Catalytic upgrading of tri-glycerides and fatty acids to transport biofuels
Benjamin
Smith
,
Hugh Christopher
Greenwell
* and
Andrew
Whiting
Department of Chemistry, South Road, Durham, DH1 3LE, United Kingdom. E-mail: chris.greenwell@durham.ac.uk; Fax: +44 1913 844737; Tel: +44 1913 342598
First published on 25th November 2008
Abstract
Fuels derived from the lipid fraction of biomass have recently received much attention for carbon neutral substitution of fossil fuels for transport use. In this article we review the current routes to catalytic upgrading of the biomass derived lipid fractions. The history and motivation for biofuels are discussed, including the link to current market trends amongst fuel prices. The sources of lipids and their chemical composition are considered. We review the current literature detailing the use of trans-esterification reactions (heterogeneous and homogeneous) which lead to oxygenated “Biodiesel”, and also decarboxylation, which leads to deoxygenated “Green Diesel”. Traditional methods are covered, as well as more recent novel research aiming to produce commercially viable fuels.
 Benjamin Smith | Benjamin Smith studied for his MChem at Durham University. He completed his research project on the self assembly of metallo–pyridyl urea systems under the supervision of Professor Judith A. K. Howard and Professor Jonathan W. Steed. At present he is carrying out his PhD at Durham University, funded by the EPSRC and KiOR, under the supervision of Dr Chris Greenwell and Dr Andy Whiting, where his research focuses on mineral catalysed decarboxylation reactions for biofuels. |
 Hugh Christopher Greenwell | Dr Chris Greenwell is the Addison Wheeler Fellow at Durham University and Honorary Research Fellow at the Centre for Computational Science, University College London. He undertook his PhD (2003) in the Materials Chemistry Group, Cambridge under the supervision of Professor W. Jones where he worked on hybrid organic–inorganic materials. His current research interests focus on the structure and behaviour of organo–mineral systems, including heterogeneous catalysis to produce biofuels. He is a consultant to both industry and government on algae biofuel technology. |
 Andrew Whiting | Dr Andy Whiting carried out his PhD studies with Professor R. J. Stoodley at Newcastle University, working on β-lactam chemistry, before moving on to postdoctoral research at Boston College, with Professor T. Ross Kelly working on natural product synthesis and the development of chiral Diels–Alder Lewis-acid catalysts. After a short period in industry with Ciba-Geigy Central Research, he moved to his first academic position as Lecturer in Chemistry at UMIST, and in 2001, moved to a Readership at Durham University. |
Broader contextWe review the use of catalysis in producing biofuels from plant and algae lipids. Plant and algae lipid fractions provide an alternative, renewable and carbon neutral source of biofuels. Lipid derived fuels offer advantages over other alternative transport fuels such as hydrogen, biogas and carbohydrate fuels, being compatible with the existing transport infrastructure. The lipid fraction consists mainly of triacylglycerides (TAGs) which can be readily converted through trans-esterification with alcohols into either fatty acid methyl esters (FAME) or fatty acid ethyl esters (FAEE). FAME and FAEE are generally described as biodiesel, and can be blended at low levels with fossil oil derived diesel and used in existing transport infrastructure to extend fossil fuel reserves. Homogeneous catalysts have conventionally been used to produce FAME and FAEE, but result in highly basic aqueous waste streams and require large excesses of alcohol. By using heterogeneous catalysis the process may be made more efficient and cleaner. Ongoing research is directed at producing deoxygenated biofuels, sometimes described as green diesel, which will be a direct replacement for current road transport fuels and amenable to production using existing petrochemical refining infrastructure. |
1. Introduction
The discovery and use of petroleum-based fuels has transformed the economies and infrastructure of societies and nations. Until recently, the cost of oil had decreased until fuels were readily available and inexpensive; no longer was it the rich-few who could afford to use this energy source and the technology required to utilise it. With the consumption of crude oil increasing by around 1 million barrels (bbl) of oil daily1 and known reserves estimated to last only another 40 years, prices have rapidly escalated over the last decade. There is a pressing need to develop alternative fuels that are compatible with existing automotive technology, rather than develop wholly new fuels that require the costly and time-consuming complete replacement of existing transport infrastructure.1.1 History and motivation for biofuels
Biofuels are by no means a modern idea, over a century ago, Rudolph Diesel invented an engine which first ran on peanut oil. It was not until many years later that crude oil-based diesel, being cheaper, became the alternative. Biofuels have also been used in times of shortage, for example during periods of conflict and civil unrest. Currently, industry and transport infrastructure across the developed world are inherently based on fossil fuels derived from crude oil. This is becoming a costly and an increasingly unsustainable source of energy, which is also responsible for the phenomena of global warming, where carbon dioxide acts as a “greenhouse” in the atmosphere, raising global temperatures. World temperatures have been increasing in recent years, with evidence suggesting a strong correlation with rising CO2 levels. For example, globally, 2005 was the warmest year on record throughout all 12 months.2 There is an increasingly recognised need to slow down this warming trend for future generations, and reduce the warming effects on Earth.Using natural, biological resources, solar energy can be captured by plants, viaphotosynthesis, to convert atmospheric carbon dioxide to biomass, from which biofuels can be produced. These types of fuels are regarded as “environmentally friendly” since they are carbon neutral, “recycling” carbon dioxide from the atmosphere, rather than releasing long-term entrapped carbon as associated with fossil fuel combustion. Although plants are not as efficient at converting light as photovoltaic solar cells, their cost of production is significantly less and they are compatible with a petrochemical based society. Every year it is estimated the equivalent of 640 billion bbl of crude oil energy is released by microbes.3 This shows the capability of natural processes in producing energy resources.
