Robust recognition of malonate and 2-amino-4-picolinium in conjunction with M(II) as a triad (M = Ni/Co/Mn): role of this highly stable hydrogen-bonded motif in driving supramolecular self-assembly†
Somnath Ray
Choudhury
a,
Biswajit
Dey
a,
Suranjana
Das
a,
Arturo
Robertazzi
b,
Atish Dipankar
Jana
*c,
Chih-Yuan
Chen
d,
Hon Man
Lee
d,
Patrick
Gamez
e and
Subrata
Mukhopadhyay
*a
aDepartment of Chemistry, Jadavpur University, Kolkata, 700 032, India. E-mail: smukhopadhyay@chemistry.jdvu.ac.in
bCNR-INFM SLACS and Dipartimento di Fisica, Università di Cagliari, S.P. Monserrato-Sestu Km 0.700, I-09042, Monserrato, Italy
cDepartment of Physics, Sripat Singh College, Jiaganj, Murshidabad, West Bengal, 742 123, India. E-mail: atishdipankar@yahoo.com
dDepartment of Chemistry, National Changhua University of Education, Changhua, Taiwan, 50058
eLeiden Institute of Chemistry, Leiden University, P.O. Box 9502, 2300, RA, Leiden, The Netherlands. E-mail: p.gamez@chem.leidenuniv.nl
First published on 20th July 2009
Abstract
The crystal lattice of the three isostructural compounds 1–3, (C6H8N2H)2[M(C3H2O4)2(H2O)2]·4H2O (C6H8N2H = protonated 2-amino-4-picoline, M = Ni/Co/Mn, C3H4O4 = malonate dianion; hereafter, malonate) is formed by supramolecular 2D layers. Hydrogen-bonding, π⋯π and lone pair⋯π interactions play crucial role in organizing monomeric [MII(mal)2(H2O)2] units into 2D sheets along the ab plane, through the self-association between two different supramolecular building blocks, namely a tetrameric water cluster including metal-coordinated water molecules, and R22(8) and R22(7) hydrogen-bonded recognition synthons between 2-amino-4-picolinium and malonate. DFT calculations clearly show that the robust 2-amino-4-picolinium/malonate hydrogen-bonded motif drives the self-assembly of the supramolecular network observed.
Introduction
Molecular recognition is not only at the heart of bio-chemical processes,1–5 but is also crucial in various other areas of science and technology, such as the design of sensor materials,6 selective catalysis,7,8host–guest systems,9 surface patterning,10 nano-scale assembly,11 and so on. The basis of molecular recognition is the appropriate complementarities between the host and the guest. Although various weak forces such as π⋯π,12–14 C–H⋯π,15–17etc. have been employed for this purpose, hydrogen bonding still remains the most reliable and widely used means for enforcing molecular recognition.18–20Self-assembly of molecular building blocks through molecular recognition has led the way in the design and preparation of various functional materials, both of organic as well as of hybrid organic–inorganic type.21,22 It is generally assumed that identical recognition patterns exist both in solution and in the crystalline solid state; in many cases, this has been unequivocally established.23 The rational synthesis of desired supramolecular materials has reached a state of the art where one relies on the most commonly observed recognition pattern between a set of molecular building blocks. However, sometimes, various unforeseen factors such as the participation of solvent molecules or the pH of the solution play an important role in the self-assembly process.Malonate has been widely used as ligand for the synthesis of crystalline coordination materials of diverse architectures, as the result of its various potential binding modes to transition metals as well as its participation in hydrogen bonding interactions.24–28 Moreover, in combination with other ligands,29–33malonate has allowed the generation of remarkable architectures. The use of malonate in association with 2-amino-4-picoline in copper(II) systems34,35 has shown that these two ligands have a preferred recognition pattern through the formation of a R22(8) hydrogen-bonding synthon. It is also worth mentioning that this recognition is enforced in situ by the protonation of 2-amino-4-picoline.
In the present study, the preparation of malonate–metal–2-amino-4-picoline ternary systems has been extended to other transition metals, namely nickel(II), cobalt(II) and manganese(II). As expected, this investigation shows that a similar recognition mode between 2-amino-4-picolinium and malonate, through in situ protonation of 2-amino-4-picoline, is responsible for the formation of supramolecular assemblies. However, contrary to the Cu(II) compounds described earlier,34,35 for which the malonate moiety acts as a bridging ligand producing 2D coordination polymers, the present systems exhibit individual mononuclear units. The crystal packing of the three isostructural compounds reveals the presence of two types of hydrogen-bonded supramolecular units. The first network is generated by the association of [MII(mal)2(H2O)2] (M = Ni/Co/Mn) synthons through hydrogen bonds involving coordinated and lattice water molecules. The second network is produced viahydrogen-bonding interactions between [MII(mal)2(H2O)2] units and protonotated 2-amino-4-picoline molecules (via the typical R22(8) hydrogen-bonding motif for malonate/2-amino-4-picolinium recognition). The combination of these two gives rise to the formation of 2D supramolecular sheets. These two different supramolecular associations have been theoretically investigated to compare their formation energies and their relative involvement in the construction of the 2D self-assembly.
Experimental
General
All reactions were carried out in aerobic conditions and in water as the solvent. Malonic acid (Lancaster), manganese(II) acetate tetrahydrate (Aldrich), cobalt(II) acetate tetrahydrate (Aldrich), nickel(II) acetate tetrahydrate (Aldrich) and 2-amino-4-picoline (Aldrich) were used as received. Doubly distilled and then freshly boiled water was used throughout. Elemental analyses (C, H, N) of compounds 1–3 were performed on a Perkin-Elmer 240C elemental analyzer. IR spectra were recorded on a Perkin-Elmer L120–00A FT-IR spectrophotometer with the sample prepared as a KBr pellet in the range 4000–600 cm−1. The TGA were carried out with a Mettler Toledo TGA/SDTA 851e system (Fig. S1, ESI†).36All the compounds exhibit almost identical IR spectra. The absorption band at 3342 cm−1 can be assigned to the stretching vibration, ν(O–H), of the hydroxyl groups in the water molecules. The band at 3058 cm−1 can be assigned to the stretching vibration, ν(N–H), of the primary aromatic amine. The next band at 2848 cm−1 corresponds to the stretching vibration, ν(C–H), of the malonate ligands. The νas(OCO) and νs(OCO) absorption bands are observed at 1579 and 1434 cm−1, respectively. The δ(OCO) absorptions are located at 796 and 742 cm−1.
