High capacity hydrogen storage in a hybrid ammonia borane–lithium amide material†
K. R.
Graham
,
T.
Kemmitt
and
M. E.
Bowden
*
Industrial Research Ltd, PO Box 31-310, Lower Hutt, New Zealand. E-mail: m.bowden@irl.cri.nz
First published on 30th March 2009
Abstract
The direct (solventless) reaction of lithium amide with ammonia borane is described and is demonstrated to form a new hybrid material approximating to LiNH2BH3NH3. This material rapidly loses 3.2 mole equivalents of hydrogen, (11.9 wt%) on heating to 250 °C, with the most rapid hydrogen release occurring at temperatures as low as 60 °C. The product was an insoluble, amorphous polymer with the general formula –(NLi–BNH2)n– which did not rehydrogenate directly on exposure to hydrogen gas pressure. The hybrid material and its decomposition products were characterised by solid state 11B NMR. Selective deuterium labelling helped to elucidate a reaction sequence for the hydrogen release using mass spectrometry of the released gases.
Broader contextHydrogen is favoured as a future fuel because of its high gravimetric energy content and the potential for low environmental impact. To be utilised as transport fuel, however, a means of safely and efficiently storing hydrogen in a relatively small volume is required. Materials containing approximately 10 wt% hydrogen are required to enable storage systems for fuel cell vehicles whose performance fulfils consumer expectations. Chemical hydrides, for example ammonia borane (NH3BH3), are seen as strong candidates to meet these demands. A successful material will ideally exhibit a high storage capacity as well as favourable release kinetics and thermodynamics at temperatures close to 100 °C, properties not realised by any material to date. The present paper demonstrates a new borane chemical environment arising from interaction between ammonia borane and lithium amide. This results in a significantly lower temperature for the release of hydrogen compared to ammonia borane, and a total of 11.2 wt% H2. The mechanism of hydrogen release has been investigated and will assist the discovery and design of future storage materials. |
Introduction
The development of a safe and efficient means of storing hydrogen is one of the major technical challenges facing the establishment of a hydrogen economy. Storage for automotive use represents one of the more demanding applications,1 where systems with a high gravimetric and volumetric hydrogen density in addition to favourable economics, durability, and charge/discharge rates are required. In a fuel cell powered vehicle, it is advantageous if the waste heat generated by the fuel cell (typically 90–100 °C) can be used to drive the release of hydrogen from the storage material.Ammonia borane (NH3BH3, AB)2–4 is a leading hydrogen storage material on account of its high gravimetric (19.6 wt%) and volumetric (0.145 kg L−1) storage densities. However the temperature for hydrogen release is higher than ideal: 1 equivalent (6.5 wt%) of hydrogen is released at approximately 100 °C, but the 2nd equivalent requires temperatures around 150 °C.5 In addition, the first equivalent of hydrogen is subject to an induction period during which the diammoniate of diborane ([(NH3)2BH2]+[BH4]−), an ionic isomer of AB, is formed as a crucial precursor to hydrogen release.6
As part of an international collaboration under the International Partnership for the Hydrogen Economy we have examined the hydrogen release characteristics of mixtures of AB with a second phase containing hydrogen in a polar or ionic form. We anticipate that an interaction between the protic N![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3BH3 or hydridic NH3B3 hydrogens and the admixed compound will result in more rapid or lower temperature hydrogen release. Xiong et al.7 have demonstrated this approach by ball-milling AB with either LiH or NaH to form alkali amidoboranes MNH2BH3. These compounds release 2 equivalents of hydrogen at ca. 90 °C, which corresponds to 10.9 wt% H2 for LiNH2BH3.
3BH3 or hydridic NH3B3 hydrogens and the admixed compound will result in more rapid or lower temperature hydrogen release. Xiong et al.7 have demonstrated this approach by ball-milling AB with either LiH or NaH to form alkali amidoboranes MNH2BH3. These compounds release 2 equivalents of hydrogen at ca. 90 °C, which corresponds to 10.9 wt% H2 for LiNH2BH3.
