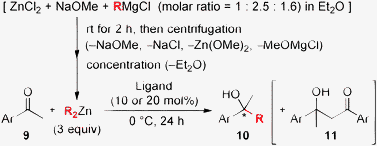Catalytic enantioselective alkyl and aryl addition to aldehydes and ketones with organozinc reagents derived from alkyl Grignard reagents or arylboronic acids†
Received
28th March 2011
, Accepted 2nd June 2011
First published on 1st July 2011
Abstract
A highly practical, catalytic enantioselective alkyl and aryl addition to aldehydes and ketones with organozinc reagents, which were prepared in situ from commercially available Grignard reagents or arylboronic acids, was developed. A chiral phosphoramide ligand was essential for promoting the addition reactions in high yields with high enantioselectivities.
1. Introduction
Catalytic enantioselective organozinc addition to aldehydes and ketones is an important synthetic approach that gives optically active secondary (2°) and tertiary (3°) alcohols, respectively, viacarbon–carbon bond-formation under mild conditions.1 However, in sharp contrast to many examples of the di(1°-alkyl)zinc addition to aldehydes, only a few examples of the di(2°-alkyl)zinc addition to aldehydes and ketones have been reported.2 Moreover, the addition of arylzinc reagents to less-reactive ketones rather than aldehydes has not been well-established. These difficulties are due to steric constraints between substrates and reagents, particularly when ketones are used. In addition, the fact that di(2°-alkyl)zinc and arylzinc reagents are not available is another serious problem. Therefore, particularly for industrial use, inexpensive, easily handled, and low-toxicity alkyl and aryl sources for organozinc reagents are needed in large quantities. In this regard, Grignard reagents and boronic acids are highly attractive not only for academic reasons but also for industry, since a variety of these reactants are commercially available or readily prepared from the corresponding halides. Recently, some useful and practical methods for preparing pure organozinc reagents in situ from these more convenient organometallic reagents have been developed.3–6 In this context, we report here a catalytic enantioselective alkyl and aryl addition to aldehydes and ketones with a variety of organozinc reagents, which are prepared in situ from alkyl Grignard reagents or arylboronic acids as convenient alkyl or aryl sources.
2. Results and discussion
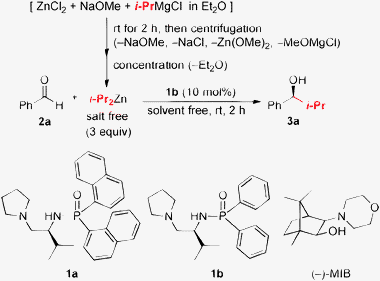
Traditional Grignard reagents are some of the most useful and safe reagents for alkylating carbonyl compounds in academic and industrial research on process chemistry. Today, more than 200 inexpensive Grignard reagents can be purchased. In particular, the transmetallation of alkyl groups in Grignard reagents to other less-reactive metal complexes such as Zn(II) should be attractive for catalytic enantioselective alkylation to carbonyl compounds because the reaction would proceed only in the presence of the catalysts. Although convenient traditional methods have been reported for the preparation of diorganozinc reagents in situ, they are usually accompanied by the generation of inorganic salts, which sometimes disturb subsequent asymmetric catalysis. In contrast, Côté and Charette recently developed a highly useful method for the preparation of salt-free di(1°-alkyl)zinc reagents from ZnCl2, NaOMe, and Grignard reagents.6 This method is highly practical for small scale application in both laboratory and industry since centrifugation is needed to separate the salt-free dialkylzinc reagents from the resultant magnesium salts. By taking advantage of Charette's procedure, we here prepared salt-free di(2°-alkyl)zinc reagents for the catalytic enantioselective 2°-alkylation of aldehydes. First, the isopropylation of benzaldehyde (2a) was investigated with chiral phosphoramide ligand 1b (10 mol%) (Table 1). By following Charette's typical procedure for the preparation of di(1°-alkyl)zinc,6 we prepared a 0.44 M solution of i-Pr2Zn in Et2O, but nearly racemic 3a was obtained in 90% yield under Et2O conditions (entry 1). We assumed that a small amount of the remaining Grignard reagent might trigger the racemic pathway, since a highly active zinc(II) ate complex for alkylation, namely [i-Pr3Zn]−[MgCl]+, would be generated in situ.7 As expected, a conservative molar ratio of 1/2/1.6 of ZnCl2/NaOMe/i-PrMgCl was effective, and 3a was obtained in 73% yield with 91% ee (entry 2). However, BnOH was also obtained as an undesired reduction byproduct in 26% yield (entry 2). Therefore, we examined the reaction under solvent-free conditions8 by using salt-free liquid i-Pr2Zn (bp 134 °C),9 which was prepared by the same method, followed by the removal of Et2Oin vacuo. As a result, 3a was obtained in an improved yield (93%) with 91% ee, along with a trace amount of BnOH (<2%) (entry 3). Moreover, with the use of 2.5 equiv. of NaOMe, 3a was obtained in 94% yield with 94% ee (entry 4). Both less-bulky 1b (10 mol%) and more-bulky 1a10 were effective in this reaction (entries 4 and 5). Côté and Charette used (2S)-(−)-3-exo-(N-morpholino)isoborneol [(−)-MIB], which is a representative chiral ligand,11 in their asymmetric 1°-alkylation to aldehydes.6 However, (−)-MIB was less effective than chiral ligand 1 in the isopropylation of 2a under solvent-free or conventional toluene conditions (entries 6 and 7).
| Entry |
Molar ratio of |
Ligand |
Yield [%] of 3a |
ee [%] of 3a |
ZnCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaOMe:i-PrMgCl :NaOMe:i-PrMgCl |
|
Unless otherwise noted, salt-free i-Pr2Zn was used under solvent-free conditions.
i-Pr2Zn (0.44 M, in Et2O) was used.
BnOH was obtained in 26% yield.
Toluene was used as a solvent.
|
| 1b |
1:2:1.9 |
1b
|
90 |
3 |
| 2b,c |
1:2:1.6 |
1b
|
73 |
91 |
| 3 |
1:2:1.6 |
1b
|
93 |
91 |
| 4 |
1:2.5:1.6 |
1b
|
94 |
94 |
| 5 |
1:2.5:1.6 |
1a
|
93 |
93 |
| 6 |
1:2.5:1.6 |
(−)-MIB
|
85 |
59 |
| 7d |
1:2.5:1.6 |
(−)-MIB
|
85 |
94 |
Encouraged by the efficient isopropylation of 2a, we next examined the catalytic enantioselective 2°-alkyl addition to various aldehydes under solvent-free conditions with salt-free di(2°-alkyl)zinc reagents derived from Grignard reagents (Table 2). The isopropylation of aromatic aldehydes (entries 1 and 2), heteroaromatic aldehydes (entries 3 and 4), and cycloaliphatic aldehydes (entries 5 and 6) proceeded, and the corresponding products were obtained in high yields with high enantioselectivities (80–>99% ee). The isopropylation of tiglic aldehyde as an α,β-unsaturated aldehyde provided only a 1,2-adduct (3h) with high enantioselectivity (97% ee) (entry 7). Catalytic enantioselective sec-butylation also proceeded for the first time, and the desired products were obtained with high enantioselectivities (94–98% ee) (entries 8 and 9). Next, as an unprecedented cyclic 2°-alkyl addition to aldehydes, the addition of (c-C5H9)2Zn and (c-C6H11)2Zn was explored. Since (c-C6H11)2Zn is viscous at room temperature, toluene (2.5 M) was used as a solvent. Fortunately, the desired products were obtained with high enantioselectivities from a variety of aromatic aldehydes (entries 10, 13–16), heteroaromatic aldehydes (entries 11 and 17), cycloaliphatic aldehydes (entries 12 and 18), α-branched or β-branched aliphatic aldehydes (entries 19 and 20), and α,β-unsaturated aldehydes (entries 21 and 22). Ligand 1a generally showed high reactivity,10 while less-bulky 1b was often less effective than bulky 1a due to reduction byproducts, especially when less-reactive aliphatic aldehydes were used (entries 9, 18, and 22). Probably, bulky ligand 1a would be essential for keeping active monomeric catalystsin situ and thus for establishing both high yield and high enantioselectivity.10 Some of the secondary alcohols (3, R′RCHOH) which have two similar cyclic and/or acyclic fragments (R′ and R) can hardly be obtained via complementary methods such as asymmetric reduction of the corresponding ketones.12 In this catalysis, optically active novel secondary alcohols with similar R′ and R were successfully obtained with high to excellent enantioselectivities.
Table 2 Enantioselective 2°-alkyl addition to aldehydes
| Entry |
Product (3) |
Yield and ee |
Entry |
Product (3) |
Yield and ee |
|
10 mol% of ligand 1b was used in place of 1a.
Temperature was 0 °C.
Toluene (2.5 M) was used as a solvent.
|
| 1a |