Recently there has been a push towards growing biomass for biofuels across the world. There are various reasons for this, not least the potential for perceived “reduction” of greenhouse gas emissions and sustainability.4 Biofuels are also lower in sulfur content compared to their petroleum counterparts.5 However, recently biofuels have received much negative press with studies suggesting that many biofuels, rather than being carbon neutral, may in fact lead to greater carbon dioxide and other greenhouse gas emissions, due to changing land use in their production.6,7 Conversely, some biofuel sources are described as carbon negative in that they lock up more carbon dioxide than is released during their production.8 Clearly, the type of biomass used as a source of biofuels is an important consideration. There is ever increasing concern about the use of agricultural land for energy generation, rather than food production, for an increasing world population.9 As a potential solution to this issue the use of algae to produce oils has been suggested since it does not compete with food for land use or water resources, leads to highly efficient yields and it is estimated that biofuels can be produced for around $50 bbl.10
Legislation is also a driver for biofuel production. The European Union (EU) has set a target of 5.75% biofuel content in its member states' fuels by the close of 2010, and also recommends that emissions are monitored on non-adapted vehicles.11 The United Kingdom (UK) government has set its own biofuel target of 5% by volume of total road transport fuel sales by 2010. The estimated amount of UK agricultural land was 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 272000 hectares in 2005, of which 10% was available for biofuel growth without adversely affecting food supplies (based on grassland less than 5 years old). To reach the UK target of 5% by 2010 1200000 hectares are required, which is achievable, whereas the EU target of 5.75% would require 1750000 hectares which is greater than that currently available.12 Countries producing their own biofuels would have reduced reliance on other states to provide crude oil for fossil fuels, resulting in energy supply security. Reliance on other nations for energy supplies can lead to tension, and drive up prices through speculation owing to uncertainty of supply. Biofuel production can also lead to increased support and employment for rural communities.9
272000 hectares in 2005, of which 10% was available for biofuel growth without adversely affecting food supplies (based on grassland less than 5 years old). To reach the UK target of 5% by 2010 1200000 hectares are required, which is achievable, whereas the EU target of 5.75% would require 1750000 hectares which is greater than that currently available.12 Countries producing their own biofuels would have reduced reliance on other states to provide crude oil for fossil fuels, resulting in energy supply security. Reliance on other nations for energy supplies can lead to tension, and drive up prices through speculation owing to uncertainty of supply. Biofuel production can also lead to increased support and employment for rural communities.9
The world production of biofuels was estimated to be over 35 billion litres in 2006.13 The transport sector, including aviation, produces about one quarter of the UK's total carbon emissions. Road transport contributes 85% of this, with passenger cars accounting for approximately half of all carbon emitted by the transport sector.14
The use of biofuels has previously been limited due to the high cost of production relative to mineral-based fuels, partly due to high feedstock costs, though with crude oil prices behaving erratically, recently rising (and then subsequently falling) past the $140 bbl mark, biofuels are becoming an increasingly viable alternative.
1.2 Transport biofuel sources
Biofuels can be produced from a wide variety of biomass sources, with bioethanol and biodiesel the main types currently in commercial production and use.First generation biofuels are defined as those fuels derived from existing food-based crops. Producing these fuels leads to competition between food and fuel crops for arable land. The sources of first generation biofuels are: sugar crops for bioethanol, such as sugar cane and sugar beets; starch crops for bioethanol, such as corn and wheat; oilseed crops for biodiesel, such as rapeseed, soybeans and palm.
There is an emerging use of second, or next, generation biofuels derived from non-food based crops consisting of cellulosic biomass made up of cellulose, hemicellulose and lignin. The sources of these include Switchgrass, Miscanthus, Willow and also crop residues. These specific energy-crops do not compete with land explicitly destined for food crops and can be grown in low quality environments. The main challenge with second generation biofuels is that the initial capital investment costs are a great deal higher than for first generation biofuels, which can be processed via more conventional methods.
In 2005 Brazil was producing 282000 bbl of ethanol a day from sugarcane,17 and the United States of America (USA) was producing 260000 bbl a day from corn (Table 1), although corn requires increased inputs of fertiliser and a greater amount of biomass feedstock per bbl.18 Achieving this output required 3.5% of the total USA water consumption.19 In view of water and fertiliser requirements, some countries are beginning to change the energy crops they grow.20 It has been found that wheat, grown in Europe during winter, was more energy efficient when fertilizer was used, with at least 5 times as much energy returned than used during the fertiliser's own production, packaging, transport and spreading energies combined.21 Contrary to this, it has been suggested that there may be an increase in nitrous oxide emissions overall, due to the increased use of fertilisers required to grow crops in the, sometimes, poorer environments.22 Clearly, for any biomass to biofuel process a full life cycle analysis must be considered before production proceeds.
Microalgae are a promising source of biomass for biofuel production, since the oil yields can be very high with certain species containing over 80% oils based on dry weight.25 It has been estimated that it would take 61% of arable farming land in the USA to meet domestic fuel requirements using first generation biomass, but only 3% of the equivalent area for microalgae, which grow extremely rapidly (some species doubling in mass in less than 24 h). Crucially, algae can be grown in photobioreactors (Fig. 1), as well as open systems, yielding biomass crop continuously year round. The oil fraction can be extracted using a solvent such as hexane, with the remaining biomass being digested under anaerobic conditions to make methane, or used as animal feed. Trans-esterification is then used to produce biodiesel.26 Use of microalgae is currently on a relatively small scale compared to the use of sugar cane and soybeans, but it has the potential to provide biofuels that do not compete with domestic food crops in the long run.
 | ||
| Fig. 1 Tubular photobioreactor system for algae production (courtesy of Joe McDonald, Varicon Aqua Solutions). | ||
Swedish Biogas have developed a fuel, mainly comprised of methane sourced from food waste and sewage. In Sweden there are tax advantages associated with the use of biogas vehicles and this fuel is currently used to power a train, with a range of 600 km and maximum speed of 130 km h−1.30
Efforts have been made to yield the maximum amount of energy from biomass sources, rather than just using the accessible oils, and thus reducing waste. Singh et al. have demonstrated the use of not only the oils of Jatropha curcas to produce biodiesel, but also the use of seed husks, seed shell and oilcake, resulting in a 300% increase in the amount of energy released, compared to using just the oil for biodiesel alone.32 This is an important issue which must be tackled during biofuel production to ensure all resources are used effectively since there is limited land set-aside for bioenergy production and process efficiency is of the utmost importance in its justification.