In this paper we report on a new hydrogen storage material prepared from LiNH2 and AB. LiNH2 has previously been studied for H2storage, most notably by Chen's group8 in combination with LiH. In this system, rapid release of hydrogen took place only at temperatures greater than 170 °C.
Experimental
LiNH2 was obtained from Sigma chemicals and had a reported purity of 95%. AB was made according to a literature method9 by reacting NaBH4 and ammonium carbonate in tetrahydrofuran. Large, needle-shaped crystals were recrystallised from propan-2-ol. Deuterated isotopologues were made using NaBD4 as a starting material for NH3BD3, or by isotopic exchange of NH3BH3 in D2O to give ND3BH3. In both cases the degree of deuteration was greater than 90%, as judged from NMR intensities in d6-DMSO solution.Caution: This method results in the generation of a significant amount of hydrogen gas, which is a potential fire hazard. It is therefore recommended that the reaction is performed with suitable ventilation. Both the starting and spent material react violently with water ; materials should be handled in an inert atmosphere where possible and care in disposal of materials is recommended.
Stoichiometric ratios of AB and LiNH2 (typically 5–10 mmol) were weighed under Ar in a glove box, and loaded into a round-bottomed flask containing a magnetic stir bar and fitted with Teflon valves. Unless otherwise noted, compositions were prepared in a 1 : 1 molar ratio. The compounds were mixed together just prior to experimental measurements. Heating experiments were conducted at a ramp rate of 1 °C min−1 using a laboratory oven in which the flask was attached to stainless steel gas lines.
The composition of evolved gas was monitored using a quadrupole mass spectrometer (Dycor Dymaxion) with a flow (10 mL min−1) of Ar carrier gas. Quantification of NH3 was carried out by bubbling the gases through 0.1 M HCl solution and back-titrating with standard NaOH solution. The total quantity of gas evolved was determined in separate experiments without a carrier gas by directing the outlet to a gas burette initially filled with paraffin oil. Correction for temperature expansion in the reaction flask was established with a blank run.
Solid state 11B NMR spectra were recorded at 11 T on a Bruker Avance 500 spectrometer without proton decoupling and referenced to BF3·Et2O (0 ppm). Samples were packed in silicon nitride rotors in the Ar glove box and spun at ca. 10 kHz or greater at the magic angle in a Doty probe. Parameters from spectra with anisotropic quadrupolar resonances were obtained using the line shape analysis module of the Bruker TopSpin software.
Powder X-ray diffraction utilised a Bruker D8 diffractometer fitted with Co Kα radiation and parallel beam optics. Samples were loaded into a custom atmosphere-protected cell inside the Ar dry box.
Rehydrogenation of the material heated to 250 °C was attempted in a stainless steel pressure vessel. After pressurising with hydrogen gas, the vessel was closed and heated to 400 °C in a tube furnace with a 1/16″ stainless tube leading to a pressure transducer in the laboratory.
Results and discussion
Initial mixing
Upon mixing, the two solid components combined in an apparently exothermic reaction, forming a liquid phase. Depending on the ambient temperature, the mixing time needed to form the liquid varied from a few minutes to about half an hour. The air-sensitivity of the materials and speed of reaction prevented us from measuring the reaction enthalpy in a calorimeter. The liquid began evolving hydrogen slowly at room temperature, eventually transforming into an amorphous solid. Only a small amount of hydrogen loss (<0.1 mol H2/mol AB) was associated with the observed change of state, and no other gases were detected at this stage. The rate of hydrogen production increased with larger LiNH2 : AB ratios; 3 : 1 samples exhibited quite vigorous reaction while those with a 1 : 2 ratio appeared relatively stable. The heat evolved was most likely a factor in these observations. X-ray diffraction showed that specimens with a molar ratio in excess of 1Li : AB contained crystalline LiNH2 while those less than 1 : 1 exhibited crystalline AB.11B NMR spectroscopy of the material in its liquid state showed a single resonance at −22 ppm, resolved into a 1 : 2 : 2 : 1 quartet (Fig. 1(A)). The chemical shift and coupling allowed a confident assigment of this resonance to sp3boron in a –BH3 environment, but one which is different from that in AB (typically −25 to −27 ppm). The coupling constant (JBH = 82 Hz) was also different to that reported6 for the new mobile AB observed at −23 ppm during decomposition at 80 °C (JBH = 94 Hz). We propose that the resonance observed here is one in which AB and LiNH2 are associated in a new hybrid phase with 1 : 1 stoichiometry.