|
95%, 96% ee |
12b |
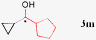
|
90%, 99% ee |
| 2 |

|
90%, 80% ee |
13 |

|
78%, 90% ee |
| 3a |
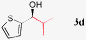
|
>99%, 95% ee |
14a,c |

|
89%, 97% ee |
| 4 |
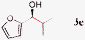
|
86%, 90% ee |
15a,c |

|
96%, 90% ee |
| 5 |

|
>99%, 96% ee |
16 |

|
91%, 95% ee |
| 6a |

|
98%, >99% ee |
17c |
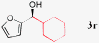
|
94%, 84% ee |
| 7b |

|
97%, 97% ee |
18c |

|
77% [36%]a, >99% ee |
| 8 |

|
76%, (dr 55/45) 94% ee /95% ee |
19b,c |
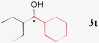
|
80%, 99% ee |
| 9 |

|
45% [15%]a, (dr 59/41) 98% ee /96% ee |
20b,c |

|
75%, 82% ee |
| 10 |
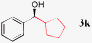
|
52%, >99% ee |
21c |

|
85%, 99% ee |
| 11 |

|
56%, 96% ee |
22c |

|
88% [42%]a, 98% ee |
To demonstrate the synthetic utility of this approach, several transformations from the obtained optically active alcohols were examined. The optically active cyclohexyl(furan-2-yl)methanol (3r) was treated with N-bromosuccinimide in THF–H2O at room temperature for 4 h,13 and the desired cyclohexyl-substituted 6-hydroxy-2H-pyran-3-one 414 was obtained via oxidative Achmatowicz rearrangement in 89% yield with a diastereomeric ratio of 73:27 with 84% ee (eqn (1)). Moreover, after TBS-protection of the hydroxy group of 3r, ozonolysis cleavage of the furan ring provided α-alkoxy carboxylic acid 515,16 in 96% yield (eqn (2)).
| |  | (1) |
| |  | (2) |
| |  | (3) |
| |  | (4) |
| |  | (5) |
By ozonolysis and subsequent Me
2S treatment,
17allyl alcohol
3v was directly converted to α-hydroxy ketone
618 in 98% yield with 99% ee (
eqn (3)). This transformation is synthetically important because compound
6 cannot be prepared directly from unstable
methylglyoxal by
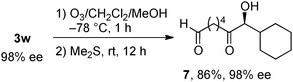
alkylation. Similarly, α-hydroxy ketone
7 bearing a terminal
formyl moiety was readily prepared from allyl alcohol
3w in 86% yield with 98% ee (
eqn (4)). For another oxidative transformation, the diastereoselective epoxidation of allyl alcohol
3w with
tert-butyl hydroperoxide (TBHP) in the presence of vanadyl acetylacetonate was examined (
eqn (5)).
19 Fortunately,
syn-epoxide
8, which is a key intermediate in the synthesis of optically active 1,3-diols with three consecutive chiral carbon centers,
20 was obtained in a diastereomeric ratio of 96
:
4 with 98% ee. Other
oxidation methods
19c such as
m-CPBA, Ti(O
i-Pr)
4/TBHP, Mo(CO)
6/TBHP were less effective than VO(acac)
2/TBHP and gave poor to moderate diastereoselectivities (up to 80/20 dr).
Finally, the 2°-alkylation of ketones in place of aldehydes was examined (Table 3). A preliminary (−)-MIB catalysis in the isopropylation of 4′-(trifluoromethyl)acetophenone (9a) did not proceed and undesired aldol byproduct 11a was obtained along with recovery of the starting ketone 9a. In sharp contrast, the isopropylation of 9a and 3′,5′-bis(trifluoromethyl)acetophenone (9b) proceeded without aldol formation at 0 °C for 24 h in the presence of 10 mol% of ligand 1a, and the desired tertiary alcohols (10a and 10b) were obtained in moderate yield with >99% ee (entries 2 and 3). Furthermore, the cyclohexylation of 9a and 9b in the presence of 20 mol% of ligand 1a provided the desired products 10c and 10d in moderate to good yield with >99% ee (entries 4 and 5). Although further improvements with regard to yield and the use of less-reactive substrates will be necessary, to the best of our knowledge, this is the first example of catalytic asymmetric tertiary alcohol synthesis via di(2°-alkyl)zinc addition to ketones.21
Table 3 Enantioselective 2°-alkyl addition to ketones
| Entry |
Ligand |
Product (Ar, R) |
Yield and ee of 10 |
Yield of 11 |
Recovery of 9 |
|
20 mol% of (−)-MIB was used. Toluene was used as a solvent.
10 mol% of 1a was used under solvent-free conditions.
20 mol% of 1a was used in 2.5 M toluene for 9.
|
| 1a |
(–)-MIB |
10a (4-CF3C6H4, i-Pr) |
0%, — |
40% |
60% |
| 2b |
1a
|
10a (4-CF3C6H4, i-Pr) |
38%, >99% ee |
<3% |
59% |
| 3b |
1a
|
10b (3,5-(CF3)2C6H3, i-Pr) |
40%, >99% ee |
<3% |
57% |
| 4c |
1a
|
10c (4-CF3C6H4, c-Hex) |
40%, >99% ee |
<3% |
57% |
| 5c |
1a
|
10d (3,5-(CF3)2C6H3, c-Hex) |
56%, >99% ee |
<3% |
41% |
2.2. A concise synthesis of (S)-(+)-ginnol based on a catalytic enantioselective long n-alkyl chain addition to aldehydes
The catalytic enantioselective long n-alkyl chain addition to aldehydes has thus far been limited, since the corresponding di(n-alkyl)zinc reagents are difficult to prepare.22 By taking advantage of the preparation of solvent-free di(2°-alkyl)zinc reagents as above, we next examined a long-chain-alkylation to aldehydes, such as n-heptylation, n-octylation, n-nonylation, and n-decylation.
First, we prepared di(n-alkyl)zinc reagents and performed the subsequent addition reaction to aromatic aldehydes such as benzaldehyde and p-anisaldehyde under the optimized conditions with the use of chiral ligand 1a (Table 4). n-Heptylation, n-octylation, and n-decylation proceeded smoothly, and the corresponding secondary alcohols (12) were obtained in almost quantitative yields with 92–96% ee. Remarkably, the use of in situ-prepared Grignard reagents (RCl + Mg + LiCl23) appeared to have no negative effect.
Table 4 Enantioselective long n-alkyl chain addition to aldehydes
| Product, yield, enantioselectivity, and alkyl source |

