Rassat, F. Zheng, K. Rappe and T. Autrey, Chem. Mater., 2008, 20, 5332 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. Butterick III, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748 CrossRef CAS.
- (a) Z. Xiong, C. K. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 CrossRef CAS; (b) Z. Xiong, G. Wu, Y. S. Chua, J. Hu, T. He, W. Xu and P. Chen, Energy Environ. Sci., 2009, 2, 360 Search PubMed.
- K. J. Fijałkowski and W. Grochala, J. Mater. Chem., 2009, 19, 2043 RSC.
- During the preparation of this manusript, Ping Chen and co-workers reported nanosized Co- and Ni-catalyzed AB dehydrogenation. T. He, Z. Xiong, G. H. Chu, T. Zhang and P. Chen, Chem. Mater., 2009, 21, 2315 Search PubMed.
- S. D. Benedetto, M. Carewska, C. Cent, P. Gislon, M. Pasquali, S. Scaccia and P. P. Prosini, Thermochim. Acta, 2006, 441, 184 CrossRef CAS.
- B. L. Li, X. Yao, C. Sun, A. Du, L. Cheng, Z. Zhu, C. Yu, J. Zou, S. C. Smith, P. Wang, H.-M. Cheng, R. L. Frost and G. Qing (Max) Lu, Adv. Funct. Mater., 2009, 19, 265 CrossRef.
- C. Gervais and F. Babonneau, J. Organomet. Chem., 2002, 657, 75 CrossRef CAS.
- M. G. Hu and R. A. Geanangel, Inorg. Chem., 1979, 18, 3297 CrossRef CAS.
- H. Landesman and R. E. Willliams, J. Am. Chem. Soc., 1961, 83, 2663 CrossRef CAS.
- L. D. Schwartz and P. C. Keller, J. Am. Chem. Soc., 1972, 94, 3015 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, powder XRD. See DOI: 10.1039/b909448m |
| ‡ Caution:Milling of anhydrous CuCl2 and ammonia borane can result in eruption of fire. |
| This journal is © The Royal Society of Chemistry 2009 |