Biofuels is a broad topic, and a rapidly developing area of research. Indeed, biofuels are regularly mentioned in news bulletins, at local, national and international levels. This review will focus on the topic of converting the lipid fraction of biomass feedstock to diesel-type fuels. In Section 2 we address the composition of oils from plants and algae. In Section 3 we briefly examine pyrolysis routes for converting biomass to oils. In Sections 4 and 5 we review the current state of the literature for upgrading the lipid fraction of biomass to ester fuels (biodiesel), or deoxygenated fuels (green diesel).
2. Vegetable oils
Crude vegetable/algae oils consist of mainly triglycerides, which are glycerol molecules esterified with three fatty acids (Fig. 2). The three fatty acids can all be the same, two the same or all different, with the most common carbon chain lengths being evenly numbered and containing either 16, 18 or 20 carbon atoms. Various mixtures of triglycerides have been characterised, depending on the oil feedstock used. Long chain free fatty acids (FFAs) may also be present in the oils, which may be saturated or unsaturated.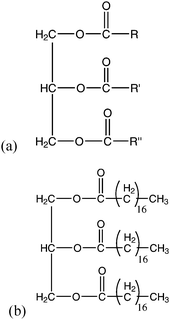 | ||
| Fig. 2 (a) Structure of a triglyceride molecule which is a glycerol molecule esterified with three fatty acids, of which R, R′ and R″ may all be the same, two the same or all different; (b) the tri-acyl glyceride of stearic acid (octadecanoic acid). | ||
The choice of oil feedstock plays an important role in the overall cost of the finished product since it constitutes up to 60–75% of the process costs.33 Feedstocks are generally area-specific, for example soybean oil in the USA and rapeseed oil in Europe. The oils comprise of fatty acids which vary in their physical properties such as density, viscosity and cold flow as chain length and degree of saturation alters, with higher quality feedstocks leading to more desirable fuel properties and increased overall costs.34
3. Pyrolysis and “whole” biomass upgrading
Cracking (or pyrolysis) is used to break down larger petroleum hydrocarbon molecules into smaller more desirable hydrocarbons, in the presence of a catalyst and the absence of oxygen. Catalysts used for this process include zeolites35 and mesoporous aluminosilicates.36 A number of 2-dimensional structures called pillared clays containing various metals have been investigated for their ability to crack vegetable oils such as canola oil, palm oil and sunflower oil into biofuels.37 These oils all have different triacylglycerol compositions, so various qualities of biofuels are produced in the final products. The triacylglycerols vary according to the three fatty acids linked by an ester link to the glycerol molecule. Catalytic cracking was believed to take place via two possible pathways for canola oil: β-elimination and γ-hydrogen transfer.4. Producing oxygenated biofuels (biodiesel) via trans-esterification reactions
Biodiesel is a term used to describe “a fuel comprised of monoalkyl esters of long-chain fatty acids that are derived from vegetable oils or animal fats”,38 although some believe it should be used in a more general nature to describe “diesel fuel that originates from renewable raw materials”.39 This can be used as a direct replacement for petroleum diesel. One of the methods used commercially to produce biodiesel is trans-esterification, as shown in Fig. 3, where triglycerides present in vegetable oils are esterified, usually with methanol (methanolysis), to yield the corresponding methyl esters and glycerol. Glycerol may then be sold as a byproduct to help make the process more economically viable. Trans-esterification is usually performed in the presence of a catalyst, although it may also be driven by supercritical conditions. Fatty acid methyl ester (FAME) or fatty acid ethyl ester (FAEE) products may be used neat as biodiesel or be blended with petroleum-based diesel, although some modifications may be required for engines to run at over 10% biodiesel content.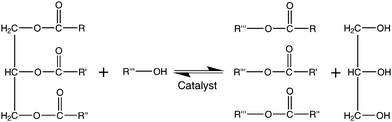 | ||
| Fig. 3 Trans-esterification of a triglyceride. Methanol is normally the alcohol used, together with an acid or base catalyst. This is a three step reaction, proceeding via the tri-,di- and monoglycerides to the alkyl esters which can be used as biodiesel. The byproduct glycerol can also be sold to recuperate production costs. | ||
4.1 Homogeneous catalysis for biodiesel production
Homogeneous acid or base catalysis has the advantage of the catalyst being in constant contact with the reaction mixture leading to increased rates, but they suffer from the requirement of a neutralisation step to remove the catalyst, and thus loss of raw material.:1 molar ratio of methanol to oil, 30 °C and a reaction time of 4 h.