|
As an application of this approach to an aliphatic aldehyde, we next examined the synthesis of a chiral long-chain aliphatic alcohol, (S)-(+)-ginnol (13),24 which is an optically active natural product that has been noted in chiral bilayers of the wax tubes of higher plants. Unfortunately, the reaction of icosanal (n-C19H39CHO) with di(n-nonyl)zinc [(n-C9H19)2Zn] in the presence of chiral ligand 1a (20 mol%) failed, and a complex mixture was obtained. In sharp contrast, our previous chiral BINOL-derived ligand 14 under toluene/THF = 2/1 conditions25 was effective, and enantio-enriched (S)-(+)-ginnol (13) was obtained in one step in 81% yield with 97% ee at room temperature within 12 h (Scheme 1). To the best of our knowledge, this is the first one-step example, and thus this is the simplest asymmetric synthesis of (S)-(+)-ginnol. Fortunately, a single recrystallization in n-hexane provided optically pure (S)-(+)-ginnol (>99% ee).
2.3.
Arylzinc addition to ketones with arylboronic acids as an aryl source
Commercially available diphenylzinc has been widely used in the catalytic enantioselective phenylation of aldehydes and ketones.26 However, diphenylzinc is not inexpensive, and other diarylzinc reagents are not readily commercially available. In this regard, some alternative methods for preparing arylzinc reagents, which are suitable for catalytic enantioselective aryl addition, have recently been developed.27 One of the most attractive methods was reported by Bolm et al., and arylzinc reagents were prepared in situ from diethylzinc (7.2 equiv.) and arylboronic acids (2.4 equiv.) as an aryl source.28 Arylboronic acids are generally non-toxic, easily handled, and have recently become commercially available in bulk quantities since these reagents are used in the Suzuki–Miyaura cross-coupling reaction. It has since been shown that triarylboroxines [(ArBO)3] could be used in place of arylboronic acids to reduce the amount of diethylzinc needed.29 By taking advantage of these developments with arylboronic acids and triarylboroxines, we next examined catalytic enantioselective arylation to ketones. Unlike aldehydes, catalytic enantioselective arylation to ketones with arylzinc reagents has not yet been well-explored due to the extremely low reactivities of ketones.30,31
We first investigated catalytic enantioselective phenylation to 4′-chloroacetophenone (9c) with phenylboronic acid (15a) (Table 5). In this reaction, triphenylboroxine (16a) can be readily prepared in situ in toluene under azeotropic reflux conditions for 3 h. According to the procedure developed by Zhao and co-workers,29a,b the amount of Et2Zn (3.0–4.5 equiv.; 15a:Et2Zn = 1:2 or 1:3) was examined. In the presence of chiral ligand 1a (0.17 M; 9c/toluene), compound 17a was obtained in 70% yield with 94% ee when 3 equiv. of Et2Zn was used (entry 1). However, self-aldol condensation was also observed, and 18 was obtained in 18% yield. In contrast, when 4.5 equiv. of Et2Zn was used, the selectivity of the reactions was improved and 17a was obtained in 86% yield with 94% ee (entry 2). The same enantioselectivity (94% ee) and reduced yield (79%) of 17a were observed when the reaction was conducted under higher concentration conditions (0.25 M; 9c/toluene) (entry 3), while the yield of 17a was significantly reduced when the reaction was conducted under lower concentration (0.14 M; 9c/toluene) conditions (entry 4). Moreover, when readily available ligand 1c (vide infra) was used, 17a was obtained in 83% yield and 94% ee, which was similar to the result with ligand 1a (entries 2 and 5).
| Entry |
Et2Zn (equiv.) |
Concentrationa/M |
Yield (%) of 17a |
ee (%) of 17a |
Yield (%) of 18 |
Concentration was based on 9c in toluene.
Chiral ligand 1c was used in place of 1a.
|
| 1 |
3.0 |
0.17 |
70 |
94 (S) |
18 |
| 2 |
4.5 |
0.17 |
86 |
94 (S) |
11 |
| 3 |
4.5 |
0.25 |
79 |
94 (S) |
9 |
| 4 |
4.5 |
0.14 |
68 |
92 (S) |
10 |
| 5b |
4.5 |
0.17 |
83 |
94 (S) |
13 |
Under the optimized reaction conditions, a variety of ketones and arylboronic acids (15) were explored (Table 6). Not only acetylarenes (see 17b–f) but also cyclic ketones such as 1-indanone (see 17g) and α-tetralone (see 17h and 17i) could be used, and the corresponding products were obtained in good to high yields with 84–98% ee. Less-reactive p-fluorophenylboronic acid was acceptable for this reaction (see 17c and 17i). With regard to the ability to tolerate heteroaromatic groups, the thienyl moiety could be used in a substrate or a reactant, and 17j and 17k were obtained in moderate to good yields in high enantioselectivities (90–93% ee). The 1,3-benzodioxolyl moiety as an oxyheterocycle was also used in a reactant (see 17l). Furthermore, α,β-unsaturated ketones such as 1-acetyl-1-cyclohexene and benzylideneacetone were examined, and the 1,2-adducts (17m and 17n) were obtained selectively as major products in good enantioselectivities (77–80% ee).