The thermogravimetric analyses of compounds 1–3 show that dehydration starts approximately at 62 °C and all water molecules are lost at 112 °C (the calculated weight loss corresponding to six water molecules is 18.33% for 1, found 17.23%; 18.32% for 2, found 17.25% and 18.45% for 3, found 17.32%). Complete decomposition of the dehydrated compounds is achieved at 400 °C. A representative TG curve (of 1) is shown in Fig. S1 (ESI†).36
Preparation of the coordination complexes
Crystallography
Single crystals having suitable dimensions for 1–3 were used for data collection using a ‘Bruker SMART APEX II’ diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) at 150(2) K. The molecular structures were solved by direct methods and refinement by full-matrix least squares on F2 using the SHELX-97 package.37 The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. H atoms of water molecules were located by difference Fourier maps and were kept fixed.Computational details
Following our recent investigations,38,39 single-point calculations and full optimizations were carried out using the Gaussian03 suite of programs,40 employing the Becke's half-and-half (BHandH) DFT functional for the stacked system (motif 2, see text).41 This functional combined with medium-sized basis sets is able to reproduce high-level calculations for systems governed by dispersive forces, leading to significant results reported in a number of recent studies.42–48 In addition, only for the H-bonded synthon (motif 1, see text), the B3LYP/6-31+G(d) level was also employed in order to double check the results obtained with BHandH (e.g. positive formation energy, vide infra). The formation energies were obtained via the super-molecule approach, i.e. calculated as the difference between the energy of the entire complex and the energies of the single components of the complex, with no correction for the basis set superposition error.The atoms-in-molecules (AIM)49,50 theory was then performed directly on the experimental and optimized structures (motif 1 and motif 2, see text) to estimate the inter-molecular interactions. In particular, AIM is based upon those critical points where the gradient of the density, ∇ρ, vanishes. In this work, we will only consider (3, −1) or bond critical points, wherein one curvature (in the internuclear direction) is positive and two (perpendicular to the bond direction) are negative. Two bonded atoms are connected with a bond path through the bond critical point. Importantly, several studies have shown that the electron density at the bond critical points correlates with the strength of the chemical bonds and interactions.42,49–53 We would like to note that, as the structures of all the three compounds reported here are very similar, we have performed DFT studies only on the Ni(II) compound, 1.
Results and discussion
Crystal structures of (C6H8N2H)2[M(mal)2(H2O)2]·4H2O (C6H8N2H = protonated 2-amino-4-picoline; mal = malonate; M = NiII (1), CoII (2), MnII (3))
Compounds 1–3 are isostructural. The three coordination compounds crystallize in the triclinic space groupPī with the asymmetric unit consisting of half of the molecular anion [M(mal)2(H2O)2]2− (with M = NiII (1), CoII (2) or MnII (3)), one C6H8N2H+ cation and two lattice water molecules. The anionic unit is centrosymmetric with the metal atoms occupying the center of inversion. A representative ORTEP diagram is shown in Fig. 1, while detailed crystal and structure refinement data of 1–3 are collected in Table 1. Two oxygen atoms (O1 and O3) from one bidentate malonate moiety and their centrosymmetric counterparts (O1* and O3*, * = 1 −x, −y, −z for 1, 1 −x, 1 −y, 1 −z for 2, 1 −x, 1 −y, −z for 3) of a symmetry-related malonate unit constitute the equatorial plane. Two water molecules occupy the trans axial positions forming a distorted octahedral MO4O/2chromophore. The M–O bond distances in the equatorial plane vary in the range 2.0198(8)–2.1843(8) Å. The lowest M–O bond length is observed for 1, where the Ni1–O1 bond length is 2.0198(8) Å, whereas the highest one is found for 3, with the Mn1–O3 bond length amounting to 2.1843(8) Å. In the same way, the M–O5 bond length corresponding to the axially bound water molecule is also found to be the lowest in 1 (2.0691(10) Å) and the highest in 3 (2.1806(11) Å). The angle subtended at the metal centers by the malonato moiety is comparable in compounds 1–3. Selected bond lengths and angles for compounds 1–3 are listed in Table 2.| Compound | 1 | 2 | 3 |
|---|---|---|---|
| Formula | C18H34N4O14Ni | C18H34N4O14Co | C18H34N4O14Mn |
| M | 589.20 | 589.42 | 585.43 |
| Crystal system | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2) (no. 2) |
P (no. 2) |
P (no. 2) |
| Crystal dimensions/mm | 0.30 × 0.40 × 0.50 | 0.09 × 0.37 × 0.50 | 0.30 × 0.31 × 0.50 |
| a/Å | 7.4284(2) | 7.5389(2) | 7.6690(6) |
| b/Å | 8.9640(2) | 9.0543(2) | 9.1074(7) |
| c/Å | 10.5124(2) | 10.4107(3) | 10.3380(8) |
| α/° | 94.669(1) | 93.349(2) | 92.776(1) |
| β/° | 92.165(1) | 93.305(2) | 92.558(1) |
| γ/° | 111.418(1) | 112.098(1) | 111.734(1) |
| F(000) | 310 | 309 | 307 |
| S(GOF) | 1.08 | 1.06 | 1.64 |
| V/Å3 | 647.78(3) | 654.86(3) | 668.41(9) |