Vicente et al. have compared the four most common homogeneous catalysts for trans-esterification – sodium hydroxide, potassium hydroxide, sodium methoxide and potassium methoxide.41 At 65 °C, 6:1 methanol:sunflower ratio sodium methoxide was found to lead to the largest yield with 99.3% with 99.7% biodiesel purity. The remaining catalysts were found to be in the yield order: potassium methoxide (98.5%), potassium hydroxide (91.7%), sodium hydroxide (86.7%). The order is in agreement with trans-esterification of used frying oils with sodium methoxide (89%) giving higher yields than both potassium hydroxide and sodium hydroxide.42
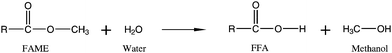 | ||
| Fig. 4 Formation of free fatty acids from hydrolysis of fatty acid methyl esters. | ||
4.2 Heterogeneous catalysis for biodiesel production
Efficient heterogeneous catalysts offer economic benefits in producing biofuels since, unlike homogeneous catalysts, they are easily separated after trans-esterification, and so can be readily recycled, lowering production costs.:Mg ratio. The co-precipitation method may be alkali free, leading to no alkaline contaminant in the catalyst. Using LDHs, the authors carried out trans-esterification in a stirred batch reactor for 3 h at 60 °C with 0.05 g calcined catalyst, with glyceryl tributyrate, methanol and hexyl ether, and the reaction was periodically sampled with gas chromatography (GC). Glyceryl tributyrate was converted into methylbutanoatevia di- and mono-glycerides. Hydrotalcite, with a Mg:Al ratio of 2.93:1, led to the highest conversion, due to increased intralayer electron density (and associated basicity) with increasing Mg content. Both Mg2+ and Al3+ can be completely or partially substituted in the layered structure by other bivalent or trivalent metal ions, respectively. Calcination of LDH materials leads to the formation of mixed metal oxides (MMOs), which are usually more basic catalysts than the corresponding parent layered samples.55 In a recent study, calcined LDH samples doped with various metal ions to replace Al3+, were tested for biodiesel production.56 10% Gallium dopants led to an increase to around 80% conversion, at 60 °C, of triacetin to the corresponding methyl esters. The use of an Fe based dopant at 5% and 10% led to even greater activity with >95% yield after 40 min at 60 °C, as compared to 1.0% weight of dopant. The surface area for this catalyst was found to be ∼50% greater than the uncalcined MgAl hydrotalcite, which is usual behavior of hydrotalcite-like or LDH materials. The mixed oxides that derive from calcination of LDHs at temperatures between ca. 400–550 °C exhibit significantly higher surface area (ca. 200–300m2 g−1) compared to the parent LDH samples (ca. 50–100 m2 g−1). Upon regeneration, through rehydration, the catalyst was extracted and re-calcined, giving only 50% of the initial activity of the original catalyst, with further regeneration yielding similar results.
A solid-base catalyst KF/Al2O3 has been utilised for the conversion of palm oil to alkyl esters by Boet al.57 The catalyst was prepared via impregnation of KF to give a supported catalyst on Al2O3. This was then dried and calcined at 600 °C. The trans-esterification was carried out at atmospheric pressure and with an optimum temperature of 65 °C; above this the volatility of methanol became an issue, leading to a decrease in the methanol:oil ratio from the desired 12:1. A catalyst ratio of KF:Al2O3 0.331 (wt/wt) using 4% catalyst (wt) over 4 h was found to lead to triglyceride production of over 90%. Interestingly, calcination of the catalyst at 600 °C led to a new phase of K3AlF6 as characterised by X-ray diffraction (XRD) and Thermogravimetric Analysis (TGA).
A superbase (as denoted by the Hammett scale) was prepared by calcination of Eu(NO3)3/Al2O3 for 2 h at 300 °C, 2 h at 550 °C and 8 h at 900 °C forming Eu2O3/Al2O3 with an optimal Eu content of >6.75%.58 This was used to trans-esterify soybean oil in a fixed bed reactor at atmospheric pressure. Again the reaction temperature was optimal at around 70 °C due to the volatility of methanol. Water was removed from the oil and methanol to prevent reaction with the catalyst. No reaction was observed for the first 30 min as observed by GC, with a steady increase in rate from 2 h and a final conversion of 63% at 8 h. The methanol:oil ratio was ≥4 for the greatest conversion, although continually increasing the methanol ratio can lead to separation problems from the prepared methyl esters, so a value of 5–6 was proposed. After 40 h of use catalyst activity had decreased, leading only to around 35% conversion, thought to be due to water and FFAs. After each subsequent regeneration the catalyst had lost surface area and its activity had decreased.
Some potential oils for biodiesel production such as deep frying oils are high in FFA content, making them unsuitable for base catalysed transesterification. In these cases a heterogeneous acid catalyst is preferred. Sulfated zirconia catalyst (S–ZrO2) has been found to catalyse soybean oil to biodiesel with 98.6% FAME yield.49 Unfortunately the catalyst is deactivated rapidly. Zinc stearate immobilised on silica gel was found to convert waste cooking oil of 15% FFA to 98% FAME with no loss of activity after four catalytic cycles, though the reaction temperature was relatively high at 200 °C.59Carbohydrate-derived heterogeneous acid catalysts have been shown to transesterify oils with up to 27.8 wt% FFA content to 92% FAME after 8 h.60 These catalysts were found to be exceptionally stable in that they were still around 93% active after 50 successive uses.
The alcohol used in trans-esterification may lead to fuels with differing properties. Usually methanol is the alcohol of choice, but Bokade et al. varied the alcohol used from methanol to n-octanol over 10 wt% of catalyst TPA/K-10.61 The reported percentage conversions were methanol (84%), ethanol (80%), n-propanol (76%) and n-octanol (72%) showing a decrease in oil conversion, possibly due to the increasing number of carbon atoms leading to a lower rate of reaction. This means less efficiency in the process and so greater costs incurred in the resulting fuel product.
It is useful to know when the trans-esterification process has reached completion and how far the reaction has progressed along its reaction profile so that no energy is wasted in the process. To monitor the process of trans-esterification, in situ viscosity measurements with an acoustic wave viscometer, have been tested.62 When the reaction had gone to completion a characteristic plateau was observed in the viscosity measurements. This was achieved successfully, both on a bench-top batch scale and at a pilot plant scale capable of 300 L/day. Using progression measurements helps increase efficiency and maximise productivity, making biodiesel ever more competitive as a fuel source.