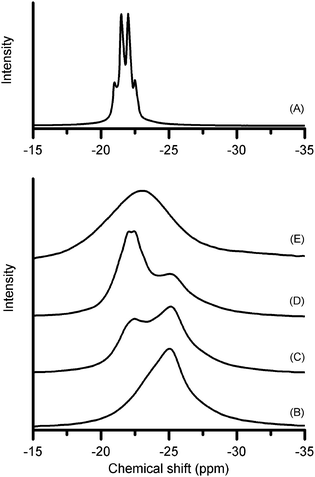 | ||
| Fig. 1 Solid-state 11B NMR Spectra of 1 : 1 LiNH2 : AB samples. Spectrum (A) was measured at room temperature from a sample mixed until the formation of a liquid phase. Spectra (B) to (E) were measured from a sample mixed for <1 min and held at 60 °C for 0 min, 15 min, 30 min, and 60 min respectively. | ||
Further evidence that the NMR resonance is distinct from AB was obtained using a sample which had been mixed for less than 1 min and had not yet formed the liquid phase. The 11B chemical shift (−25 ppm, Fig. 1(B)) was initially the same as the –BH3group in pure AB. Reaction between AB and LiNH2 took place in this partially-mixed sample when the temperature was raised, indicating that the hybrid phase can be formed by either thorough mixing at room temperature or by an increase in temperature. We observed a decrease in the intensity of the −25 ppm signal with time and the growth of the new signal at −22 ppm (Fig. 1(C)–(E)). The width of the resonance was noticeably larger in this sample and B–H coupling barely discernable even under favourable circumstances (Fig. 1(D)). This is presumably a consequence of whether the material is in a primarily liquid or solid state.
The change in the NMR spectra shown in Fig. 1(B)–(E) was not associated with appreciable loss of hydrogen. Samples prepared in this way had a similar hydrogen release profile to those prepared by thorough room temperature mixing, but shifted to higher temperature (see ESI†).
Gas evolution on heating
Further hydrogen was evolved when the sample was heated. EGA traces recorded under increasing temperature (Fig. 2) showed two stages of hydrogen evolution to 250 °C. The first stage exhibited a maximum rate at approximately 60 °C, while a second less intense evolution of hydrogen took place between 120 and 250 °C.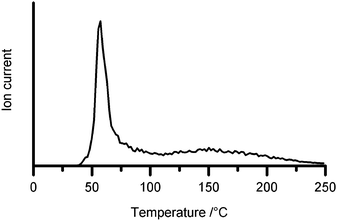 | ||
| Fig. 2 Hydrogen evolution from heating of hybrid material detected by mass spectrometry (m/z 2 signal). | ||
Ammonia was also detected by mass spectrometry in temperature range 60–120 °C, with an intensity that varied from sample to sample. We have been unable to determine the cause of this variability, which may be due to the precise sample history including the degree of mixing, and the temperatures of the laboratory and oven during sample loading. Several titration experiments however determined the amount of ammonia to be less than 1.5 vol% of the total gas evolved. No other gaseous contaminants were observed by mass spectrometry.