| Z | 1 | 1 | 1 |
| T/K | 150(2) | 150(2) | 150(2) |
| θ Range/° | 2–28.6 | 2–28.7 | 2–28.6 |
| D c/Mg m−3 | 1.510 | 1.495 | 1.454 |
| λ(Mo-Kα)/Å | 0.71073 | 0.71073 | 0.71073 |
| μ(Mo-Kα)/mm−1 | 0.823 | 0.729 | 0.566 |
| R1, I > 2σ(I) (all) | 0.0226 | 0.0259 | 0.0278 |
| wR2, I > 2σ(I) (all) | 0.0610 | 0.0673 | 0.0804 |
| Total reflections | 9820 | 9535 | 7406 |
| Unique data (Rint) | 3285 (0.017) | 3376 (0.017) | 3390 (0.014) |
| Observed data [I > 2σ(I)] | 3133 | 3045 | 3070 |
| Δρmax, min/e Å−3 | −0.31, 0.48 | −0.44, 0.39 | −0.38, 0.57 |
| 1 | |||
|---|---|---|---|
| C(1)–O(1) | 1.2640(13) | C(3)–O(4) | 1.2528(14) |
| C(1)–O(2) | 1.2579(14) | Ni(1)–O(1) | 2.0198(8) |
| C(1)–C(2) | 1.5221(16) | Ni(1)–O(3) | 2.0532(8) |
| C(2)–C(3) | 1.5233(16) | Ni(1)–O(5) | 2.0691(10) |
| C(3)–O(3) | 1.2698(14) | ||
| O(2)–C(1)–O(1) | 123.09(11) | O(1)–Ni(1)–O(3) | 90.86(3) |
| O(1)–C(1)–C(2) | 118.88(10) | O(1)–Ni(1)–O(5) | 93.50(4) |
| C(1)–C(2)–C(3) | 114.94(10) | O(1)–Ni(1)–O(3)i | 89.14(3) |
| C(2)–C(3)–O(4) | 117.30(10) | O(1)–Ni(1)–O(5)i | 86.50(4) |
| C(2)–C(3)–O(3) | 119.23(10) | O(3)-Ni(1)-O(5) | 90.37(4) |
| Ni(1)–O(1)–C(1) | 125.09(8) | O(3)–Ni(1)–O(5)i | 89.63(4) |
| Ni(1)–O(3)–C(3) | 124.47(8) | O(3)i–Ni(1)–O(5)i | 90.37(4) |
| 2 | |||
|---|---|---|---|
| C(1)–O(1) | 1.2598(14) | C(3)–O(4) | 1.2496(16) |
| C(1)-O(2) | 1.2569(15) | Co(2)–O(1) | 2.0546(8) |
| C(1)–C(2) | 1.5260(18) | Co(2)–O(3) | 2.0895(9) |
| C(2)–C(3) | 1.5251(18) | Co(2)–O(5) | 2.0781(12) |
| C(3)–O(3) | 1.2679(16) | ||
| O(2)–C(1)–O(1) | 123.24(11) | O(1)–Co(2)–O(3) | 89.37(3) |
| O(1)–C(1)–C(2) | 118.74(10) | O(1)–Co(2)–O(5) | 91.99(4) |
| C(1)–C(2)–C(3) | 114.57(11) | O(1)–Co(2)–O(3)ii | 90.63(3) |
| C(2)–C(3)–O(4) | 117.72(11) | O(1)-Co(2)-O(5)ii | 88.01(4) |
| C(2)–C(3)–O(3) | 118.49(11) | O(3)–Co(2)–O(5) | 94.15(6) |
| Co(2)–O(1)–C(1) | 127.03(8) | O(1)ii–Co(2)–O(3) | 90.63(3) |
| Co(2)–O(3)–C(3) | 125.52(8) | O(3)–Co(2)–O(5)ii | 85.85(6) |
| 3 | |||
|---|---|---|---|
| a i = 1 −x, −y, −z; ii = 1 −x, 1 −y, 1 −z; iii = 1 −x, 1 −y, –z. | |||
| C(1)–O(1) | 1.2620(14) | C(3)–O(4) | 1.2491(15) |
| C(1)-O(2) | 1.2553(14) | Mn(1)–O(1) | 2.1172(8) |
| C(1)–C(2) | 1.2691(15) | Mn(1)–O(3) | 2.1843(8) |
| C(2)–C(3) | 1.5274(16) | Mn(1)–O(5) | 2.1806(11) |
| C(3)–O(3) | 1.2691(15) | ||
| O(2)–C(1)–O(1) | 123.07(11) | O(1)–Mn(1)–O(3) | 86.80(3) |
| O(1)–C(1)–C(2) | 118.65(10) | O(1)–Mn(1)–O(5) | 88.90(4) |
| C(1)–C(2)–C(3) | 114.50(11) | O(1)–Mn(1)–O(3)iii | 93.20(3) |
| C(2)–C(3)–O(4) | 117.79(10) | O(1)–Mn(1)–O(5)iii | 91.10(4) |
| C(2)–C(3)–O(3) | 118.32(10) | O(3)–Mn(1)–O(5) | 84.06(4) |
| Mn(1)–O(1)–C(1) | 128.63(8) | O(1)iii–Mn(1)–O(3) | 93.20(3) |
| Mn(1)–O(3)–C(3) | 125.65(7) | O(3)–Mn(1)–O(5)iii | 95.94(4) |
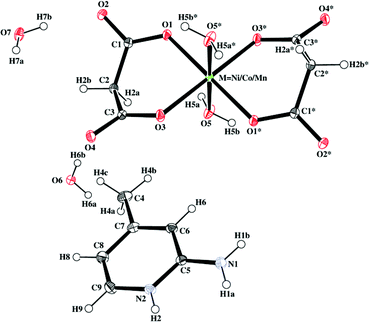 | ||
| Fig. 1 Representative ORTEP diagram of compounds 1–3. Thermal ellipsoids are drawn at the 30% probability level. Atoms marked with an asterisk (*) are at equivalent position (* = 1 −x, −y, −z for 1; 1 −x, 1 −y, 1 −z for 2;1 −x, 1 −y, –z for 3). | ||
The comparison of the present bond lengths and angles with those reported earlier for analogous compounds reveals that the axial bonds are shorter in 1–3.26,29,54–57 The equatorial bonds and angles subtended at the metal centers are comparable, which indicates that the MO4O/2 octahedra in 1–3 are axially compressed.58–65
Compounds 1–3 exhibit identical crystal lattices. The 3D supramolecular self-assembly in the solid-state structures of the three compounds can be regarded as the stacking of successive metal–organic and organic layers. 2-amino-4-picoline molecules constituting the organic layer are sandwiched between metal–malonate layers. The spatial organization of the monomeric [M(mal)2(H2O)2]2− units, generating the metal–malonate layer, appears to be driven by lattice water molecules (O7). As is evidenced in Fig. 2, the coordinated water molecules (O5) from successive [M(mal)2(H2O)2]2− anions and lattice water molecules O7 along with its symmetry related counterpart O7* form a tetrameric water cluster. These tetrameric units connect [M(mal)2(H2O)2]2− units, producing a 2D layer in the ab plane. The NbO-type topology of the layer is probably templated by the topology of the water tetramer itself. Adjacent [M(mal)2(H2O)2]2− coordination units along the a direction on the 2D sheet are doubly linked to each other through the two arms of the tetramer (O5–H5A⋯O7 = 2.750, 2.724 and 2.718 Å for 1, 2 and 3 and O5–H7B⋯O7 = 2.758, 2.750 and 2.751 Å for 1, 2 and 3) hydrogen bonds (see Tables 3, 4 and 5 for hydrogen bonding parameters), forming a supramolecular chain along the a-axis. Each such chain is linked to adjacent chains by means of hydrogen-bonding contacts. These supramolecular interactions give rise to a shifting of the successive chains, by a/2 with respect to each other, to facilitate the hydrogen bonding of the malonate (O2) atom at the other corner of the water tetramer, where O7 acts as a donor for O2, producing a supramolecular layer. The organization of this layer is such that the carboxylate terminals of the chelated malonate ligands project outwards of this plane and make an angle of about 45° with the mean plane.