| Product, yield, and enantioselectivity |
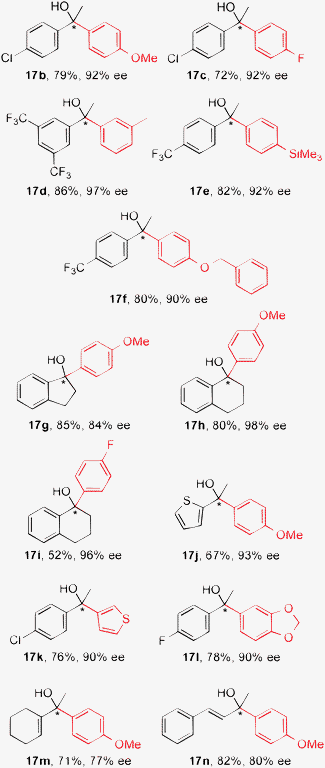
|
2.4. Preparation of L-valine-derived chiral phosphoramide ligands
Chiral phosphoramide ligand 1b was readily prepared in multigram-scale quantities from commercially available N-Boc-L-valine in four steps, as shown in Scheme 2. First, the dehydrative amidation of N-Boc-L-valine (147 g, 0.677 mol) with pyrrolidine by treatment with N,N′-dicyclohexylcarbodiimide (DCC) and 1-hydroxybenzotriazole (HOBt) provided 19 in 89% yield (166 g). After deprotection of the Boc group by AcCl in methanol, diamine 20 was obtained in 52% yield (50 g) by reduction using LiAlH4. Remarkably, one equivalent of LiAlH4 was sufficient, and the reaction proceeded smoothly at 0 °C to room temperature within 5 h. Moreover, the use of ethyl acetate could control a safety work-up procedure without the generation of gaseous hydrogen even in a multigram-scale synthesis. Finally, phosphoramidation of 20 with commercially available diphenylphosphinic chloride was conducted in the presence of triethylamine, and 1b was obtained in 92% yield (4.9 g) in a 15 mmol-scale synthesis.
On the other hand, for the synthesis of ligand 1a, di(1-naphthyl)phosphinic chloride (24) was prepared on a >100 g-scale as shown in Scheme 3. Compound 24 was obtained almost quantitatively in three steps, such as the Grignard addition reaction of diethyl phosphite with 21, followed by oxidation of 22 with H2O2, and chlorination of 23 with SOCl2.
However, in the phosphoramidation of 20 (5 mmol scale) with sterically bulky 24, the reaction in the presence of triethylamine proceeded sluggishly, and 1a was not obtained in reproducible yields in several examinations (0–24%) (Scheme 4). A lithium amide reagent prepared in situ from 20 and 1.2 equivalents of n-BuLi also showed low reactivity. In sharp contrast, the use of 2.3 equivalents of n-BuLi significantly improved the reactivity, and 1a was obtained in 76% yield (1.7 g). In this ligand synthesis, dianion species may be involved and/or the corresponding acidic phosphoramide N–H may be deprotonated by the remaining starting materials. Although it was difficult to completely control the strong exothermic reaction with 24 in a multigram-scale synthesis from 20 (30 g, 191 mmol), 1a was obtained in 31% yield (27 g).
With the optimized method with 2.3 equiv. of n-BuLi or t-BuLi to 20, some promising chiral phosphoramide ligands were prepared on a 4–6 mmol scale, and the yields in phosphoramidation as a key step are shown in Fig. 1. A variety of sterically bulky chiral phosphoramide ligands were synthesized in yields of 38–83%. Exceptionally, 1c was scarcely detected (<10% yield) in the resulting complex mixture when 1.2 or 2.3 equiv. of n-BuLi was used, since bis(4-methoxynaphthalen-1-yl)phosphinic chloride was probably too reactive under these conditions.
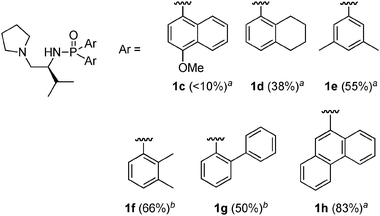 |
| | Fig. 1 Preparation of chiral phosphoramide ligands with a dianion method. aYields in the phosphoramidation step with n-BuLi (2.3 equiv.). bYields in the phosphoramidation step with t-BuLi (2.3 equiv.). | |
Fortunately, however, 1c was synthesized again by using triethylamine (2.2 equiv.) in 70% yield, as shown in Scheme 5. Since ligand 1c, like ligand 1a, is valuable (vide supra), a convenient synthesis under mild reaction conditions such as triethylamine in place of n-BuLi is highly important, particularly when bulk quantities are needed for industrial use.
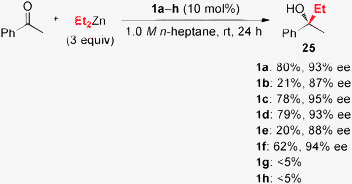
Chiral ligands 1a–h were briefly evaluated in enantioselective diethylzinc addition to acetophenone as a probe reaction, as shown in Scheme 6. As well as ligands 1a and 1c, ligand 1d, which contains tetrahydronaphthyl moieties, was effective. Ligands 1b and 1e–g, which include substituted-phenyl moieties, and ligand 1h, which includes 9-phenanthryl moieties, showed low reactivity and/or enantioselectivity.
3. Conclusion