| Author | Year | Catalyst | Reaction conditionsa | Product | Reported yields (%) |
|---|---|---|---|---|---|
| a t = reaction time. | |||||
| Vicente41 | 2004 | Sodium hydroxide | 65 °C, 6:1 methanol:oil. 1% catalyst (wt%) t = 4 h |
FAME | 86.7 |
| Vicente41 | 2004 | Potassium hydroxide | 65 °C, 6:1 methanol:oil. 1% catalyst (wt%) t = 4 h |
FAME | 91.7 |
| Vicente41 | 2004 | Sodium methoxide | 65 °C, 6:1 methanol:oil. 1% catalyst (wt%) t = 4 h |
FAME | 99.3 |
| Vicente41 | 2004 | Potassium methoxide | 65 °C, 6:1 methanol:oil. 1% catalyst (wt%) t = 4 h |
FAME | 98.5 |
| Miao40 | 2006 | Sulfuric acid | 30 °C, 56:1 molar ratio of methanol to oil. 100% catalyst (based on oil weight) t = 4 h |
FAME | >60 |
| Zafiropolous45 | 2007 | Sodium methoxide | 50 °C, 3:1 methanol:oil. 0.3 catalyst (wt%) t = 2 h |
FAME | 99 |
| Cantrell54 | 2005 | MgAl layered double hydroxide | 60 °C, 30:1 molar ratio of methanol to oil. 0.05g calcined catalyst, t = 3 h |
Methylbutanoate | 74.8 |
| Macala55 | 2008 | MgAl layered double hydroxide with Fe 10% | 80 °C, 6:1 methanol:oil. 1% calcined catalyst (wt%) t = 1 h |
trans-Esterified soybean oil | 38 |
| Bo 57 | 2007 | KF/Al2O3 0.331 (wt/wt) | 65 °C, 12:1 methanol:oil. 4% calcined catalyst (wt%) t = 3 h. |
FAME | >90 |
| Li 58 | 2007 | Eu2O3/Al2O3 with Eu 0.45–9.00 wt% | 70 °C, 6:1 methanoloil. 4% calcined catalyst (wt%) t = 8 h |
FAME | 63 |
| Bokade 61 | 2007 | TPA/K-10 (dodecatungst-ophosphoric acid) | 170 °C, 5:1 methanol:oil. 10% TPA/K-10 catalyst (wt%) t = 8 h |
FAME | 84 |
| Azcan63 | 2008 | Potassium hydroxide | 50 °C, 6:1 methanol:oil, 1% catalyst (wt%), microwave heating, t = 5 min |
FAME | 93.7 |
| Azcan63 | 2008 | Sodium hydroxide | 40 °C, 6:1 methanol:oil. 1% catalyst (wt%), microwave heating, t = 3 min |
FAME | 92.7 |
| Barakos47 | 2008 | MgAl-CO3 hydrotalcite layered double hydroxide | 200 °C, 6:1 methanol:oil. 1% catalyst (wt%), high initial FFA content, t = 3 h |
FAME | 99 |
| (final FFA < 1%) | |||||
| Jacobsen59 | 2008 | Zinc stearate on silica gel | 200 °C, 18:1 methanol:oil. 3% catalyst (wt%) t = 10 h |
FAME | 98 |
| Kouzu48 | 2008 | CaO | Reflux, 12:1 methanol:oil. 14 mmol catalyst, t = 1 h |
FAME | 93 |
| Lou60 | 2008 | Starch-derived catalyst (CH0.85O0.23S0.032) | 200 °C, 30:1 methanol:oil. 10% catalyst (wt%) t = 8 h |
FAME | 92 |
| Garcia49 | 2008 | S–ZrO2 | 120 °C, 20:1 methanol:oil. 5% catalyst (wt%) t = 1 h |
FAME | 98.6 |
| Saydut50 | 2008 | Sodium hydroxide | 60 °C, 6:1 methanol:oil. 0.5% catalyst (wt%) t = 2 h |
FAME | 74 |
| da Silva51 | 2008 | Co(II) ions adsorbed in chitosan | 70 °C, 6:1 methanol:oil. 2% catalyst (wt%) t = 3 h, pH 8.5 |
FAME | 94 |
5. De-oxygenated biofuels (green diesel) – decarboxylation/hydrogenation catalysis.
Petroleum hydrocarbons are believed to have formed in nature, possibly by the decarboxylation of fatty acids in the presence of natural clay catalysts present in formation rocks. Trans-esterification results in fuels which are oxygenated. If the fuels produced can be deoxygenated, then biomass derived fuels can be produced which could be used as a direct replacement for petroleum sourced hydrocarbons. Such fuels have been termed green diesel. This is an emerging area, with deoxygenation usually being performed via decarboxylation/hydrogenation reactions.Early studies into mineral decarboxylation by various workers, including Almon and Johns,66 were conducted to understand fossil oil formation. The decarboxylation of fatty acids during fossil fuel formation appeared to be via a free-radical mechanism, with increased reaction rates when free-radical initiators are present, such as H2O2, and decreased rates in the presence of oxygen, a free-radical inhibitor. The presence of H2O2 led to a 43% increase in reaction rate (K = 7.84 × 10−6sec−1). In the presence of O2 this led to a 41% decrease in rate (with K = 3.23 × 10−6sec−1). The presence of water was found to lead to different product distributions compared to the anhydrous reaction.
Pyrolysis is the normal method for deoxygenating vegetable feedstocks,67 using zeolites,68 but this can lead to a lower energy fuel. Bertram published work using a homogeneous catalyst over selenium to decarboxylate stearic acid to heptadecane in 1936.69 Recently, using a commercial activated carbon supported catalyst, n-heptadecane was found to be the main product when model compounds stearic acid, ethyl stearate and tristearine were deoxygenated.70 The decarboxylation of stearic acid resulted in production of heptadecenes which decreased over the reaction time, suggesting that they are intermediates. At 300 °C and at a pressure of 17 bar, stearic acid was found to have a higher percentage conversion with a reaction atmosphere of 5% hydrogen and 95% argon (by volume) when compared to reaction atmospheres of 100% helium and hydrogen. Ethyl stearate was found to convert into stearic acid before decarboxylating to n-heptadecane, best achieved in a similar reaction atmosphere of 5% hydrogen and 95% argon (by volume), although selectivity to n-heptadecane decreased from 300–360 °C when aromatics started to be produced instead, which are unsuitable in diesel fuel. The reaction kinetics for ethyl stearate and stearic acid decarboxylation over palladium/carbon (Pd/C) catalyst have been studied.71 With ethyl stearate the rate of reaction increased from 300–360 °C, with an activation energy of 57.3 kJ mol−1 from first order kinetics (K = 6.27 × 10−12 min−1 at 300 °C). With the major intermediate product, stearic acid, the reaction order is almost zero with the Pd/C catalyst deactivated at high concentrations of stearic acid.