Gas burette measurements indicated that the total amount of hydrogen evolved up to 250 °C was approximately 3.2 mol H2 per mol of LiNH2⋯AB (11.9 wt%). The loss of 3 mol H2 would leave a product with the empirical formula LiBN2H2, and appears to be a reasonable approximation for the amount of hydrogen evolved during the lower temperature event. No crystalline phases were detected by X-ray diffraction in 1 : 1 samples heated at any temperature up to 600 °C. Samples containing an excess of LiNH2 retained unreacted LiNH2 to ca. 400 °C, which reacted at higher temperatures to form β-Li3BN2.10
Direct rehydrogenation of material which has lost hydrogen does not appear to take place. A sample of the 1 : 1 composition heated to 250 °C was subsequently heated to 400 °C in high pressure hydrogen gas. The H2 pressure reached 16 MPa at 400 °C, but no reduction in pressure was observed during the experiment which would signal rehydrogenation. This is similar to the behaviour of pure AB, which also cannot be rehydrogenated directly and for which indirect chemical schemes involving digestion of the spent material have been proposed.2,11
Solid-state 11B NMR spectroscopy of heated samples
More detailed information about the chemical changes accompanying hydrogen evolution was sought using solid-state 11B NMR. Our results showed that the –BH3 resonance in the hybrid material (−22 ppm) decreased with hydrogen evolution, and was replaced by a new resonance with a positive chemical shift (Fig. 3). This new peak had a quadrupolar line shape with a maximum near +20 ppm, however lineshape analysis (see example in the ESI†) showed that the isotropic chemical shift, δiso, of this resonance was +30 ppm. The transformation occurred without any observed intermediate resonances, except for a few very weak signals at the outset of hydrogen loss. An example of one of these weak intermediate signals, at −7 ppm, is shown in the ESI.† The resonance at δiso +30 ppm was ascribed to tricoordinate sp2 hybridised boron in a BN3 environment on the basis of a previous report12 of a similar environment in polyborazilene with δiso +27 ppm.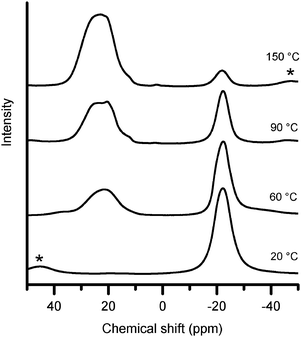 | ||
| Fig. 3 11B NMR spectra of the hybrid material quenched from the temperatures indicated after heating at 1 °C min−1. Asterisks denote spinning side bands. | ||
The transformation in 11B NMR spectra correlated with the proposed loss of 3 equivalents of hydrogen from a 1 : 1 sample at temperatures below 120 °C. A plot of the fraction of NMR intensity at −22 ppm against the quantity of gas evolved is shown in Fig. 4. A linear trend was observed which matched that expected for the release of 3 gas equivalents, corresponding to the complete loss of –BH3 and formation of BN3. This observation strengthened our assignment of the NMR resonance at δiso +30 ppm to BN3, and not to BN2H which has a reported12 shift of δiso +31 ppm.
