Carbon dioxide versus energy balances for transportation fuels
Huw O.
Pritchard
*
Department of Chemistry, York University, Toronto, Canada M3J 1P3
First published on 3rd April 2009
Abstract
Tables of energy content per litre and of CO2 production per unit energy are given for a representative selection of molecules, as an aid to quantifying the advantages, or otherwise, of using ethanol and/or butanol as transportation fuels.
Broader contextRigorously correct values for energy content and CO2 production are tabulated for a selection of substances that may be found in liquid fuels, together with illustrations of how these numbers should be used. At the present time, numbers cited in various publications, from newspapers, magazines, on up to technical reports, etc., are often inconsistent, occasionally incorrect, and hard numbers (sometimes wrong) are commonly mixed indiscriminately with guessed ones. The purpose of this Note is to set a logical base upon which to build a rational discussion of the merits of ethanol and/or butanolversus hydrocarbons as transportation fuels, and to aid in the diminution of widespread ill-informed comments. |
It is widely believed that the use of biofuels, in particular ethanol, will reduce the production of CO2 from transportation sources while, at the same time, improving the quality of tail-pipe emissions. The purpose of this Note is to summarise the basic data required for a systematic analysis of the former assertion. Present-day arguments are often clouded by a failure to separate hard facts from imponderables: common examples of imponderables cited by corn–ethanol detractors include fuel for tractors, energy used producing and transporting fertiliser, water consumption, etc.; opposing arguments are often similarly diffuse.
For any given mode of transportation (e.g. bus, train, 8-, 6-, or 4-cylinder car, motor cycle, etc.), it is axiomatic that energy per unit volume (e.g.MJ L−1) correlates with distance travelled per unit volume (e.g. miles per gallon or km L−1) for that specific vehicle. Table 1 gives a set of data for a range of liquid hydrocarbons typically found in automotive, diesel, and jet fuels. Parenthetically, butane is included as it is commonly added to gasoline in Canada to increase volatility as a cold-starting aid in winter. Values of the heats of combustion are taken from the extensive tabulation of Cox and Pilcher,1 averaged over available isomers and rounded to the nearest integer. However, it is not the standard heat of combustion ΔH°c that is required in these calculations but that of the process
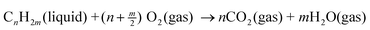
| Molecule | ΔH°c | ρ | Mol | ΔH‡c | Energy | CO2 |
|---|---|---|---|---|---|---|
| kcal mol−1 | g mL−1 | wt | kcal mol−1 | MJ L−1a | g MJ−1b | |
| a MJ L−1 = (ΔH‡c × 4.184 × ρ)/[Mol wt]. b g MJ−1 = (nC × 44 × 1000)/(4.184 × ΔH‡c). c estimated. d average over 0–20 °C. | ||||||
| Paraffin hydrocarbons CnH2n + 2 | ||||||
| n = 4 | 682c | 0.59d | 58 | 629 | 26.8 | 66.8 |
| n = 5 | 840 | 0.626 | 72 | 777 | 28.3 | 67.7 |
| n = 6 | 995 | 0.660 | 86 | 921 | 29.6 | 68.5 |
| n = 7 | 1150 | 0.683 | 100 | 1066 | 30.5 | 69.1 |
| n = 8 | 1307 | 0.700 | 114 | 1212 | 31.2 | 69.4 |
| n = 9 | 1463 | 0.717 | 128 | 1358 | 31.8 | 69.7 |
| n = 10 | 1620 | 0.730 | 142 | 1504 | 32.4 | 69.9 |
| n = 11 | 1776 | 0.740 | 156 | 1650 | 32.7 | 70.1 |
| n = 12 | 1933 | 0.748 | 170 | 1796 | 33.1 | 70.2 |
| n = 13 | 2092 | 0.756 | 184 | 1945 | 33.4 | 70.3 |
| n = 14 | — | 0.763 | 198 | — | — | — |