Further studies into heterogeneous decarboxylation were carried out by Snare et al.72 The uncatalysed reaction was performed first and found to only lead to a <5% conversion. Similarly, using direct current plasma atomic emission spectroscopy (DCP-AES) with a Pd/C catalyst it was proved that the reaction was indeed heterogeneous and not homogeneous. A range of catalysts were tested, supported on carbon and metal oxides (Ir, Mo, Ni, Os, Pd, Pt, Rh and Ru) as well as a Raney nickel catalyst. Side reactions were observed over 6 h of reaction (300 °C, 6 bar, helium) such as isomerisation, dehydrogenation, aromatics and shorter hydrocarbons by cracking). The initial rate was greatest for 5% Pd/C (1.9 mmol s−1gmet−1) with carbon supported catalysts in general leading to higher rates, possibly due to catalyst structure. It was found that some side products produced using Ru/C and Rh/C catalysts were selective towards unsaturated side products, which could have lead to their deactivation. 5% Pd/C was found to be the preferred catalyst for decarboxylation of stearic acid, with Pt/C giving best performance for decarbonylation, followed by Ni, Rh, Ir, Ru and Os. Additionally, further work with unsaturated renewables has led to diesel-like hydrocarbons73
Further work with Pd supported on Sibunit (a porous carbon–carbon composite material with a high mesopore volume) as a catalyst, using dodecane, at a pressure of 17 bar helium has been carried out.74 This process was carried out in a semi-batch reactor with 300 mL volume. Using 4 wt% catalyst, stearic acid was deoxygenated with increasing initial reaction rate and decreasing time for 100% conversion as temperature increased from 270 °C to 300 °C to 330 °C. At 270 °C it was found that the there was a lag time of 60 min before stearic acid conversion progressed.
Green diesel has been produced by catalytic saturation, hydrodeoxygenation, decarboxylation and hydroisomerisation reactions.75 Using hydrogen at around 1.5–3.8% in the reactor produced green diesel yields between 88–99% depending on the catalyst used. The resulting product was comparable in its properties to ultra-low sulfur diesel, and superior to oxygenated biodiesel. Production from palm oil is estimated as feasible at a crude oil price of $52 bbl and soybean oil at $67 bbl. In this study green diesels were found to have positively higher net energy balances than petroleum diesel and biodiesel, which translates to lower fossil fuel inputs during their production. They also lead to considerably lower greenhouse gas emissions per energy equivalent compared to petroleum diesel, being slightly less than comparable amounts of biodiesel. These results suggest that green diesel is a superior bio-energy source, in terms of sustainability, compared to oxygenated biodiesel. A summary of some of the decarboxylation reactions which have been trialled is shown in Table 3.
| Author | Year | Catalyst | Reaction conditionsa | Main product | Reported yields (%) |
|---|---|---|---|---|---|
| a t = reaction time. | |||||
| Almon66 | 1975 | Ca–montmorillonite | 250 °C, excess water | n-Heneicosane | |
| Lestari74 | 2008 | Pd/C (Sibunit) | 330 °C, 17 bar helium, 4% catalyst (wt%) t = 20 min | n-Heptadecane | 100 |
| Kubrickova70 | 2008 | Activated carbon supported palladium (5 wt%, Aldrich) | 300 °C, 17 bar hydrogen (5 vol%) + argon (95 vol%), 4% catalyst (wt%) t = 360 min | n-Heptadecane | 62 |
| Snare72 | 2006 | 5% Pd/C | 300 °C, 6 bar helium, 1 g catalyst, t = 360 min | n-Heptadecane | 100 |
6. The future
Savage et al. recently looked at how synthetic biology can help the production of modern biofuels. With the current sources of ethanol a great deal of plant biomass (cellulose) is not utilized during refinement. Engineered plants could be used to overcome this; also new chemical processes and enzymes to break down these unusable carbohydrate sources. Novel fatty acid synthases could be produced to build up fatty acid groupsvia addition of –COCH3 to a chain until the required length is obtained.76Glycerol is the main byproduct of current trans-esterification processes. If suitable processes/conversions of glycerol could make it a useful commodity, then this could lead to it moving away from being a byproduct, with reduced overall production costs for biodiesel. Example processes include steam-reforming of glycerol to form hydrogen,77pyrolysis of glycerol to produce syngas, methane and ethane.78 Also, it can be used to produce 1,3-propanediol which is a precursor for fibre synthesis, usually obtained from petrochemical origins.79
To make a true biofuel all sources of raw materials need to be derived from biomass. Methanol used in trans-esterification processes is usually produced from fossil fuel sources. Impure methanol may be produced from wood gasification, which can then be used in the synthesis of biodiesel. Impurities can also be converted into esters and again used as fuel components resulting in a truer biofuel.80
7. Conclusions
Transport fuels are a required commodity in the world today, but their traditional source, crude oil, will expire eventually. Other issues with energy security and oil prices are ever on the increase. Thus, there is a need to develop renewable replacements for petrol (gasoline) and diesel, which can be used with conventional transport technology, already in place. Fuels derived from biomass can solve this problem but they in turn have their own advantages and disadvantages. The cycling of carbon having no net gain effect is of great importance on an environmental scale, with the current issues of rising carbon dioxide levels and global warming.First generation biofuels are currently in commercial production, utilising energy from food-based crops, either by fermenting sugar to form bioethanol or extracting oils for conversion to biodiesel. Biodiesel is an oxygenated fuel and there is a need to develop decarboxylation processes over the current trans-esterification processes to produce deoxygenated fuel, so-called green diesel.
There are ethical issues with using biofuels whose feedstocks are food derived or that take up land destined for food crops or rainforests. There is a need to develop next generation biofuels to meet biofuel targets.81 “Extracting” potential energy from the whole crop, for example including the stalks of energy crops, is required, thus leading to the highest levels of efficiency and sustainability. Algae are a promising source of oils for upgrading to diesel-type fuel. They are capable of doubling their mass multiple times a day, photosynthesise more efficiently than soil-medium crops and take up far less land for corresponding amounts of fuels. Life cycle analysis of any process is likely to be critical to ensure economic feasibility.