In summary, we have developed a highly practical technology, catalytic enantioselective alkyl and aryl addition to aldehydes and ketones with organozinc reagents, which were prepared in situ from Grignard reagents or arylboronic acids. Grignard reagents and boronic acids are highly attractive alkyl and aryl sources for both academic and industrial purposes, since a variety of these items are commercially available or readily prepared from the corresponding halides. In the Grignard reagent-derived di(2°-alkyl)zinc addition to aldehydes and ketones, refined Charette's reaction conditions with ZnCl2/NaOMe/RMgCl in a molar ratio of 1/2.5/1.6 in the presence of our chiral phosphoramide was essential. Optically active novel 2°-alcohols with two similar cyclic and/or acyclic fragments could be successfully synthesized and transformed to synthetically useful γ-hydroxy-β-pyrone, α-alkoxy carboxylic acid, α-hydroxy ketone, and 2,3-epoxyalcohol. Moreover, we achieved a concise synthesis of (S)-(+)-ginnol by taking advantage of a catalytic enantioselective long n-alkyl chain addition to aldehydes with Grignard reagent-derived dialkylzincs. Furthermore, we developed an arylzinc addition to ketones with arylboronic acids as an aryl source. In addition to arylboronic acids, heterocyclic boronic acids could also be used, and the corresponding optically active 3°-biaryl carbinols were obtained in good to high enantioselectivities. Chiral phosphoramide ligands in this report were readily synthesized on a 1–30 gram scale, and the key to the development of unprecedented 2°-alkylation to aldehydes and arylation to less-reactive ketones is likely the high reactivity of these chiral ligands.
4. Experimental section
4.1. General procedure for the preparation of di(2°-alkyl)zinc reagents (Tables 1 and 2)
To a test tube equipped with a magnetic stirrer charged with ZnCl2 (682 mg, 5 mmol) and NaOMe (676 mg, 12.5 mmol) was added Et2O (5 mL) at room temperature under a nitrogen atmosphere. The suspension was stirred for 20 min and cooled to 0 °C for another 10 min. RMgCl in 2.0 M Et2O solution (titrated, 4 mL, 8 mmol) was added dropwise with vigorous stirring over 10 min at 0 °C, and the suspension was allowed to stir at room temperature for 2 h. The mixture was centrifuged for 10 min (4000 rpm) and the di(2°-alkyl)zinc reagent (0.44 M in Et2O) was gently transferred via a cannula into a well-dried Pyrex Schlenk tube to be stored before use. Caution!Centrifugation of pyrophoric organometallic reagents in high spinning should be done with a safety electromechanical centrifugal separator, particularly when large quantities are prepared.
4.2. General procedure for the catalytic enantioselective addition of di(2°-alkyl)zinc reagents to aldehydes (Tables 1 and 2)
A well-dried Pyrex Schlenk tube was charged with 1a (22.8 mg, 0.05 mmol) and the di(2°-alkyl)zinc reagent (0.44 M in Et2O) (3.4 mL, 1.5 mmol) at room temperature under a nitrogen atmosphere. Et2O was removed under reduced pressure to generate the solvent free di(2°-alkyl)zinc reagent containing 1ain situ. After the removal of Et2O, toluene (0.4 mL) was added when (c-Hex)2Zn was used. Aldehyde 2 (0.5 mmol) was added to the mixture at room temperature. The resulting mixture was stirred at room temperature for 2 h. Ether or EtOAc (10 mL) and sat. NH4Cl aqueous solution (10 mL) were poured into the mixture at 0 °C. The product was extracted with ether (10 mL × 3) and washed with brine (10 mL). The combined extracts were dried over MgSO4. The organic phase was concentrated under reduced pressure and the crude product was purified by neutral silica gel column chromatography (eluent: n-hexane/Et2O or n-pentane/Et2O) to give the desired products (3). The enantiomeric purity was determined by GC or HPLC on a chiral column.
4.3. General procedure for the catalytic enantioselective addition of di(2°-alkyl)zinc reagents to ketones (Table 3)
A well-dried Pyrex Schlenk tube was charged with 1a (22.8 mg, 0.05 mmol) and the di(2°-alkyl)zinc reagent (0.44 M in Et2O) (3.4 mL, 1.5 mmol) at room temperature under a nitrogen atmosphere. Et2O was removed under reduced pressure to generate the solvent free di(2°-alkyl)zinc reagent containing 1ain situ. After the removal of Et2O, toluene (0.4 mL) was added when (c-Hex)2Zn was used, and the mixture was cooled to 0 °C. Ketone (9) (0.5 mmol) was added to the mixture (in the case of 9b, it was added dropwise at 0 °C over 12 h). The resulting mixture was stirred at 0 °C for 24 h. Ether or EtOAc (10 mL) and sat. NH4Cl aqueous solution (10 mL) were poured into the mixture at 0 °C. The product was extracted with ether (10 mL × 3) and washed with brine (10 mL). The combined extracts were dried over MgSO4. The organic phase was concentrated under reduced pressure and the crude product was purified by neutral silica gel column chromatography (eluent: n-hexane/Et2O = 20/1–8/1) to give the desired product (10). To determine the enantioselectivity by GC or HPLC on a chiral column, compound 10 was readily transformed to the corresponding acetate derivative by using Ac2O/Et3N/DMAP in CH2Cl2.
4.4. General procedure for the catalytic enantioselective long n-alkyl chain addition to aldehydes (Table 4)
