Harnessing electric power from monosaccharides—a carbohydrate–air alkaline fuel cell mediated by redox dyes†
Daniel
Scott‡
and
Bor Yann
Liaw
*
Hawaii Natural Energy Institute, School of Ocean and Earth Science and Technology, University of Hawaii at Manoa, 1680 East-West Road, POST 109, Honolulu, HI 96822, USA. E-mail: bliaw@hawaii.edu; Fax: +1 (808) 956-2336; Tel: +1 (808) 956-2339
First published on 19th June 2009
Abstract
Although carbohydrates play a central role in the biochemical pathways of biological systems, current technologies do not allow us to seriously consider the direct oxidation of monosaccharides such as glucose as a prominent source of power for electronic devices. Here we show a simple, inexpensive approach to harness chemical energy from glucose, converting it directly to electric power without a precious metal, enzyme or microorganism catalyst to promote monosaccharide oxidation. The design of this abiotic anode using inexpensive chemical dyes in alkaline solutions with high-surface-carbon materials is capable of harnessing electrical power from glucose. In conjunction with a commercial air-breathing electrode the resulting cell can generate maximum power at about 0.3 V and more than 9 mA cm−2; thus more than 2.5 mW cm−2. This power density surpasses any existing biotic or abiotic design. This approach might open the door to a broader possibility in using such monosaccharides in energy storage and harvesting to power small devices.
Broader contextAlthough there has been great interest in high energy content bio-fuels such as carbohydrates, prior attempts have been limited by cost-prohibitive catalysts that are sensitive to operating conditions. Here we show a simple and relatively inexpensive approach to harness chemical energy from glucose and other monosaccharides, converting such a chemical energy directly to electric power without a precious metal, enzyme or microorganism catalyst involved in the monosaccharide oxidation. Such an anode configured with an air-breathing cathode under alkaline conditions produces power capable of powering small devices. |
Introduction
It has long been recognized that carbohydrates are important and somewhat ubiquitous carriers in supplying energy to living organisms. As such it seems “natural” that this carbohydrate fuel would be the most environmentally friendly. In biological metabolism processes, harnessing carbohydrate energy needs to rely on enzymes. To harness such energy in abiotic devices, only limited success has been demonstrated in fuel cell configurations1 that permit the direct conversion of electricity from a high energy carbohydrate such as glucose. Two classifications of fuel cell designs1–13 have been reported, distinguished by the type of catalyst used. In attempts to mimic nature, biotic designs using catalysts (such as enzymes and microorganisms) are favored for commercialization, as exemplified by the recent work of Sakai et al.13 The other class utilizing inorganic or precious metal catalysts in an abiotic design1 is primarily for glucose sensing and medical implant applications.Prior pursuits in abiotic fuel cell designs1 using carbohydrate fuels have been hampered by the inability of the precious metal-based catalysts to sustain power production due to poisoning. Biotic designs are thus favored to date using either microbial2–5 or enzymatic systems.6–13 Low power density, short lifetime, and complex electrode design are impeding the progress in most of these biotic systems.14 Most of the prior art dealing with aqueous fuels has to operate in acidic or neutral pH conditions; very little has been explored in the alkaline solutions. Here we present an abiotic design (Fig. 1) using inexpensive chemical dyes in alkaline solutions with high surface carbon materials, capable of harnessing electrical power from various carbohydrates including glucose. In conjunction with a commercial air breathing electrode, the resulting cell generates maximum power at about 0.3 V and more than 9 mA cm−2 or more than 2.5 mW cm−2 (geometric area) (see Fig. 2).
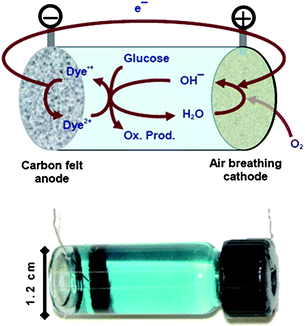 | ||
| Fig. 1 Reaction scheme of the glucose cell that takes advantage of the reactive glucose intermediate under strongly basic conditions. | ||
The power density from our rudimentary design surpasses any existing biotic or abiotic designs. More importantly, our anodic configuration does not call for any expensive catalysts; therefore, no poisoning or other impeding issues are associated with the longevity of cell operation. All materials used in the cell design are readily available as off-the-shelf components.