| D–H⋯A | d(D⋯A)/Å | ∠(D–H⋯A)/° |
|---|---|---|
| a Symmetry codes: a = 1 −x, −y, 1 −z; b = x− 1 + y, z; c = 1 −x, 1 −y, –z; d = −1 + x, −1 + y, z; e = −1 + x, y, z; f = 1 + x, y, z; g = x, y, 1 + z. | ||
| N1–H1A⋯O4a | 2.791(1) | 170 |
| N1–H1B⋯O6b | 2.834(1) | 164 |
| N2–H2⋯O3a | 2.834(1) | 177(2) |
| O5–H5A⋯O7c | 2.750(1) | 141 |
| O5–H5B⋯O7d | 2.758(1) | 167(2) |
| O6–H6A⋯O4e | 2.778(1) | 171.3(2) |
| O6–H6B⋯O2c | 2.720(1) | 169(2) |
| O7–H7A⋯O6f | 2.766(1) | 170(2) |
| O7–H7B⋯O2 | 2.737(1) | 172(2) |
| C9–H9⋯O1g | 3.253(2) | 130 |
| D–H⋯A | d(D⋯A)/Å | ∠(D–H⋯A)/° |
|---|---|---|
| a Symmetry codes: a = 1 −x, 1 −y, −z; b = 1 −x, 1 −y, 1 −z; c = 1 + x, y, z; d = −x, −y, 1 −z; e = −x, 1 −y, 1 −z; f = x, 1 + y, z; g = x, y, −1 + z. | ||
| N1–H1A⋯O4a | 2.805(2) | 177 |
| N1–H1B⋯O6b | 2.838(2) | 173 |
| N2–H2⋯O3a | 2.831(2) | 175 |
| O5–H5A⋯O7b | 2.724(2) | 170 |
| O5–H5B⋯O7c | 2.750(2) | 173 |
| O6–H6A⋯O4d | 2.780(1) | 174 |
| O6–H6B⋯O2 | 2.732(2) | 168 |
| O7–H7A⋯O6e | 2.770(1) | 168 |
| O7–H7B⋯O2f | 2.704(1) | 174 |
| C9–H9⋯O1g | 3.249(2) | 139 |
| D–H⋯A | d(D⋯A)/Å | ∠(D–H⋯A)/° |
|---|---|---|
| a Symmetry codes: a = 1 −x, 1 −y, 1 −z; b = 2 −x, 1 −y, −z; c = −1 + x, −1 + y, z; d = 1 −x, 1 −y, −z; e = 1 + x, 1 + y, z; f = x, −1 + y, z; g = x, y, 1 + z. | ||
| N1–H1A⋯O4a | 2.818(1) | 166 |
| N1–H1B⋯O6 | 2.834(1) | 173 |
| N2–H2⋯O3a | 2.822(1) | 175 |
| O5–H5A⋯O7 | 2.718(1) | 171 |
| O5–H5B⋯O7b | 2.751(1) | 175 |
| O6–H6A⋯O4c | 2.778(1) | 172 |
| O6–H6B⋯O2d | 2.740(1) | 165 |
| O7–H7A⋯O6e | 2.768(1) | 170 |
| O7–H7B⋯O2f | 2.694(1) | 171 |
| C9–H9⋯O1g | 3.275(2) | 138 |
![Organization of the monomeric [M(mal)2(H2O)2]2− units (M = Ni/Co/Mn) into a 2D layer in the ab plane in which the water tetramer acts as a supramolecular subunit.](/image/article/2009/DT/b905127a/b905127a-f2.gif) | ||
| Fig. 2 Organization of the monomeric [M(mal)2(H2O)2]2− units (M = Ni/Co/Mn) into a 2D layer in the ab plane in which the water tetramer acts as a supramolecular subunit. | ||
The protonated 2-amino-4-picoline molecules are attached to the layer from either side of it and bind to the malonate terminals through the formation of the characteristic R22(8) recognition motif (Fig. 3). The binding of the 2-amino-4-picoline molecules to the layer is further strengthened by an additional C–H⋯O hydrogen bond with the nearest oxygen atom of the adjacent malonate terminal across the metal, forming a R22(7) supramolecular pattern. These two motifs act in unison and a tripodal recognition of the protonated 2-amino-4-picoline ligands enhance the stability of the supramolecular assembly.
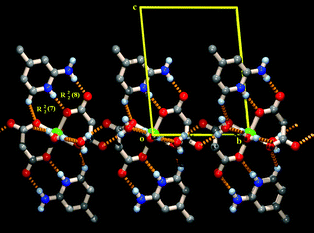 | ||
| Fig. 3 Tripodal recognition between protonated 2-amino-4-picoline molecules and malonate moieties through the formation of R22(8) and R22(7) hydrogen bonding motifs on both sides of the water assembled layer. | ||
Successive 2-amino-4-picoline-studded layers pack along the crystallographic c-axis through interdigitation which is facilitated by lone pair⋯π66–68 and π⋯π interactions (Table S1, ESI†)36 between the 2-amino-4-picoline moieties attached to adjacent layers (Fig. 4).