A well-dried Pyrex Schlenk tube was charged with 1a (22.8 mg, 0.05 mmol) and the R2Zn reagent (prepared from ZnCl2, NaOMe, and RMgCl as above, 0.4–0.6 M in Et2O) (1.5 mmol) at room temperature under a nitrogen atmosphere. Et2O was removed under reduced pressure to generate the solvent free R2Zn reagent containing 1ain situ.Aldehyde (0.5 mmol) was added to the mixture at room temperature. The resulting mixture was stirred at room temperature for 2 h. Ether (or n-hexane, toluene, EtOAc, etc.) (10 mL) and sat. NH4Cl aqueous solution (10 mL) were poured into the mixture at 0 °C. The product was extracted with ether (10 mL × 3) and washed with brine (10 mL). The combined extracts were dried over MgSO4. The organic phase was concentrated under reduced pressure and the crude product was purified by neutral silica gel column chromatography (eluent: n-hexane/Et2O) to give the desired products (12). The enantiomeric purity was determined by GC on a chiral column.
4.5.
Catalytic enantioselective synthesis of (S)-(+)-ginnol (13) (Scheme 1)
A well-dried Pyrex Schlenk tube was charged with 14 (68.7 mg, 0.10 mmol) and the (n-C9H19)2Zn reagent (0.44 M in Et2O) (3.4 mL, 1.5 mmol) at room temperature under a nitrogen atmosphere. Et2O was removed under reduced pressure to generate the solvent free (n-C9H19)2Zn reagent containing 14in situ.Toluene (1.5 mL) and THF (0.7 mL) were then added, and the mixture was stirred at room temperature for 1 h. Icosanal (148.3 mg, 0.5 mmol) was added. The resulting mixture was stirred at room temperature for 12 h. After hydrolysis with 10 mL of saturated NH4Cl aqueous solution, the product was extracted with ethyl acetate (10 mL × 3) and washed with brine (10 mL). The combined extracts were dried over MgSO4. The organic phase was concentrated under reduced pressure and the crude product was purified by neutral silica gel column chromatography (eluent: n-hexane/Et2O) to give ginnol (172.5 mg, 81% yield). Enantioselectivity was confirmed by HPLC analysis of the diastereotopic (R)-MTPA-esters of the resulting ginnol. Chiral HPLC, Daicel chiralpack AD-3 × 2 at 4 °C, n-hexane/i-PrOH = 2000/1, flow rate = 0.1 mL min−1, tR = 100.3 min [major, (S)-derivative], 103.5 min [minor, (R)-derivative].
4.6. General procedure for the catalytic enantioselective arylzinc addition to ketones with arylboronic acids as an aryl source (Tables 5 and 6)
A solution of arylboronic acid (1.5 mmol) in toluene (3 mL) in a Pyrex Schlenk tube was stirred at room temperature for 10 min. The solution was heated under azeotropic reflux conditions with the removal of water. Water was removed through a pressure-equalized addition funnel that contained a cotton plug and 4 Å molecular sieves (pellets) and functioned as a Soxhlet extractor. After heating for 3 h at reflux temperature (bath temp.; 140 °C), the reaction mixture was allowed to cool to ambient temperature. After the removal of volatiles under reduced pressure (<5 Torr), the corresponding triarylboroxine (16) was obtained in situ and could be used without purification. Et2Zn (1.1 M in toluene) (4.1 mL, 4.5 mmol) and toluene (2 mL) were added at room temperature under a nitrogen atmosphere. The mixture was stirred at 60 °C for 12 h. Ligand 1a (45.6 mg, 0.10 mmol) was added to the mixture and the solution was stirred for 10 min. Ketone 9 (1.0 mmol) was then added. The resulting mixture was stirred at room temperature for 24 h. After hydrolysis with 10 mL of sat. NH4Cl aqueous solution, the product was extracted with ether (10 mL × 3) and washed with brine (10 mL). The combined extracts were dried over MgSO4. The organic phase was concentrated under reduced pressure and the crude product was purified by neutral silica gel column chromatography (eluent: n-hexane/Et2O) to give the desired products (17). The enantiomeric purity was determined by HPLC on a chiral column.
Acknowledgements
Financial support was provided by JSPS. KAKENHI (20245022), MEXT. KAKENHI (21750094, 21200033), and the Global COE Program of MEXT. We are grateful to Tosoh Finechem Corp. for providing organometallic reagents.
Notes and references
- For reviews:
(a) K. Soai and S. Niwa, Chem. Rev., 1992, 92, 833 CrossRef CAS;
(b) L. Pu and H.-B. Yu, Chem. Rev., 2001, 101, 757 CrossRef CAS;
(c) M. Hatano, T. Miyamoto and K. Ishihara, Curr. Org. Chem., 2007, 11, 127 CrossRef CAS;
(d) M. Hatano and K. Ishihara, Synthesis, 2008, 1647 CAS.
-
(a) K. Soai, T. Hayase, K. Takai and T. Sugiyama, J. Org. Chem., 1994, 59, 7908 CrossRef CAS;
(b) K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS;
(c) T. Hayase, T. Sugiyama, M. Suzuki, T. Shibata and K. Soai, J. Fluorine Chem., 1997, 84, 1 CrossRef CAS;
(d) T. Shibata, H. Tabira and K. Soai, J. Chem. Soc., Perkin Trans. 1, 1998, 177 RSC;
(e) M. Asami, H. Watanabe, K. Honda and S. Inoue, Tetrahedron: Asymmetry, 1998, 9, 4165 CrossRef CAS;
(f) W. K. Yang and B. T. Cho, Tetrahedron: Asymmetry, 2000, 11, 2947 CrossRef CAS;