The potential of offering such a cell is of great interest, as it is simple, inexpensive, and efficient. This cell is also free of precious metals at the anode, open-to-air§, and does not contain a membrane. Cell operation is at room temperature, and power is relatively stable over long hours at the above power outputs before the fuel is replenished (in our current batch operation).
Experimental
Materials
Methyl viologen (MV), indigo carmine, and all other dyes, KOH and glucose (and other carbohydrates) were purchased from Sigma-Aldrich and were used without further purification. Carbon felt (product # 43199) with a thickness 3.18 mm was purchased from Alfa Aesar. An air-breathing oxygen-reduction cathode (silver-plated nickel screen electrode with 0.6 mg cm−2 TM loading using 10% Pt on Vulcan XC-72 with a micro-porous fluorocarbon backing) was purchased from BASF and used from the package without treatment.Instrumentation/fuel cell apparatus and assembly
A Bio-Logic 16-channel VMP3 potentiostat/galvanostat was used to conduct all electrochemical measurements. In the cell the air-breathing cathode material was exposed to the open air without any additional air flow or oxygen enrichment. The electrolyte in the cells was only the stated components (KOH, glucose, MV) differing in concentrations. For the experiment in Fig. 2, concentrations were 2 M glucose, 3 M KOH and 28 mM MV. For the experiments in Fig. 3, concentrations were the same as for the experiment in Fig. 2 except that the concentration of one of the constituents was varying in three different experiments in each graph.Other redox dyes perform comparably and their cell voltage varies according to dye potentials. An example using indigo carmine (6 mM with 2 M glucose and 3 M KOH) is shown in Fig. S2.† No stirring or agitation of the solution was applied to the cell during power production (Fig. 2 and Fig. 3). We also conducted experiments with cells under mechanical stirring, and the performance of the cells was greatly improved (Fig. S3†). All experiments were conducted at room temperature and ambient pressure. Pt wire was used for electrode contact to minimize corrosion interference.
Results
In this work, a number of commonly available dyes, such as methyl viologen (MV), methylene blue, Meldola's blue, etc., were used as electron mediators. By properly connecting these cells in series or parallel (Fig. S4†), we were able to generate power from glucose and other monosaccharides sustainably (Table S1†). Fig. 2 displays the performance of a glucose alkaline fuel cell with 2 M glucose in 3 M KOH and 28 mM MV solution at room temperature. In this rudimentary cell design, we can easily achieve an open circuit voltage of 0.65 V, with little dependence on the glucose concentration. In this case, the anode was at −0.63 V versus Ag/AgCl, which is close to what is expected from the formal redox potential of MV,15,16 whereas the air-breathing cathode was at 0.02 V against Ag/AgCl. Fig. 3 depicts the polarization curves (steady-state cell voltage versus polarization current density profiles), showing the dependence of cell performance on the concentration of (a) glucose, (b) MV, and (c) KOH. A steady voltage after each current step can be reached in about 20–30 min if not shorter. In each case, the baseline concentration for the three constituent reagents are as follows: glucose : MV : KOH = 2 M : 28 mM : 3 M, which is the same as that of the experiment shown in Fig. 2. Each inset in Fig. 3 is a plot of (short-circuit) limiting current density versus the constituent reagent's concentration. In a cell containing only KOH and dye, we did not detect any sustainable power generation.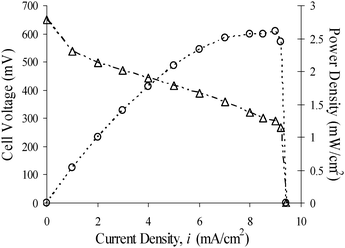 | ||
| Fig. 2 Polarization curve and power profile for a cell of 2 M glucose in 28 mM MV and 3 M KOH solution. | ||
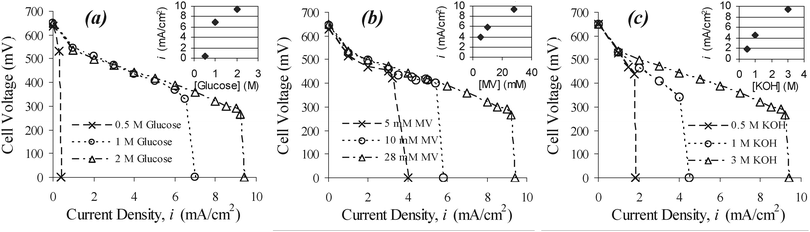 | ||
| Fig. 3 Dependence of current generation on concentration of (a) glucose (with 28 mM MV and 3 M KOH), (b) MV (with 2 M glucose and 3 M KOH), and (c) KOH (with 2 M glucose and 28 mV MV). | ||