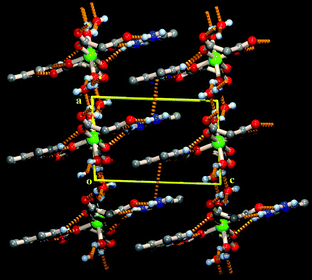 | ||
| Fig. 4 Interdigitation of organic layers through lone pair⋯π and π⋯π interactions along the crystallographic c axis in a penultimate 3D supramolecular organization of the complexes. | ||
Understanding and controlling the self-assembly of molecular building blocks is at the crux of designed synthesis of functional materials. Though it is possible to preconceive certain preferred recognition motifs between a set of building blocks, it is still not possible to a large extent to completely predict the self-assembly of crystalline materials. In this regard, the most disturbing aspect is the near impossibility to predict the role of the solvent in the self-assembly process. For instance, the role of water, one of the most common solvents, has been extensively analyzed in the assembly of crystalline materials. Although no concrete rule has yet been established, it is generally assumed that water normally plays two types of role: it fills the void space in porous materials and it acts as balancing agent for the number of donors and acceptors when there is a mismatch between a set of hydrogen-bonding partners.69
In recent years, there have been a number of reports of crystalline materials encapsulating water clusters of various nuclearities70–75 and morphologies,76–79 either remaining in the void space or as a constituent of the supramolecular network itself. The water tetramer is one of the important water motifs that has been observed in crystalline materials in more instances than the other water motifs.80,81 This suggests that water molecules have an inherent tendency to self-organize themselves especially in the form of the water tetramer even in the presence of other molecular building blocks. In the gas phase it has a calculated binding energy of ca. −27 kcal mol−1 (quite strong)82 and it would not be wrong to treat it as a kind of stable supramolecular synthon. The influence of this synthon on the geometry of a number of coordination environments has been established beyond doubt through neutron diffraction studies.83 In the gas phase, the highest formation energy for the water tetramer has been found for the hydrogen-bonded motif R44(8).84 In the present system, the water tetramer exhibits a R22(8) motif as a result of the coordination of two of the water molecules to metal ions and this is expected to be less stable than the gas phase water tetramer cluster (R44(8) hydrogen bonded motif). The presently observed R22(8) motif has been reported previously81 in other trans-aquo ligated transition metal coordination compounds. Also, when two of the water molecules in this cluster are replaced by two acceptor atoms of coordinating ligand molecules, the R22(8) motif remains intact.81,85,86
To saturate the dangling hydrogen-bonding sites of the tetramer, additional lattice water molecules (O6) also take part in the hydrogen-bonding network, further stabilizing the assembly (Tables 3, 4 and 5), and filling the remaining void space.
Previous studies on copper(II) complexes prepared from malonate and 2-amino-4-picolinium have clearly shown that the preferred recognition mode between these two molecules is the formation of a R22(8) hydrogen-bonded pattern.34,35 This motif is found as well in the present compounds. In the copper(II) complexes, the malonate units act as bridging ligands to form a 2D framework. In contrast, for the compounds herein described, the malonates function as chelating ligands, resulting in mononuclear coordination compounds, which are involved in hydrogen-bonding interactions. The significant structural differences observed for the copper(II) complexes may arise from the allowed Jahn–Teller distortion, usually occurring in CuII octahedral coordination environments. Indeed, a syn–anti bridging coordination mode between Cu centers is possible, which gives rise to a 1D coordination polymer, in contrast to the monomeric coordination units formed in the present compounds.
It is important to mention that, while the recognition between 2-amino-4-picolinium units and the Cu-malonate coordination polymer takes place through the same R22(8) hydrogen-bonding motif, a subtle difference exists. Actually, in the compounds described in the present paper, the N2 atom of the 2-amino-4-picoline acts as donor (donating H2) to the malonate oxygen atom O1 that is directly coordinated to the metal center, and the N1 atom of the 2-amino-4-picoline donates H1A to the uncoordinated oxygen atom O2 of the malonate (Fig. 3). For the Cu complexes, the reverse situation is observed.34,35 Indeed, the N1 atom of 2-amino-4-picoline donates its hydrogen to the inner oxygen atom of the malonate which is directly coordinated to the Cu center, and the N2 atom of 2-amino-4-picoline donates its hydrogen to the bridging malonate oxygen that is axially bound to the nearest Cu center (Fig. S2).36 Effectively, in comparison to the present compounds, the malonate in the Cu complexes has rotated 180° around an axis normal to the metal–malonate plane. Nevertheless, the R22(8) hydrogen-bonding pattern between 2-amino-4-picolinium and malonate is maintained, which is indicative of its robust nature. It is to be further noted that in all these compounds (including the Cu complexes), the 2-amino-4-picoline gets protonated in situ, which is a prerequisite for this robust recognition motif to prevail. Moreover, regardless of the transition metal salts used (acetate, nitrate, chloride or carbonate salt), the same molecular structures are obtained, therefore showing that the particular supramolecular features observed are not anion dependent.
As mentioned above, the supramolecular network characterizing compounds 1–3 is built through the formation of two types of hydrogen-bonded synthons. These two supramolecular motifs, i.e.motif 1 (water tetramer) and motif 2 (2-amino-4-picolinium–malonate recognition), are represented in Fig. 5.
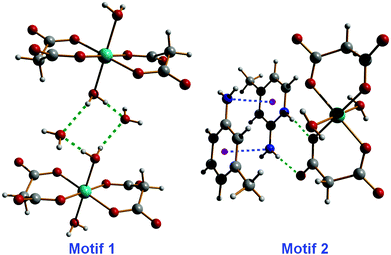 | ||
| Fig. 5 Supramolecular synthons generating a 2D network in compounds 1–3, by means of hydrogen-bonding, lone pair⋯π and π⋯π interactions. | ||
The formation energy of motif 1 and motif 2 (Fig. 5) has been calculated to investigate their relative involvement in the generation of the 2D supramolecular network.
First, the stability of the isolatedH-bonded system, i.e.motif 1, has been determined by running both B3LYP/6-31+G(d) and BHandH/6-31+G(d) single-point calculations, which generate a positive formation energy (Table S2, ESI†).36 To corroborate these results, the full optimization of the system has been carried out with both DFT functionals. These additional calculations confirm that the H-bonded structure observed experimentally may not exist on its own, as the two metal–malonate units fall apart during optimization.
Certainly the most interesting system is that constituted by the association between the metal–malonate compound and two protonated 2-amino-4-picoline molecules, namely motif 2. Single-point calculations and full optimization have been performed at the BHandH/6-31+G(d) level of theory (Fig. S3 (ESI†)36 and see computational details). The single-point calculations suggest that the supramolecular complex is thermodynamically stable, i.e., the formation energy equals −208.7 kcal mol−1 (Table S2, ESI†).36 The optimization carried on the experimental structure confirms the first data, as the resulting complex (slightly distorted compared to the initial configuration, see Fig. S3, ESI†)36 shows a formation energy of −274.0 kcal mol−1 (Table S2, ESI†).36 The AIM analysis has been initially carried out directly on the experimental structure to visualize and estimate the intermolecular interactions between the single molecules constituting the whole system. Remarkably, this analysis reveals an intricate network of supramolecular interactions (Table S3 (ESI†),36Fig. 6). The aminopicoline rings show two fairly strong and virtually symmetrical interactions involving the amino nitrogen atoms (N⋯N, blue lines in Fig. 6) with ρ values of 0.00637 and 0.00604 au.