A considerable body of research is currently being undertaken by many organisations around the world, on large and small scales, to produce high grade renewable fuels of sustainable origin. Breakthroughs in heterogeneous catalysis for decarboxylation reactions that allow the use of existing petrochemical processing and refining infrastructure, and produce a product that is compatible with existing transport are likely to make biofuels a viable alternative energy source.
Acknowledgements
The authors would like to thank the Engineering and Physical Sciences Research Council and KiOR (http://www.kior.com) for an industrial case award. We are grateful to Paul O'Connor (KiOR) for his assistance in the preparation of this manuscript and valuable comments.References
- http://www.guardian.co.uk/business/2008/jun/11/commodities.bp accessed 13/06/08.
- K. A. Shein, A. M. Waple and M. J. Menneet Al., Bull. Am. Meteorol. Soc., 2006, 87, S6–S102 CrossRef.
- L. P. Wackett, Curr. Opin. Chem. Biol., 2008, 12, 187–193 CrossRef CAS.
- Editorial, Nature Geosci., 2008, 1, 281 Search PubMed.
- A. K. Agarwal, Prog. Energy Combust. Sci., 2007, 33, 233–271 CrossRef CAS.
- T. Searchinger, R. Heimlich, R. A. Houghton, F. X. Dong, A. Elobeid, J. Fabiosa, S. Tokgoz, D. Hayes and T. H. Yu, Science, 2008, 319, 1238–1240 CrossRef CAS.
- J. Fargione, J. Hill, D. Tilman, S. Polasky and P. Hawthorne, Science, 2008, 319, 1235–1238 CrossRef CAS.
- D. Tilman, J. Hill and C. Lehman, Science, 2006, 314, 1598–1600 CrossRef CAS.
- J. Goldemberg, Energy Environ. Sci., 2008 10.1039/b814178a.
- P. J. L. Williams, Nature, 2007, 450, 478–478 CrossRef CAS.
- Directive 2003/30/EC of 8 May 2003 on the promotion of the use of biofuels or other renewable fuels for transport (OJ L 123, 17.5.2003).
- G. P. Hammond, S. Kallu and M. C. McManus, Appl. Energy, 2008, 85, 506–515 CrossRef CAS.
- An EU Strategy for Biofuels, 2006, Communication from the Commission, SEC, 2006, 142, p. 3 Search PubMed.
- Our energy future – creating a low carbon economy, Cm 5761, February 2003, 5.1, Energy White Paper, DTI Search PubMed.
- S. Atsumi, T. Hanai and J. C. Liao, Nature, 2008, 451, 86–89 CrossRef CAS.
- Biofuels for transport: global potential and implications for energy and agriculture, Worldwatch Institute, London, 2007, pp. 1 Search PubMed.
- E. Marris, Nature, 2006, 444, 670–672 CrossRef CAS.
- K. Sanderson, Nature, 2006, 444, 673–676 CrossRef CAS.
- C. W. King, A. S. Holma and M. E. Webber, Nature Geosci., 2008, 1, 283–286 CrossRef CAS.
- News in Brief, Nature, 2007, 447, p. 897 Search PubMed.
- J. Kuesters and J. Lammel, Eur. J. Agron., 1999, 11, 35–43 CrossRef.
- L. Ruth, Embo Rep., 2008, 9, 130–133 CrossRef CAS.
- A. S. Ramadhas, S. Jayaraj and C. Muraleedharan, Renewable Energy, 2004, 29, 727–742 CrossRef CAS.
- F. R. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- Y. Chisti, Tr. Biotechnol., 2008, 26, 126–131 Search PubMed.
- Y. Chisti, Biotechnol. Adv., 2007, 25, 294–306 CrossRef CAS.
- M. L. Ghirardi, J. P. Zhang, J. W. Lee, T. Flynn, M. Seibert, E. Greenbaum and A. Melis, Tr. Biotechnol., 2000, 18, 506–511 Search PubMed.
- A. Melis, L. P. Zhang, M. Forestier, M. L. Ghirardi and M. Seibert, Plant Physiol., 2000, 122, 127–135 CrossRef CAS.
- R. Surzycki, L. Cournac, G. Peltiert and J. D. Rochaix, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17548–17553 CrossRef CAS.
- http://www.handsontv.info/series7/01_energy_wise_reports/report4.html accessed 01/07/08.
- J. Hill, E. Nelson, D. Tilman, S. Polasky and D. Tiffany, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11206–11210 CrossRef CAS.
- R. N. Singh, D. K. Vyas, N. S. L. Srivastava and M. Narra, Renewable Energy, 2008, 33, 1868–1873 CAS.
- K. Narasimharao, A. Lee and K. Wilson, J. Biobased Mater. Bioenergy, 2007, 1, 19–30 Search PubMed.
- M. J. Ramos, C. M. Fernández, A. Casas, L. Rodríguez and Á. Pérez, Bioresour. Technol., 2009, 100, 261–268 CrossRef CAS.
- F. A. Twaiq, N. A. M. Zabidi and S. Bhatia, Ind. Eng. Chem. Res., 1999, 38, 3230–3237 CrossRef.
- F. A. Twaiq, A. R. Mohamed and S. Bhatia, Microporous Mesoporous Mater., 2003, 64, 95–107 CrossRef CAS.
- J. T. Kloprogge, L. V. Duong and R. L. Frost, Environ. Geol., 2005, 47, 967–981 CrossRef CAS.
- Z. B. Zhao, Ind. Eng. Chem. Res., 2006, 45, 6874–6874 CrossRef CAS.
- M. Snare and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 6875–6875 CrossRef.
- X. L. Miao and Q. Y. Wu, Bioresour. Technol., 2006, 97, 841–846 CrossRef CAS.
- G. Vicente, M. Martinez and J. Aracil, Bioresour. Technol., 2004, 92, 297–305 CrossRef CAS.