| n = 15 | 2402 | 0.768 | 212 | 2234 | 33.9 | 70.6 |
| n = 16 | 2557 | 0.773 | 226 | 2378 | 34.0 | 70.7 |
| Aromatic hydrocarbons | ||||||
| Benzene | 780 | 0.876 | 78 | 748 | 35.2 | 84.3 |
| Toluene | 934 | 0.867 | 92 | 892 | 35.2 | 82.5 |
| Xylenes | 1088 | 0.864 | 106 | 1035 | 35.3 | 81.2 |
| Ethylbenzene | 1090 | 0.867 | 106 | 1037 | 35.5 | 81.1 |
| Propylbenzene | 1247 | 0.862 | 120 | 1184 | 35.6 | 79.9 |
| Butylbenzene | 1401 | 0.860 | 134 | 1327 | 35.6 | 79.2 |
| Indonaphthene | 1146 | 0.996 | 116 | 1104 | 39.7 | 85.7 |
| Tetralin | 1334 | 0.970 | 132 | 1271 | 39.1 | 82.7 |
| Molecule | ΔH°c | ρ | Mol | ΔH‡c | Energy | CO2 |
|---|---|---|---|---|---|---|
| kcal mol−1 | g mL−1 | wt | kcal mol−1 | MJ L−1a | g MJ−1b | |
| a MJ L−1 = (ΔH‡c × 4.184 × ρ)/[Mol wt]. b g MJ−1 = (nC × 44 × 1000)/(4.184 × ΔH‡c). c anhydrous: engines using neat (95%) ethanol will show ∼5% reduction in MJ L−1, but g MJ−1 does not change. | ||||||
| Methanol | 173 | 0.791 | 32 | 152 | 15.7 | 69.2 |
| Ethanol c | 327 | 0.789 | 46 | 295 | 21.2 | 71.2 |
| n-Propanol | 483 | 0.804 | 60 | 441 | 24.7 | 71.5 |
| n-Butanol | 639 | 0.808 | 74 | 586 | 26.8 | 71.7 |
| Diethylether | 649 | 0.714 | 74 | 596 | 24.1 | 70.5 |
| MTBE | 803 | 0.752 | 88 | 740 | 26.5 | 71.1 |
| DTBP | 1275 | 0.794 | 146 | 1180 | 26.9 | 71.3 |
| Acetone | 428 | 0.790 | 58 | 396 | 22.6 | 79.6 |
| Ethylene glycol | 285 | 1.109 | 62 | 253 | 19.0 | 83.0 |
| Glycerol | 396 | 1.261 | 92 | 356 | 20.4 | 88.6 |
Several simple observations can be made from Table 1: (i) that the energy content per litre of aromatic fuels is significantly greater than for aliphatic fuels of similar molecular weight (or volatility); this arises mainly from the more compact nature of aromatic molecules which enables a more efficient packing in the liquid structure; (ii) likewise, there is a strong correlation between density and energy content in the aliphatic series; (iii) that the CO2 formation per MJ is roughly constant within each class of molecule, but is considerably greater for aromatics than for aliphatics; this is because unsaturated hydrocarbons have smaller heats of combustion than their parent saturated molecules since less water is formed in the combustion process.
Underlying these observations are the well-known concepts of bond additivity2 and of constant bond energy terms.1
Gasolines and diesel fuels typically comprise hydrocarbons lying in the C7–C9 and C12–C22 boiling ranges respectively and, depending upon their source, can have very different aromatic–aliphatic ratios.3,4 Hence, we cannot use the numbers from Table 1 without knowing an approximate value of this ratio, whence adequate estimates of MJ L−1 and g MJ−1 can be made by assuming a linear mixture rule (implying no heat or volume change on mixing). Aromatic fuels tend to be more resistant to spontaneous ignition, an advantage for spark-ignition engines (high Octane Number), but not for compression-ignition engines (low Cetane Number).
Table 2 gives a similar set of data for some oxygenated liquids that may occur in transportation fuels (although some only in trace amounts). We see a much smaller energy content per litre than for hydrocarbons because these molecules are, in effect, already partially oxidised hydrocarbons. Also, a virtual invariance of the CO2 production with energy content, except in the last three cases, where acetone is deficient in H atoms and the other two are more strongly hydrogen-bonded (high ρ and ΔH°v for the liquid, high ΔHa for the gas).