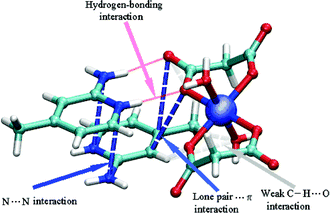 | ||
| Fig. 6 Schematic view of the AIM analysis, revealing the occurrence of N⋯N and lone pair⋯π interactions (dotted blue lines), weak C–H⋯O interactions (dotted light gray lines) and hydrogen bonds (red lines). Both lines schematically represent the bond path connecting two interacting atoms through the bond critical points as obtained through the AIM analysis (see Computational details for further information). For the sake of clarity, critical points are not displayed. | ||
Two significant lone pair⋯π interactions between aromatic carbons and the oxygen atoms of a coordinate malonate are found as well, with ρ values of 0.00206 and 0.00266 au (blue lines in Fig. 6). As expected from the crystal structure, two strong N–H⋯O bonds (red lines) also contribute to the relatively large formation energy of motif 2, with a total electron density of 0.0537 au. Finally, very weak C–H⋯O interactions between the hydrogen atoms H4 and H6 (from the C4 and C6 carbon atoms of 2-amino-4-picoline) and the malonate oxygen atoms O1 and O4, respectively, could also contribute to the formation energy of motif 2. These hydrogen-bonding associations have a total electron density equal to 0.00666 au (gray lines in Fig. 6). Interestingly, the optimization strengthens these interactions, increasing in all cases the electron density at the bond critical points (see Table S3, ESI†).36 This effect is, perhaps, not surprising as the optimization carried out in vacuo maximizes all possible interactions with respect to those occurring in solid state structures and calculated via single-point calculations.
The DFT calculations confirm that the stacked structure (corresponding to motif 2, Fig. 5), observed experimentally, is stable upon optimization. Several supramolecular interactions, particularly N⋯N and lone pair⋯π interactions, contribute to the relatively large formation energy.
Conclusions
In summary, we have shown that the malonate dianion have a robust recognition mode towards 2-amino-4-picoline which gets protonated in situ to enact this R22(8) hydrogen-bonded synthon. This recognition pattern leads to a layered assembly constituted of successive organic layers sandwiched between hybrid organic–inorganic layers. The study also revealed that in the supramolecular assembly of the metal–malonate layers, solvent water molecules (most likely in synergistic combination with the previous 2-amino-4-picoline/malonate motif, as well as π⋯π and lone pair⋯π interactions) are involved in the organization of individual coordination monomers, through their inherent tendency to self-assemble into tetrameric water clusters. The present study, besides establishing the robust nature of the recognition between malonate and 2-amino-4-picolinium generating a R22(8) hydrogen-bonded supramolecular species, also reveals the importance of dispersion forces, such as lone pair⋯π interactions, in the molecular packing of crystalline materials.Acknowledgements
Financial assistance received by S. M. from the Department of Chemistry, Jadavpur University under the UGC-CAS programme is thankfully acknowledged. H. M. L. is grateful to the National Science Council of Taiwan for financial support. A. D. J. acknowledges UGC (India) for granting leave in FIP programme. Computational resources were granted by CINECA (INFM grant). This work makes use of results produced by the Cybersar Project managed by the Consorzio COSMOLAB, a project cofunded by the Italian Ministry of University and Research (IMUR) within the Programma Operativo Nazionale 2000–2006 “Ricerca Scientifica, Sviluppo Tecnologico, Alta Formazione per le Regioni Italiane dell'Obiettivo 1 (Campania, Calabria, Puglia, Basilicata, Sicilia, Sardegna) Asse II, Misura II.2 Societa dell'Informazione, Azione a Sistemi di calcolo e simulazione ad alte prestazioni”. More information is available at http://www.cybersar.it.Notes and references
- M. J. Hannon, Chem. Soc. Rev., 2007, 36, 280–295 RSC.
- E. Kimura, Acc. Chem. Res., 2001, 34, 171–179 CrossRef CAS.
- J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, 1995 Search PubMed.
- J. L. Atwood and J. W. Steed, Supramolecular Chemistry, VCH Verlag GmbH, Weinheim, Germany, 2000 Search PubMed.
- G. R. Desiraju and V. K. Sharma, Perspectives in Supramolecular Chemistry, Wiley, Chichester, 1995 Search PubMed.
- I. Yoshimura, Y. Miyahara, N. Kasagi, H. Yamane, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 12204–12205 CrossRef CAS.
- J. M. Thomas, Angew. Chem., Int. Ed., 1999, 38, 3589–3628 CrossRef.
- B. Cornils, W. A. Hermann, R. Scholgl and C.-H. Wong, Catalysis from A to Z: A Concise Encylopedia, John Wiley & Sons, New York, 2000 Search PubMed.
- L. R. Nassimbeni, Acc. Chem. Res., 2003, 36, 631–637 CrossRef CAS.
- L. M. A. Perdigão, N. R. Champness and P. H. Beton, Chem. Commun., 2006, 538–540 RSC.
- A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409–3424 RSC.
- A. J. Blake, N. R. Champness, A. N. Khlobystov, D. A. Lemenovskii, W. S. Li and M. Schroder, Chem. Commun., 1997, 1339–1340 RSC.
- C. Janiak, Angew. Chem., Int. Ed. Engl., 1997, 36, 1431–1434 CrossRef CAS.
- K. Muller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143–167 CrossRef CAS.
- F. A. Quiocho and N. K. Vyas, Nature, 1984, 310, 381–386 CrossRef CAS.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction, Wiley-VCH, New York, 1998 Search PubMed.
- H. Takahashi, S. Tsuboyama, Y. Umezawa, K. Honda and M. Nishio, Tetrahedron, 2000, 56, 6185–6191 CrossRef CAS.
- Y. Zhang, Z. M. Yang, F. Yuan, H. W. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028–15029 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 Search PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed.
- K. Nørgaard and T. Bjornholm, Chem. Commun., 2005, 1812–1823 RSC.