- D. Y. C. Leung and Y. Guo, Fuel Process. Technol., 2006, 87, 883–890 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- M. Canakci and H. Sanli, J. Ind. Microbiol. Biotechnol., 2008, 35, 431–441 CrossRef CAS.
- N. A. Zafiropoulos, H. L. Ngo, T. A. Foglia, E. T. Samulski and W. B. Lin, Chem. Commun., 2007, 3670–3672 RSC.
- M. J. Haas, S. Bloomer and K. Scott, US Pat., 6399800, 2002 Search PubMed.
- N. Barakos, S. Pasias and N. Papayannakos, Bioresour. Technol., 2008, 99, 5037–5042 CrossRef CAS.
- M. Kouzu, T. Kasuno, M. Tajika, S. Yamanaka and J. Hidaka, Appl. Catal., A, 2008, 334, 357–365 CrossRef CAS.
- C. M. Garcia, S. Teixeira, L. L. Marciniuk and U. Schuchardt, Bioresour. Technol., 2008, 99, 6608–6613 CrossRef CAS.
- A. Saydut, M. Z. Duz, C. Kaya, A. B. Kafadar and C. Hamamci, Bioresour. Technol., 2008, 99, 6656–6660 CrossRef CAS.
- R. B. da Silva, A. F. L. Neto, L. S. S. dos Santos, J. R. D. Lima, M. H. Chaves, J. R. dos Santos, G. M. de Lima, E. M. de Moura and C. V. R. de Moura, Bioresour. Technol., 2008, 99, 6793–6798 CrossRef.
- B. M. Choudary, M. L. Kantam, C. V. Reddy, S. Aranganathan, P. L. Santhi and F. Figueras, J. Mol. Catal. A: Chem., 2000, 159, 411–416 CrossRef CAS.
- A. Corma, S. Iborra, S. Miquel and J. Primo, J. Catal., 1998, 173, 315–321 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS.
- F. Cavani, F. Trifirò and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- G. S. Macala, A. W. Robertson, C. L. Johnson, Z. B. Day, R. S. Lewis, M. G. White, A. V. Iretskii and P. C. Ford, Catal. Lett., 2008, 122, 205–209 CrossRef CAS.
- X. Bo, G. M. Xiao, L. F. Cui, R. P. Wei and L. J. Gao, Energy Fuels, 2007, 21, 3109–3112 CrossRef CAS.
- X. Li, G. Z. Lu, Y. Guo, Y. Q. Wang, Z. G. Zhang, X. H. Liu and Y. S. Wang, Catal. Commun., 2007, 8, 1969–1972 CrossRef CAS.
- K. Jacobson, R. Gopinath, L. C. Meher, A. K. Dalai, Appl. Catal., B, 2008, DOI:10.1016/j.apcatb.2008.07.005.
- W.-Y. Lou, M.-H. Zong and Z.-Q. Duan, Bioresour. Technol., 2008, 99, 8752–8758 CrossRef CAS.
- V. V. Bokade and G. D. Yadav, Proc. Saf. Environ. Prot., 2007, 85, 372–377 Search PubMed.
- N. Ellis, F. Guan, T. Chen and C. Poon, Chem. Eng. J., 2008, 138, 200–206 CrossRef CAS.
- N. Azcan and A. Danisman, Fuel, 2008, 87, 1781–1788 CrossRef CAS.
- EN 14214-Automotive fuels - Fatty acid methyl esters (FAME) for diesel engines - Requirements and test methods, 2003.
- A. H. West, D. Posarac and N. Ellis, Bioresour. Technol., 2008, 99, 6587–6601 CrossRef CAS.
- W. R. Almon and W. D. Johns, Advances in Organic Geochemistry, 7th Int. Meeting, 1975, 157–171 Search PubMed.
- A. Demirbas, Fuel Process. Technol., 2007, 88, 591–597 CrossRef CAS.
- A. Demirbas, Energy Educ. Sci. Technol., 2008, 21, 1–59 Search PubMed.
- S. H. Bertram, Chem. Weekbl., 1936, 33, 457–459 Search PubMed.
- I. Kubickova, M. Snare, K. Eranen, P. Maki-Arvela and D. Y. Murzin, Catal. Today, 2005, 106, 197–200 CrossRef CAS.
- M. Snare, I. Kubickova, P. Maki-Arvela, K. Eranen, J. Warna and D. Y. Murzin, Chem. Eng. J., 2007, 134, 29–34 CrossRef CAS.
- M. Snare, I. Kubickova, P. Maki-Arvela, K. Eranen and D. Y. Murzin, Ind. Eng. Chem. Res., 2006, 45, 5708–5715 CrossRef.
- M. Snare, I. Kubickova, P. Maki-Arvela, D. Chichova, K. Eranen and D. Y. Murzin, Fuel, 2008, 87, 933–945 CrossRef CAS.
- S. Lestari, I. Simakova, A. Tokarev, P. Maki-Arvela, K. Eranen and D. Y. Murzin, Catal. Lett., 2008, 122, 247–251 CrossRef CAS.
- T. Kalnes, T. Marker and D. R. Shonnard, Int. J. Chem. React. Eng., 2007, 5 Search PubMed.
- D. F. Savage, J. Way and P. A. Silver, Chem. Biol., 2008, 3, 13–16 CAS.
- M. Slinn, K. Kendall, C. Mallon and J. Andrews, Bioresour. Technol., 2008, 99, 5851–5858 CrossRef CAS.
- T. Valliyappan, N. N. Bakhshi and A. K. Dalai, Bioresour. Technol., 2008, 99, 4476–4483 CrossRef CAS.
- S. Hirschmann, K. Baganz, I. Koschik and K. D. Vorlop, Landbauforsch. Volkenrode, 2005, 55, 261–267 Search PubMed.
- Y. Isayama and S. Saka, Bioresour. Technol., 2008, 99, 4775–4779 CrossRef CAS.
- Editorial, Nature, 2008, 451, p. 499 Search PubMed.
| This journal is © The Royal Society of Chemistry 2009 |