The lower energy output per litre is clearly reflected in fuel economy: the official Canadian Government statistics show that for three 2008 “Flex-Fuel” vehicles (Chevrolet Impala 3.5 and 3.9 L, Lincoln Town Car 4.6 L) the volumetric fuel consumption is from 1.3–1.45 greater for E85 (85% ethanol) than for hydrocarbon fuel.5 Likewise, the 10% ethanol blend commonly available in some countries carries a 3% penalty in volumetric fuel consumption.
The overall reaction that forms ethanol from glucose is
| C6H12O6 → 2C2H5OH + 2CO2 |
With a different set of enzymes, a more complex fermentation process (often known by the acronym ABE) occurs, forming acetone, butanol and ethanol.6 The proportions of these three products (and of other side-products) are not fixed, but can be manipulated depending upon a variety of experimental conditions.6,7 This reaction was pioneered by Weizmann for the production of acetone for explosives during the First World War, to the extent that due to the threat of starvation in Britain, production was moved over to North America towards the end of the war.6
As is the case with the ethanol reaction, large amounts of CO2 are produced as a byproduct, typically6 around 4 moles of CO2 per mole of butanol, together with about 2 moles of H2. Hence, the CO2 production per MJ should roughly be doubled to around 130–150 g MJ−1.
During the 1980s, the principal fates of the CO2 were stated to be dry-ice manufacture and the carbonation of soft drinks;6 the latter is still sometimes cited as a benefit but, in fact, both are really only delayed release mechanisms. There were plenty of uses for the H2 including ammonia synthesis and hydrogenation of edible oils, together with studies of the economic viabilty of converting the CO2/H2 mixture into methanol,6 which might also be used as a fuel. Were this to be done in 100% yield, it would still amount to delayed release, but the transportation CO2 burden would fall to ∼70.5 g MJ−1; however, it takes 12 moles of H2 to convert 4 moles of CO2 to methanol,
| 4CO2 + 12H2 → 4CH3OH + 4H2O |
On the other side, there are some serious advantages to the use of oxygenated fuels: unlike hydrocarbon fuels, they are largely free of contaminants, particularly sulfur and nitrogen which end up as SOx and NOx; being already partially oxidised, they have a lower peak combustion temperature, thus reducing further the nitric oxide formation in the engine exhaust;8 and under conditions of incomplete combustion, pure oxygenates are less prone to form soot and other PAHs, but when blended with hydrocarbon, it is difficult to predict the outcome. Of course, any CO2 formed from combustion of a biofuel or as a fermentation side product (if not captured and buried) simply replaces that from which the sugar feedstock was formed by photosynthesis in the recent past; in contrast CO2 created by burning traditional hydrocarbon fuel generates gas that was trapped from the atmosphere millions of years ago.
The balance between these alternatives may well be chosen differently in different jurisdictions or in the light of new developments.9,10 Preferably, these choices should be guided by basic thermochemical data, such as those outlined in Tables 1 and 2.
References
- J. D. Cox and G. Pilcher, Thermochemistry of Organic and Organometallic Compounds, Academic Press, London, 1970 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, 1945, pp. 47–50 Search PubMed.
- P. Q. E. Clothier, B. D. Aguda, A. Moise and H. O. Pritchard, How do diesel-fuel ignition improvers work?, Chem. Soc. Rev., 1993, 22, 101–108 RSC.
- J. F. Griffiths and J. A. Ballard, Flame and Combustion, Blackie, London, 1995 Search PubMed.
- Fuel Consumption Guide 2008, Government of Canada, Ottawa Search PubMed.
- D. T. Jones and D. R. Woods, Acetone–butanol fermentation revisited, Microbiol. Rev., 1986, 50, 484–524 CAS.
- P. Durre, Biobutanol: An attractive biofuel, Biotechnol. J., 2007, 2, 1525–1534 Search PubMed.
- R. A. Craig and H. O. Pritchard, Nitric oxide abatement in jet-aircraft engines, Can. J. Chem., 1977, 55, 1599–1608 CrossRef CAS.
- J. Goldemberg, The challenge of biofuels, Energy Environ. Sci., 2008, 1, 523–525 RSC.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Biofuels: A technological perspective, Energy Environ. Sci., 2008, 1, 542–564 RSC.
| This journal is © The Royal Society of Chemistry 2009 |