- C. J. Kepert, Chem. Commun., 2006, 695–700 RSC.
- S. Parveen, R. J. Davey, G. Dent and R. G. Pritchard, Chem. Commun., 2005, 1531–1533 RSC.
- C. Ruiz-Pérez, Y. Rodríguez-Martin, M. Hernández-Molina, F. S. Delgado, J. Pasán, J. Sanchiz, F. Lloret and M. Julve, Polyhedron, 2003, 22, 2111–2123 CrossRef CAS.
- Y. Rodríguez-Martin, M. Hernández-Molina, F. S. Delgado, J. Pasán, C. Ruiz-Pérez, J. Sanchiz, F. Lloret and M. Julve, CrystEngComm, 2002, 440–446 RSC.
- F. S. Delgado, J. Sanchiz, C. Ruiz-Pérez, F. Lloret and M. Julve, CrystEngComm, 2003, 5, 280–284 RSC.
- F. S. Delgado, J. Sanchiz, C. Ruiz-Pérez, F. Lloret and M. Julve, CrystEngComm, 2004, 6, 443–450 Search PubMed.
- Y. Rodríguez-Martin, M. Hernández-Molina, F. S. Delgado, J. Pasán, C. Ruiz-Pérez, J. Sanchiz, F. Lloret and M. Julve, CrystEngComm, 2002, 522–535 RSC.
- T. K. Maji, S. Sain, G. Mostafa, T. H. Lu, J. Ribas, M. Monfort and N. R. Chaudhuri, Inorg. Chem., 2003, 42, 709–716 CrossRef CAS.
- F. S. Delgado, J. Sanchiz, C. Ruiz-Pérez, F. Lloret and M. Julve, Inorg. Chem., 2003, 42, 5938–5948 CrossRef CAS.
- M. Liang, Q. L. Wang, L. H. Yu, D. Z. Liao, Z. H. Jiang, S. P. Yan and P. Cheng, Polyhedron, 2004, 23, 2203–2208 CrossRef CAS.
- J. Pasán, F. S. Delgado, Y. Rodríguez-Martin, M. Hernández-Molina, C. Ruiz-Pérez, J. Sanchiz, F. Lloret and M. Julve, Polyhedron, 2003, 22, 2143–2153 CrossRef , and references therein.
- J. Pasán, J. Sanchiz, C. Ruiz-Pérez, F. Lloret and M. Julve, Inorg. Chem., 2005, 44, 7794–7801 CrossRef CAS.
- S. R. Choudhury, A. D. Jana, E. Colacio, H. M. Lee, G. Mostafa and S. Mukhopadhyay, Cryst. Growth Des., 2007, 7, 212–214 CrossRef.
- S. R. Choudhury, A. D. Jana, C.-Y. Chen, A. Dutta, E. Colacio, H. M. Lee, G. Mostafa and S. Mukhopadhyay, CrystEngComm, 2008, 10, 1358–1363 RSC.
- See ESI†.
- G. M. Sheldrick, SHELXL97: Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- S. R. Choudhury, P. Gamez, A. Robertazzi, C. Y. Chen, H. M. Lee and S. Mukhopadhyay, Cryst. Growth Des., 2008, 8, 3773–3784 CrossRef CAS.
- S. R. Choudhury, B. Dey, S. Das, P. Gamez, A. Robertazzi, K.-T. Chan, H. M. Lee and S. Mukhopadhyay, J. Phys. Chem. A, 2009, 113, 1623–1627 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, and J. Montgomery, J. A. Vreven, T. Kudin, K. N. Burant, J. C. Millam, J. M. Iyengar, S. S. Tomasi, J. Barone, V. Mennucci, B. Cossi, M. Scalmani, G. Rega, N. Petersson, G. A. Nakatsuji, H. Hada, M. Ehara, M. Toyota, K. Fukuda, R. Hasegawa, J. Ishida, M. Nakajima, T. Honda, Y. Kitao, O. Nakai, H. Klene, M. Li, X. Knox, J. E. Hratchian, H. P. Cross, J. B. Adamo, C. Jaramillo, J. Gomperts, R. Stratmann, R. E. Yazyev, O. Austin, A. J. Cammi, R. Pomelli, C. Ochterski, J. W. Ayala, P. Y. Morokuma, K. Voth, G. A. Salvador, P. Dannenberg, J. J. Zakrzewski, V. G. Dapprich, S. Daniels, A. D. Strain, M. C. Farkas, O. Malick, D. K. Rabuck, A. D. Raghavachari, K. Foresman, J. B. Ortiz, J. V. Cui, Q. Baboul, A. G. Clifford, S. Cioslowski, J. Stefanov, B. B. Liu, G. Liashenko, A. Piskorz, P. Komaromi, I. Martin, R. L. Fox, D. J. Keith, T. Al-Laham, M. A. Peng, C. Y. Nanayakkara, A. Challacombe, M. Gill, P. M. W. Johnson, B. Chen, W. Wong, M. W. Gonzalez, C. and J. A. Pople, Gaussian, Inc., Pittsburgh PA, 2003.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- M. P. Waller, A. Robertazzi, J. A. Platts, D. E. Hibbs and P. A. Williams, J. Comput. Chem., 2006, 27, 491–504 CrossRef CAS.
- W. Z. Wang, M. Pitonak and P. Hobza, ChemPhysChem, 2007, 8, 2107–2111 CrossRef CAS.
- A. Magistrato, A. Robertazzi and P. Carloni, J. Chem. Theory Comput., 2007, 3, 1708–1720 CrossRef CAS.
- J. Overgaard, M. P. Waller, R. Piltz, J. A. Platts, P. Emseis, P. Leverett, P. A. Williams and D. E. Hibbs, J. Phys. Chem. A, 2007, 111, 10123–10133 CrossRef CAS.
- X. L. Dong, Z. Y. Zhou, L. J. Tian and G. Zhao, Int. J. Quantum Chem., 2005, 102, 461–469 CrossRef CAS.
- M. Meyer, T. Steinke and J. Suhnel, J. Mol. Model., 2007, 13, 335–345 CrossRef CAS.
- C. Luiz Antonio Sodre, R. R. Willian, B. D. A. Wagner and F. D. S. Helio, J. Chem. Phys., 2003, 118, 10584–10592 CrossRef.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules-A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- R. J. Boyd and S. C. Choi, Chem. Phys. Lett., 1986, 129, 62–65 CrossRef CAS.