A control experiment using 2 M glucose and 3 M KOH in which the cell is opperated without the dye produces a notable amount of current, but it is still significantly limited without the dye acting as a mediator (Fig. S1(a)†). Results for a control experiment in which the cell is operated in 2 M glucose, 3 M KOH and 28 mM MV but in the presence of only the Pt wire that collects current from the carbon felt are given in Fig. S1(b).† The galvanostatic polarization curves for various concentrations of glucose and dye show that the cell voltage follows a common polarization profile. This common profile exhibits a rather low activation polarization loss, indicating that the polarization is likely dominated by the electrolyte conductivity, as evident in the dependence of the KOH concentration, until the reaction reaches a purported mass transport limitation.17 In Fig. 4 the power profiles reflect two distinct patterns in the behavior between dye (and glucose) and KOH. In the case of dye (and glucose), as shown in Fig. 4(a), the power profiles superimpose on one another at current densities below the onset of mass transport derived limiting currents; thus, they follow a common power generation profile over a wide range of dye (and glucose) concentration, which determines the (short-circuit) limiting polarization current density. The mediator concentration varies from 5 mM to 28 mM and the glucose concentration from 0.5 M to 2 M in 3 M KOH solution. The common power profile depicts that the maximum power and limiting current monotonically increase with the dye or glucose concentration (Fig. 4(a)), ranging from 0.53 V, 0.3 mA cm−2, and 0.16 mW cm−2 at 0.5 M glucose and 28 mM MV to 0.29 V, 9 mA cm−2, and 2.6 mW cm−2 at 2 M glucose and 28 mM MV. In contrast, the power profiles of different KOH concentrations do not overlap (Fig. 4(b)). The polarization curves also show different slopes in the Ohmic regime (Fig. 3(c)), indicating that the ionic conductivity of the electrolyte is critical to power generation. We also noticed that the transition from the maximum power generation to short-circuit limiting current operation is quite rapid, as shown in all figures. This transition seems faster than one would expect from a conventional mass-transport-limiting regime.17 To determine a total coulombic efficiency of glucose oxidation, long polarization trials were performed in cells of various conditions. An example of a cell that was allowed to produce power from a 1.6 mL solution of 400 mM glucose in 6 mM MV and 1 M KOH is shown in Fig. S5.† Current was drawn from the cell at 0.7 mA cm−2 and the cell voltage remained above 0.5 V for longer than 15 h. This experiment illustrated the feasibility of sustainable power generation with an overall coulombic efficiency of about 37%.
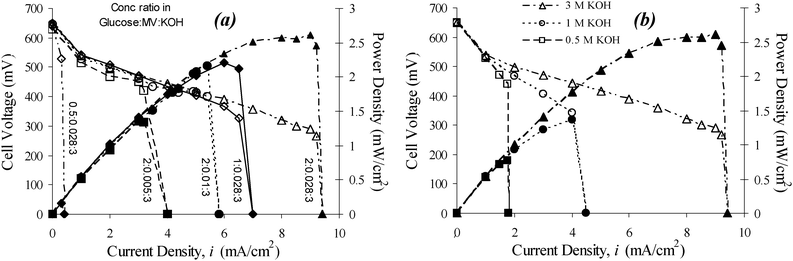 | ||
| Fig. 4 Polarization curves and power profiles for cells with (a) different concentration ratios of glucose versus MV in 3 M KOH, and (b) different KOH concentrations at 0.5, 1, and 3 M, respectively with 2 M glucose and 28 mM MV. | ||
Discussion
From the literature, it is known that in enzymatic bio-fuel cells, glucose oxidation occurs in a two-electron process,10,18 due to the selective nature of the enzyme catalysis, as follows:On the anode:
| β-D-glucose → δ-gluconolactone + 2H+ + 2e− | (1) |
On the cathode:
| O2 + 4H+ + 4e− → 2H2O | (2) |
Cell:
| 2β-D-glucose + O2 → 2δ-gluconolactone + 2H2O | (3) |
In microbial bio-fuel cells, the metabolism in the living cells can drive the glucose oxidation completely to CO2 according to:3
On the anode:
| β-D-glucose + 6H2O → 6CO2 + 24H+ + 24e− | (4) |
On the cathode:
| O2 + 4H+ + 4e− → 2H2O | (5) |
Cell:
| β-D-glucose + 6O2 → 6CO2 + 6H2O | (6) |
In abiotic cells where a Pt-based anode is used, the glucose is partially oxidized to gluconic acid in a two-electron process:
On the anode:
| β-D-glucose → gluconic acid + 2H+ + 2e− | (7) |
These reactions are for acidic or neutral pH conditions. In alkaline cells (disregarding the presence of MV), the glucose oxidation should occur through a partial oxidation pathway:19
In the solution:
| β-D-glucose + 2MV2+ + 2OH− → δ-gluconolactone + 2H2O + 2MV˙+ | (8) |
The reduced MV (MV˙+) will shuttle the electron and re-oxidize on the anode surface according to:11,16
On the anode:
| MV˙+ ↔ MV2+ + e− | (9) |
On the cathode:
| O2 + 4H2O + 4e− → 4OH− | (10) |
Cell:
| 2β-D-glucose + O2 → 2δ-gluconolactone | (11) |