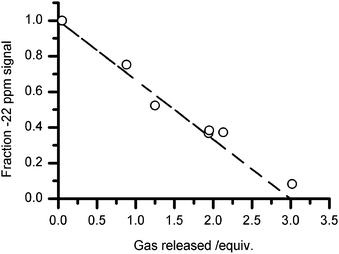 | ||
| Fig. 4 Fraction of total NMR signal attributed to –BH3 (δ −22 ppm) as a function of gas volume. The dotted line is drawn to indicate the expected trend for the release of 3 mole equivalents of H2 per –BH3. | ||
Isotopic composition of the evolved gas
The mechanism of hydrogen loss was also investigated using selectively deuterated AB coupled with mass spectral analysis of the evolved gases. Significant differences were observed in the isotopic composition of hydrogen released below 120 °C. When NH3BD3 was utilised, the gas was predominantly HD (Fig. 5a). In contrast, a mixture comprising approximately twice as much H2 as HD was observed when ND3BH3 samples were used (Fig. 5b). These results suggest that 2/3 of the hydrogen gas arises from dehydrocoupling between LiNH2 and 2 hydrogen atoms initially bonded to boron in AB. The remaining 1/3 arises from the final hydrogen bonded to boron and one bonded to N in AB.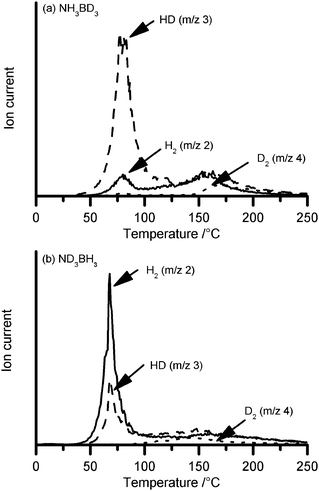 | ||
| Fig. 5 Mass spectra of gases evolved from heating 1 : 1 hybrid material formed from LiNH2 with (a) NH3BD3, and (b) ND3BH3. | ||
The isotopic composition of gas released above 120 °C was a mixture of H2, HD, and D2, indicating significant isotopic exchange occurred during the reaction which precluded further interpretation. Similarly, the small amount of H2 observed between 50 and 120 °C in samples derived from NH3BD3 (Fig. 5a) may have arisen from isotopic scrambling or an alternate mechanism in addition to that proposed. The ammonia released between 60 and 120 °C (discussed above) was detected as NH3; no deuterated analogues were detected by mass spectrometry irrespective of starting AB isotopologue.
Reaction pathway
The combination of 11B NMR, isotopic mass spectrometry, and gas volume measurements led us to propose the reaction pathway outlined in Scheme 1. The 11B NMR data showed that hydrogen loss is accompanied by the formation of new B–N bonds and a progressive change from boron in a BH3N to a BN3 environment. The first stage in our proposed reaction pathway is therefore the formation of a new B–N bond between AB and LiNH2 accompanied by the release of molecular hydrogen. An intermediate compound with the chemical formula LiNHBH2NH3 is formed as a result. Further hydrogen release and B–N bond formation leads to a polymeric [–LiN–BHNH3–]n compound formed by either condensation of LiNHBH2NH3 units, or by successive addition of LiNH2 and BH3NH3 to LiNHBH2NH3. The weak −7 ppm signal observed in occasional 11B NMR experiments may be attributed to one of these intermediate compounds (LiNHBH2NH3 or [–LiN–BHNH3–]n). The third equivalent of molecular hydrogen released arises from reaction between the remaining boron hydrogen and the –NH3group to leave a polymer of alternating LiN and BNH2 entities. The sp2 hybridised BN3 environment in this polymer is consistent with the δiso +30 ppm 11B NMR resonance observed after 3 equivalents of hydrogen had been released (Fig. 3).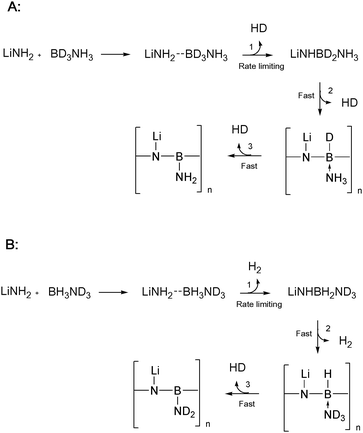 | ||
| Scheme 1 Reaction pathway for hydrogen release from LiNH2…AB hybrid materials. Separate pathways for (A) NH3BD3 and (B) ND3BH3 are shown for comparison with the isotopic gas analysis shown in Fig. 5. | ||
The isotopic composition of gas released from this proposed reaction pathway is also consistent with the observed data. Scheme 1A (starting with NH3BD3) predicts the formation of HD in each of the 3 gas release steps. Scheme 1B (starting with ND3BH3) predicts two molecules of H2 and one of HD. Both of the predictions agree with the mass spectrometry data shown in Fig. 5. Although deuterium labelling experiments provide good evidence for the decomposition pathway, we are planning 15N NMR experiments in order to further characterise the reaction.