- S. J. Grabowski, Chem. Phys. Lett., 2001, 338, 361–366 CrossRef CAS.
- S. T. Howard and O. Lamarche, J. Phys. Org. Chem., 2003, 16, 133–141 CAS.
- S. Konar, P. S. Mukherjee, M. G. B. Drew, J. Ribas and N. R. Chaudhuri, Inorg. Chem., 2003, 42, 2545–2552 CrossRef CAS.
- F. S. Delgado, M. Hernández-Molina, J. Sanchi, C. Ruiz-Pérez, Y. Rodríguez-Martin, T. López, F. Lloret and M. Julve, CrystEngComm, 2004, 6, 106–111 RSC.
- C. Ruiz-Pérez, M. Hernández-Molina, J. Sanchiz, T. López, F. Lloret and M. Julve, Inorg. Chim. Acta, 2000, 298, 245–250 CrossRef CAS.
- S. Sain, T. K. Maji, G. Mostafa, T. H. Li and N. R. Chaudhuri, Inorg. Chim. Acta, 2003, 351, 12–20 CrossRef CAS.
- D. Chattopadhyay, S. K. Chattopadhyay, P. R. Lowe, C. H. Schwalbe, S. K. Mazumder, A. Rana and S. Ghosh, J. Chem. Soc., Dalton Trans., 1993, 913–916 RSC.
- I. G. de Muro, F. A. Mautner, M. Insausti, L. Lezama, M. I. Arriortua and T. Rojo, Inorg. Chem., 1998, 37, 3243–3251 CrossRef CAS.
- C. Ruiz-Pérez, J. Sanchiz, M. Hernández-Molina, F. Lloret and M. Julve, Inorg. Chem., 2000, 39, 1363–1370 CrossRef CAS.
- C. Ruiz-Pérez, M. Hernández-Molina, P. Lorenzo-Luis, F. Lloret, J. Cano and M. Julve, Inorg. Chem., 2000, 39, 3845–3852 CrossRef CAS.
- Y. Rodríguez-Martin, J. Sanchiz, C. Ruiz-Pérez, F. Lloret and M. Julve, Inorg. Chim. Acta, 2001, 326, 20–26 CrossRef CAS.
- Y. Rodríguez-Martin, C. Ruiz-Pérez, J. Sanchiz, F. Lloret and M. Julve, Inorg. Chim. Acta, 2001, 318, 159–165 CrossRef CAS.
- J. Sanchiz, Y. Rodríguez-Martin, C. Ruiz-Pérez, A. Mederos, F. Lloret and M. Julve, New J. Chem., 2002, 26, 1624–1628 RSC.
- S. Sain, T. K. Maji, G. Mostafa, T. H. Lu and N. R. Chaudhuri, New J. Chem., 2003, 27, 185–187 RSC.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- Z. L. Lu, P. Gamez, I. Mutikainen, U. Turpeinen and J. Reedijk, Cryst. Growth Des., 2007, 7, 1669–1671 CrossRef CAS.
- T. J. Mooibroek, S. J. Teat, C. Massera, P. Gamez and J. Reedijk, Cryst. Growth Des., 2006, 6, 1569–1574 CrossRef CAS.
- G. R. Desiraju, J. Chem. Soc., Chem. Commun., 1991, 426–428 RSC.
- Y. Q. Sun, J. Zhang, Z. F. Ju and G. Y. Yang, Aust. J. Chem., 2005, 58, 572–577 CrossRef CAS.
- M. T. Ng, T. C. Deivaraj, W. T. Klooster, G. J. McIntyre and J. J. Vittal, Chem.–Eur. J., 2004, 10, 5853–5859 CrossRef CAS.
- J. N. Moorthy, R. Natarajan and P. Venugopalan, Angew. Chem., Int. Ed., 2002, 41, 3417–3420 CrossRef CAS.
- S. Supriya, S. Mankumari, P. Raghavaiah and S. K. Das, New J. Chem., 2003, 27, 218–220 RSC.
- S. K. Ghosh and P. K. Bharadwaj, Angew. Chem., Int. Ed., 2004, 43, 3577–3580 CrossRef CAS.
- Q. Y. Liu and L. Xu, CrystEngComm, 2005, 7, 87–89 RSC.
- J. P. Naskar, M. G. B. Drew, A. Hulme, D. A. Tocher and D. Datta, CrystEngComm, 2005, 7, 67–70 RSC.
- B. Q. Ma, H. L. Sun and S. Gao, Chem. Commun., 2004, 2220–2221 RSC.
- J. M. Zheng, S. R. Batten and M. Du, Inorg. Chem., 2005, 44, 3371–3373 CrossRef CAS.
- R. Luna-García, B. M. Damián-Murillo, V. Barba, H. Höpfl, H. I. Beltrán and L. S. Zamudio-Rivera, Chem. Commun., 2005, 5527–5529 RSC.
- M. Mascal, L. Infantes and J. Chisholm, Angew. Chem., Int. Ed., 2006, 45, 32–36 CrossRef.
- L. Infantes and S. Motherwell, CrystEngComm, 2002, 454–461 Search PubMed.
- S. S. Xantheas, J. Chem. Phys., 1994, 100, 7523–7534 CrossRef CAS.
- D. R. Turner, M. Henry, C. Wilkinson, G. J. McIntyre, S. A. Mason, A. E. Goeta and J. W. Steed, J. Am. Chem. Soc., 2005, 127, 11063–11074 CrossRef CAS.
- F. N. Keutsch and R. J. Saykally, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10533–10540 CrossRef CAS.
- L. R. Falvello, D. Ferrer, M. Piedrafita, T. Soler and M. Tomas, CrystEngComm, 2007, 9, 852–855 RSC.
- C. Biswas, A. D. Jana, M. G. B. Drew, G. Mostafa and A. Ghosh, Polyhedron, 2009, 28, 653–660 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1: TG curve of 1. Table S1: π–π interaction data for compounds 1–3. Fig. S2: Recognition between protonated 2-amino-4-picoline molecules and malonate moieties in some copper complexes. Table S2: Formation energies of Motif 1 and Motif 2. Fig. S3: Experimental and calculated structure of Motif 2. Table S3: Electron density analysis on the experimental and optimized structures of Motif 2. CCDC reference numbers 676210 (1), 676211 (2) and 676212 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b905127a |
| This journal is © The Royal Society of Chemistry 2009 |