(g) I. Sato, R. Kodaka, K. Hosoi and K. Soai, Tetrahedron: Asymmetry, 2002, 13, 805 CrossRef CAS.
- Textbooks for the preparation of dialkylzinc reagents:
(a)
M. Schlosser, Organometallics in Synthesis: A Manual, Wiley, Chichester, 2nd edn, 2001 Search PubMed;
(b)
H. Yamamoto and K. Oshima, Main Group Metals in Organic Synthesis, Wiley-VCH, Weinheim, 2004 Search PubMed;
(c)
P. Knochel, Handbook of Functionalized Organometallics, Wiley-VCH, Weinheim, 2005 Search PubMed.
- Recent selected papers for the preparation of dialkylzinc reagents:
(a) P. Wipf and W. Xu, Tetrahedron Lett., 1994, 35, 5197 CrossRef CAS;
(b) B. H. Lipshutz, M. R. Wood and R. Tirado, J. Am. Chem. Soc., 1995, 117, 6126 CrossRef CAS;
(c) S. Berger, F. Langer, C. Lutz, P. Knochel, T. A. Mobley and C. K. Reddy, Angew. Chem., Int. Ed. Engl., 1997, 36, 1496 CrossRef CAS;
(d) C. Lutz, P. Jones and P. Knochel, Synthesis, 1999, 312 CrossRef CAS;
(e) C. Bolm, N. Hermanns, J. P. Hildebrand and K. Muñiz, Angew. Chem., Int. Ed., 2000, 39, 3465 CrossRef CAS;
(f) S. Dahmen and S. Bräse, Org. Lett., 2001, 3, 4119 CrossRef CAS;
(g) A. Rimkus and N. Sewald, Org. Lett., 2002, 4, 3289 CrossRef CAS;
(h) S.-J. Jeon, H. Li and P. J. Walsh, J. Am. Chem. Soc., 2005, 127, 16416 CrossRef CAS;
(i) J. G. Kim and P. J. Walsh, Angew. Chem., Int. Ed., 2006, 45, 4175 CrossRef CAS;
(j) L. Salvi, J. G. Kim and P. J. Walsh, J. Am. Chem. Soc., 2009, 131, 12483 CrossRef CAS.
- Harada and co-workers reported a catalytic enantioselective 1°-alkylation and arylation to aldehydes with Grignard reagent/Ti(Oi-Pr)4.
(a) Y. Muramatsu and T. Harada, Angew. Chem., Int. Ed., 2008, 47, 1088 CrossRef CAS;
(b) Y. Muramatsu and T. Harada, Chem.–Eur. J., 2008, 14, 10560 CrossRef CAS;
(c) Y. Muramatsu, S. Kanehira, M. Tanigawa, Y. Miyawaki and T. Harada, Bull. Chem. Soc. Jpn., 2010, 83, 19 CrossRef CAS. Recently, they reported a catalytic enantioselective synthesis of allylic alcohols from aldehydes and alkenylboron reagents.
(d) T. Shono and T. Harada, Org. Lett., 2010, 12, 5270 CrossRef CAS.
- A. Côté and A. B. Charette, J. Am. Chem. Soc., 2008, 130, 2771 CrossRef.
-
(a) M. Hatano, S. Suzuki and K. Ishihara, J. Am. Chem. Soc., 2006, 128, 9998 CrossRef CAS;
(b) M. Hatano, S. Suzuki and K. Ishihara, Synlett, 2010, 321 CAS.
- P. J. Walsh, H. Li and C. A. de Parrodi, Chem. Rev., 2007, 107, 2503 CrossRef CAS.
- H. Soroos and M. Morgana, J. Am. Chem. Soc., 1944, 66, 893 CrossRef CAS.
-
(a) M. Hatano, T. Miyamoto and K. Ishihara, Org. Lett., 2007, 9, 4535 CrossRef CAS;
(b) M. Hatano, T. Mizuno and K. Ishihara, Synlett, 2010, 2024 CAS;
(c) M. Hatano, T. Mizuno and K. Ishihara, Chem. Commun., 2010, 46, 5443 RSC;
(d) M. Hatano, T. Mizuno and K. Ishihara, Tetrahedron, 2011, 67, 4417 CrossRef CAS.
-
MIB is an advantageous alternative to Noyori's DAIB [3-exo-(dimethylamino)isoborneol].
(a) M. Kitamura, S. Suga, K. Kawai and R. Noyori, J. Am. Chem. Soc., 1986, 108, 6071 CrossRef CAS;
(b) R. Noyori, S. Suga, K. Kawai, S. Okada, M. Kitamura, N. Oguni, M. Hayashi, T. Kaneko and Y. Matsuda, J. Organomet. Chem., 1990, 382, 19 CrossRef CAS;
(c) W. A. Nugent, Chem. Commun., 1999, 1369 RSC;
(d) R. Rosner, P. J. Sears, W. A. Nugent and D. G. Blackmond, Org. Lett., 2000, 2, 2511 CrossRef.
- Reviews and perspectives for the catalytic asymmetric hydrogenation and transfer hydrogenation of ketones:
(a) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS;
(b) W. S. Knowles, Angew. Chem., Int. Ed., 2002, 41, 1998 CrossRef CAS;
(c) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS;
(d) R. Noyori, M. Kitamura and T. Ohkuma, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5356 CrossRef CAS;
(e) W. S. Knowles and R. Noyori, Acc. Chem. Res., 2007, 40, 1238 CrossRef CAS.
- K. Cheng, A. R. Kelly, R. A. Kohn, J. F. Dweck and P. J. Walsh, Org. Lett., 2009, 11, 2703 CrossRef CAS.
- N. L. Holder, Chem. Rev., 1982, 82, 287 CrossRef CAS.
- M. Kusakabe, Y. Kitano, Y. Kobayashi and F. Sato, J. Org. Chem., 1989, 54, 2085 CrossRef CAS.
- H. Gröger, Adv. Synth. Catal., 2001, 343, 547 CrossRef.
- K. M. Miller, W.-S. Huang and T. F. Jamison, J. Am. Chem. Soc., 2003, 125, 3442 CrossRef CAS.
-
(a) F. A. Davis and B. C. Chen, Chem. Rev., 1992, 92, 919 CrossRef CAS;
(b) L. A. Paquette, S. V. O'Neil, N. Guillo, Q. Zeng and D. G. Young, Synlett, 1999, 1857 CrossRef CAS;
(c) P. Hoyos, J.-V. Sinisterra, F. Molinari, A. R. Alcántara and P. D. de María, Acc. Chem. Res., 2010, 43, 288 CrossRef CAS.
-
(a) K. B. Sharpless and R. C. Michaelson, J. Am. Chem. Soc., 1973, 95, 6136 CrossRef CAS;
(b) C. M. Marson, A. J. Walker, J. Pickering, S. Harper, R. Wrigglesworth and S. J. Edge, Tetrahedron, 1993, 49, 10317 CrossRef CAS;
(c) W. Adam and T. Wirth, Acc. Chem. Res., 1999, 32, 703 CrossRef CAS;
(d) M. M. Hussain and P. J. Walsh, Acc. Chem. Res., 2008, 41, 883 CrossRef CAS.