It is important to note that the redox MV basically functions as an electron shuttle medium, while the partial oxidation of glucose actually occurs in the solution in the presence of hydroxide ions. The detailed electron transfer mechanism is still under investigation in our laboratory. A similar experiment has been reported recently by Hansen et al.20 who proposed a complete glucose oxidation pathway as described in (4). As their results are preliminary, there is no reason at this point to question previous studies which suggest that OH− interacts with glucose in the formation of the glucose-ene-diol.21–23 This ene-diol likely serves as the electron transfer intermediate. The product of this reaction would be gluconolactone.19 It is unlikely that further oxidation would occur,19 because the lactone and its derivative are quite stable. We also question whether the complete oxidation of glucose, as suggested by Hansen et al., can be achieved with a single mediator like viologen. Such a pathway requires a number of C–C bonds to be broken down by the mediator. It is also worth noting that it is of interest to determine the purity of the gluconolactone as the resulting product of glucose oxidation. Glucolactone can be a chemical of value for use in food additive as acidifier, sequestrant or curing agent, depending on its purity. It is however difficult to speculate any utility or down stream value of this by-product, or if a separation method should be developed to exploit such value at this juncture.
The common power generation profile shared by glucose and dye over a wide range of concentration is a unique feature that we think deserves some elaboration. As we observed, the anode exhibits the redox potential of MV at open circuit, and such potential is independent of glucose concentration. It is evident that the anodic reaction indeed involves the oxidation of reduced MV (MV˙+) as described in (9). Such MV redox reaction and its applications in providing electron shuttling between substrate reaction at a bioelectroactive center and current-collecting electrode surface have been often used in mediated bio-electrodes for biosensing and bio-fuel cell applications.11,15 Katz et al.11 have discussed such a mediated process in bio-fuel cell applications. Cooney and Liaw15 use viologen as an example to explain mediated bio-electrode reaction potential in enzymatic biofuel cell operation. Niessen and Notten16 even used MV redox in the study of oxygen influence on hydride storage mechanism. The insensitivity of open circuit potential to glucose concentration also implies that glucose is not electroactive on the anode surface. As we showed that the current/power generation would only occur in the presence of glucose, it is therefore evident that glucose is the fuel. Variations of glucose or dye concentration in 3 M KOH solution will affect the electron transfer rate thus altering the amount of reduced MV available for current/power generation. Our experiments indicate a commonly shared power profile for glucose and dye; therefore, the current appears to reflect the flux of reduced dye arriving at the anode. We can further infer that the maximum power is contingent on the flux of the dye at its mass transport limit for a specific concentration; and, once the imposed galvanostatic polarization exceeds such mass transport limit, the cell will run to short circuit, which explains the rapid drop-off of the cell voltage to the short-circuit condition beyond the maximum power. Fig. S3† provides additional evidence of such mass transport limitation. By mechanical stirring, better mass transport was facilitated and resulted in a longer, more gradual tail in the polarization curve and a higher limiting current density. Finally, it is important to point out that ene-diol formation occurs in the solution. Through electron transfer between the ene-diol intermediate and the mediator, which migrates to the anode surface to complete the half-cell reaction, we were able to harness the chemical energy from the partial oxidation of glucose. This mechanism is very different from those in traditional fuel cells where the fuel redox reactions occur on the electrode surfaces (or electrolyte/electrode interface) and require help with catalysts.