The near absence of intermediate NMR resonances between –BH3 and –BN3 suggests that the first reaction between LiNH2 and AB to release hydrogen is rate limiting and that the other steps proceed relatively rapidly. A possible explanation for this may be found in the charge density calculations presented by Wuet al.13 These showed a substantial increase in the hydridic nature of the boron hydrogens (−0.13 e) in LiNH2BH3 compared to those in AB (−0.05 e). If an analogous effect occurred in the intermediates proposed here, this would promote stronger interaction with the protic amine hydrogens for steps 2 and 3 (Scheme 1).
Conclusions
Solid-state reaction between LiNH2 and AB takes place to yield a new hybrid material which differs from a simple physical mixture of the two compounds. This material does not have a crystalline diffraction pattern and contains –BH3groups with a different 11B NMR chemical shift than AB.Hydrogen is released from this material when it is heated with the maximum rate of hydrogen evolution occurring at approximately 60 °C. 3.2 equivalents (11.9 wt%) of hydrogen are released from the hybrid material when it is heated to 250 °C. The dehydrogenated product is an amorphous material containing boron in a BN3 environment. No reaction was observed when this product was exposed to hydrogen gas at 400 °C and 16 MPa. A reaction pathway for hydrogen release has been proposed based on the results of 11B NMR spectroscopy and mass spectrometry of selectively deuterated starting materials.
Acknowledgements
The authors gratefully acknowledge the contributions made from discussions with our International Partnership for the Hydrogen Economy collaborators, especially Prof. Ping Chen (National University of Singapore), Drs Tom Autrey (Pacific Northwest National Lab.), Tony Burrell (Los Alamos National Lab.) and Bill David (Rutherford-Appleton Lab.). Funding for this work was kindly provided by the NZ Foundation for Research, Science and Technology.References
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef.
- F. H. Stephens, V. Pons and R. T.Baker, Dalton Trans., 2007, 2613 RSC.
- D. J. Heldebrant, A. Karkamkar, J. C. Linehan and T. Autrey, Energy Environ. Sci., 2008, 1, 156 RSC.
- B. Peng and J. Chen, Energy Environ. Sci., 2008, 1, 479 RSC.
- F. Baitalow, J. Baumann, G. Wolf, K. Jaenicke-Rößler and G. Leitner, Thermochim. Acta, 2002, 391, 159 CrossRef CAS.
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 9, 1831 RSC.
- Z. Xiong, C. K. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 CrossRef CAS.
- P. Chen, Z. Xiong, J. Luo, J. Lin and K. L. Tan, Nature, 2002, 420, 302 CrossRef CAS.
- M. G. Hu, J. M. van Paasschen and R. A. Geanangel, J. Inorg. Nucl. Chem., 1977, 39, 2147 CrossRef CAS.
- H. Yamane, S. Kikkawa and M. Koizumi, J. Solid State Chem., 1987, 71, 1 CrossRef CAS.
- S. Hausdorf, F. Baitalow, G. Wolf and F. O. R. L. Mertens, Int. J. Hydrogen Energy, 2008, 33, 608 CrossRef CAS.
- C. Gervais, J. Maquet, F. Babonneau, C. Duriez, E. Framery, M. Vaultier, P. Florian and D. Massiot, Chem. Mater., 2001, 13, 1700 CrossRef CAS.
- H. Wu, W. Zhou and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 14834 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Example NMR spectra for line-shape fitting of anisotropic resonance and for weak resonance observed at −7 ppm. See DOI: 10.1039/b901082c |
| This journal is © The Royal Society of Chemistry 2009 |