- A. H. Hoveyda, D. A. Evans and G. C. Fu, Chem. Rev., 1993, 93, 1307 CrossRef CAS.
- Recent asymmetric synthesis of tertiary alcohols with 2°-nucleophiles.
(a) K. Oisaki, D. Zhao, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2006, 128, 7164 CrossRef CAS;
(b) J. L. Stymiest, V. Bagutski, R. M. French and V. K. Aggarwal, Nature, 2008, 456, 778 CrossRef CAS;
(c) F. Wang, X. Liu, Y. Zhang, L. Lin and X. Feng, Chem. Commun., 2009, 7297 RSC.
-
(a) F. Langer, J. Waas and P. Knochel, Tetrahedron Lett., 1993, 34, 5261 CrossRef CAS;
(b) C. Eisenberg and P. Knochel, J. Org. Chem., 1994, 59, 3760 CrossRef CAS;
(c) R. Ostwald, P.-Y. Chavant, H. Stadtmueller and P. Knochel, J. Org. Chem., 1994, 59, 4143 CrossRef CAS;
(d) S. Vettel, A. Vaupel and P. Knochel, Tetrahedron Lett., 1995, 36, 1023 CrossRef CAS;
(e) S. Vettel, A. Vaupel and P. Knochel, J. Org. Chem., 1996, 61, 7173 CrossRef;
(f) C. Lutz, C.-D. Graf and P. Knochel, Tetrahedron, 1998, 54, 10317 CrossRef CAS;
(g) A. Fürstner, J. Mlynarski and M. Albert, J. Am. Chem. Soc., 2002, 124, 10274 CrossRef;
(h) A. Fürstner, M. Albert, J. Mlynarski, M. Matheu and E. DeClercq, J. Am. Chem. Soc., 2003, 125, 13132 CrossRef;
(i) S.-J. Jeon, H. Li, C. García, L. K. LaRochelle and P. J. Walsh, J. Org. Chem., 2005, 70, 448 CrossRef CAS.
- It has been reported that LiCl generally improves the activity of Grignard reagents.
(a) A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS;
(b) D. R. Armstrong, P. García-Álvarez, A. R. Kennedy, R. E. Mulvey and J. A. Parkinson, Angew. Chem., Int. Ed., 2010, 49, 3185 CrossRef CAS.
-
(a) S. Beckmann and H. Schühle, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1968, 23, 471 CAS;
(b) J.-H. Fuhrhop, T. Bedurke, A. Hahn, S. Grund, J. Gatzmann and M. Riederer, Angew. Chem., Int. Ed. Engl., 1994, 33, 350 CrossRef;
(c) T. Kusumi, H. Takahashi, T. Hashimoto, Y. Kan and Y. Asakawa, Chem. Lett., 1994, 1093 CrossRef CAS;
(d) L. Schwink and P. Knochel, Tetrahedron Lett., 1994, 35, 9007 CrossRef CAS;
(e) F. Langer, L. Schwink, A. Devasagayaraj, P.-Y. Chavant and P. Knochel, J. Org. Chem., 1996, 61, 8229 CrossRef CAS;
(f) T. Berkenbusch and R. Brückner, Tetrahedron, 1998, 54, 11471 CrossRef CAS;
(g) A. Dommisse, J. Wirtz, K. Koch, W. Barthlott and T. Kolter, Eur. J. Org. Chem., 2007, 3508 CrossRef CAS.
-
(a) M. Hatano, T. Miyamoto and K. Ishihara, Adv. Synth. Catal., 2005, 347, 1561 CrossRef CAS;
(b) M. Hatano, T. Miyamoto and K. Ishihara, Synlett, 2006, 1762 CAS;
(c) M. Hatano, T. Miyamoto and K. Ishihara, J. Org. Chem., 2006, 71, 6474 CrossRef CAS.
- Pioneering report of the catalytic enantioselective addition of Ph2Zn to ketones.
(a) P. I. Dosa and G. C. Fu, J. Am. Chem. Soc., 1998, 120, 445 CrossRef CAS;
(b) C. García and P. J. Walsh, Org. Lett., 2003, 5, 3641 CrossRef;
(c) H. Li, C. García and P. J. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5425 CrossRef CAS.
- For a review of catalytic enantioselective arylation.
(a) C. Bolm, J. P. Hildebrand, K. Muñiz and N. Hermanns, Angew. Chem., Int. Ed., 2001, 40, 3284 CrossRef CAS;
(b) F. Schmidt, R. T. Stemmler, J. Rudolph and C. Bolm, Chem. Soc. Rev., 2006, 35, 454 CAS;
(c) M. W. Paixão, A. L. Braga and D. S. Lüdtke, J. Braz. Chem. Soc., 2008, 19, 813 CrossRef.
-
(a) C. Bolm and J. Rudolph, J. Am. Chem. Soc., 2002, 124, 14850 CrossRef CAS;
(b) J. Rudolph, F. Schmidt and C. Bolm, Synthesis, 2005, 840 CAS.
-
(a) C. Jimeno, S. Sayalero, T. Fjermestad, G. Colet, F. Maseras and M. A. Pericàs, Angew. Chem., Int. Ed., 2008, 47, 1098 CrossRef CAS;
(b) X. Wu, X. Liu and G. Zhao, Tetrahedron: Asymmetry, 2005, 16, 2299 CrossRef CAS;
(c) Z. Chai, X.-Y. Liu, X.-Y. Wu and G. Zhao, Tetrahedron: Asymmetry, 2006, 17, 2442 CrossRef CAS;
(d) C. Liu, Z.-L. Guo, J. Weng, G. Lu and A. S. C. Chan, Chirality, 2010, 22, 159 CrossRef CAS.
-
Catalytic enantioselective arylation of ketones using arylboronic acids or triarylboroxines and Et2Zn.
(a) O. Prieto, D. J. Ramón and M. Yus, Tetrahedron: Asymmetry, 2003, 14, 1955 CrossRef CAS;
(b) V. J. Forrat, O. Prieto, D. J. Ramón and M. Yus, Chem.–Eur. J., 2006, 12, 4431 CrossRef CAS.
-
Catalytic enantioselective rhodium and palladium-catalyzed addition of arylboronic acids to ketones.
(a) S. L. X. Martina, R. B. C. Jagt, J. G. de Vries, B. L. Feringa and A. J. Minnaard, Chem. Commun., 2006, 4093 RSC;
(b) G. Liu and X. Lu, J. Am. Chem. Soc., 2006, 128, 16504 CrossRef CAS;
(c) P. He, Y. Lu, C.-G. Dong and Q.-S. Hu, Org. Lett., 2007, 9, 343 CrossRef CAS;
(d) H.-F. Duan, J.-H. Xie, X.-C. Qiao, L.-X. Wang and Q.-L. Zhou, Angew. Chem., Int. Ed., 2008, 47, 4351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and product characterization. See DOI: 10.1039/c1cy00108f |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.