Given the multi-body interactions involved in the electron transfer, the stepwise reaction mechanism should be further investigated to provide more insight on the rate limiting step in order to push the extent of glucose oxidation for better use of the fuel and efficiency. As the partial oxidation two-electron process in (8) prevails, we have derived the coulombic charge transfer efficiency on the order of 30–40% based on several long galvanostatic polarization experiments. The depletion of glucose in the solution was inferred by the decrease in voltage at the end of the cell operation under the galvanostatic condition. The voltage of the expended cell would not respond to additional KOH or dye. Only the addition of glucose to the expended cell would cause the cell to produce power again.
The cause of parasitic loss in coulombic efficiency remains ambiguous at this time. One possible loss could be the formation of peroxides24 because the reactive ene-diol form of glucose may interact with dissolved O2 which could poison the cathode reaction. Another possible cause is related to the stability of the dye in the charge transfer cycle. It has been reported16 that overly-reduced MV to MV˙˙ may become inactive to electron transfer. Another cause of parasitic loss may be due to oxygen, as it is known for its affinity to oxidizing reduced dyes quickly. We have also noted§ the possible interference of CO2 in the air and the associated bicarbonate–carbonate formation and shuttle process which may lead to efficiency loss. However, our preliminary results show no noticeable difference in the maximum power generation between aerobic and anaerobic conditions.
Conclusions
We have demonstrated an intriguing approach to produce electrical power directly from monosaccharides that does not rely on precious metal or difficult-to-manage bio-catalysts to assist glucose oxidation. The resulting current and power density surpasses any existing glucose fuel cell designs. It is simple to assemble and operate with a variety of inexpensive raw materials. The process uses materials that are abundant and therefore is not projected to be resource limited. Our results suggest that the partial oxidation of glucose occurs in the bulk solution, while the anode half-cell reaction relies on a mediator shuttle process. This mechanism may allow the cell operating at an optimal volume-to-surface ratio to achieve the maximum power generation.Acknowledgements
We would like to thank the funding support for DS under the Intelligence Community Postdoctoral Fellow Research Program (National Geospatial-Intelligence Agency, HM1582-04-I-2013). Further gratitude goes to the AFOSR funded MURI program team on “Fundamentals & Bioengineering of Enzyme Fuel Cells” (FA9550-06-1-0264) for their stimulating discussions and active collaborations. Additional gratitude also goes to Dr. Richard Rocheleau who provides partial support under the Hawaii Energy and Environmental Technology (HEET) initiative funded by ONR (N00014-06-1-1055 and N00014-07-1-1094).References
- S. Kerzenmacher, J. Ducrée, R. Zengerle and F. von Stetten, Energy harvesting by implantable abiotically catalyzed glucose fuel cells, J. Power Sources, 2008, 182, 1–17 CrossRef CAS.
- R. M. Allen and H. P. Bennetto, Microbial fuel cells: electricity production from carbohydrates, Appl. Biochem. Biotechnol., 1993, 39/40, 27–40 Search PubMed.
- S. K. Chaudhuri and D. R. Lovley, Electricity generation by direct oxidation of glucose in mediatorless microbial fuel cells, Nat. Biotechnol., 2003, 21, 1229–1232 CrossRef.
- H. Richter, K. McKarthy, K. P. Nevin, J. P. Johnson, V. M. Rotello and D. R. Lovley, Electricity generation by Geobacter sulfurreducens attached to gold electrodes, Langmuir, 2008, 24, 4376–4379 CrossRef CAS.
- B. E. Logan, P. Aelterman, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, W. Verstraete and K. Rabaey, Microbial fuel cells: Methodology and technology, Environ. Sci. Technol., 2006, 9, 5181–5192 CrossRef.
- R. A. Bullen, T. C. Arnot, J. B. Lakeman and F. C. Walsh, Biofuel cells and their development, Biosens. Bioelectron., 2006, 21, 2015–2045 CAS.
- S. Topcagic and S. D. Minteer, Development of a membraneless ethanol/oxygen biofuel cell, Electrochim. Acta, 2007, 51, 2168–2172.
- T. Chen, S. C. Barton, G. Binyamin, Z. Gao, Y. Zhang, H.-H. Kim and A. Heller, A miniature biofuel cell, J. Am. Chem. Soc., 2001, 123, 8630–8631 CrossRef CAS.
- N. Mano, F. Mao and A. Heller, Characteristics of a miniature compartment-less glucose-O2 biofuel cell and its operation in a living plant, J. Am. Chem. Soc., 2003, 125, 6588–6594 CrossRef CAS.
- A. Heller, Miniature biofuel cells, Phys. Chem. Chem. Phys., 2004, 6, 209–216 RSC.
- E. Katz, A. N. Shipway and I. Willner, Biochemical fuel cells, in Handbook of Fuel Cells-Fundamentals, Technology, Applications, ed. W. Vielstich, A. Gasteiger and A. Lamm, John Wiley & Sons, Chichester, PA, 2003, vol. 1(4), ch. 21, pp. 355–381 Search PubMed.
- S. C. Barton, J. Gallaway and P. Atanassov, Enzymatic biofuel cells for implantable and microscale devices, Chem. Rev., 2004, 104, 4867–4886 CrossRef CAS.
- H. Sakai, T. Nakagawa, Y. Tokita, T. Hatazawa, T. Ikeda, S. Tsujimura and K. Kano, A high-power glucose/oxygen biofuel cell operating under quiescent conditions, Energy Environ. Sci., 2009, 2, 133–138 RSC.
- S. D. Minteer, B. Y. Liaw and M. J. Cooney, Enzyme-based biofuel cells, Curr. Opin. Biotechnol., 2007, 18, 228–234 CrossRef CAS.
- M. Cooney and B. Y. Liaw, In situ characterization techniques for design and evaluation of micro and nano enzyme-catalyzed power sources, in Biomolecular Catalysis: Nanoscale Science and Technology, ed. J. Kim, S. H. Kim and P. Wang, ACS symposium series, 986, Washington, DC., the American Chemical Society, Cleveland, OH, 2008, pp. 289–333 Search PubMed.
- R. A. H. Niessen and P. H. L. Notten, The influence of O2 on the electrochemistry of thin film, hydrogen storage, electrodes, Electrochim. Acta, 2005, 50, 2959–2965 CrossRef CAS.
- M. Winter and R. J. Brodd, What are batteries, fuel cells, and supercapacitors?, Chem. Rev., 2004, 104, 4245–4269 CrossRef CAS.
- J. R. Rao, in Bioelectrochemistry. I. Biological Redox Reactions, ed. G. Milazzo and M. Blank, Plenum Press, New York, 1983, pp. 283–335 Search PubMed.
- S. B. Aoun, G. S. Bang, T. Koga, Y. Nonaka, T. Sotomura and I. Taniguchi, Electrocatalytic oxidation of sugars on silver-UPD single crystal gold electrodes in alkaline solutions, Electrochem. Comm., 2003, 5, 317–320 CrossRef CAS.
- D. Hansen, G. Watt, J. Nichols, M. Andrus, D. Wheeler and S. Choi, Viologen catalyst for direct-carbohydrate fuel cell, ECS Transactions, 2008, 16, 2057–2063 Search PubMed.
- H. F. Cui, J. S. Ye, X. Liu, W. D. Zhang and F. S. Sheu, Pt–Pb alloy nanoparticle/carbon nanotube nanocomposite: a strong electrocatalyst for glucose oxidation, Nanotechnology, 2006, 17, 2334–2339 CrossRef CAS.
- S. Itoh, M. Mure and Y. Ohshiro, Oxidation of D-glucose by coenzyme PQQ. 1,2-Enediolates as substrates for PQQ oxidation, J. Chem. Soc., Chem. Comm., 1987, 20, 1580–1581 Search PubMed.
- S. Shinkai, T. Kunitake and T. C. Bruice, Importance of 1,2-enediols in the reduction of lumiflavin by α-ketols, J. Am. Chem. Soc., 1974, 96, 7140–7142 CrossRef CAS.
- A. Weissberger, J. E. LuValle and D. S. Thomas, Jr., Oxidation processes. XVI. The autoxidation of ascorbic acid, J. Am. Chem. Soc., 1943, 65, 1934–1939 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experiments (Fig. S1–S6 and Table S1). See DOI: 10.1039/b906770a |
| ‡ Current address: Department of Chemistry, Brigham Young University-Hawaii, 55-220 Kulanui Street, Laie, HI 96762, USA. E-mail: mailto:sdanielm@byuh.edu; Fax: +1 (808) 675-3825; Tel: +1 (808) 675-3813. |
| § Although it is known that CO2 in the air and the bicarbonate/carbonate formation interfere cell operation in alkaline fuel cell and metal-air batteries, we have not observed any significant interference in the cell operation so far. This issue certainly needs to be further assessed and quantified. |
| This journal is © The Royal Society of Chemistry 2009 |