Spectroscopy and conformational preferences of gas-phase helices
Jaime A.
Stearns
,
Caroline
Seaiby
,
Oleg V.
Boyarkin
and
Thomas R.
Rizzo
*
Laboratoire de Chimie Physique Moléculaire, Ecole Polytechnique Fédérale de Lausanne, EPFL SB ISIC LCPM, Station 6, CH-1015, Lausanne, Switzerland
First published on 6th November 2008
Abstract
We describe here a study of the spectroscopy of two peptides that we expect to be helical, Ac-Phe-(Ala)5-Lys-H+ and Ac-Phe-(Ala)10-Lys-H+, and one that we expect to be globular, Ac-Lys(H+)-Phe-(Ala)10, with the goal of identifying the spectral features characteristic of their secondary structure. Conformation-specific IR-UV double resonance spectroscopy in a cold ion trap, together with nitrogen-15 isotopic substitution, allow us to identify four conformers of the smaller helix. Infrared spectra in the OH and amide NH stretch regions, together with theoretical calculations, provide diagnostics of the presence of helical structure as well as details of the specific hydrogen bonding patterns within the helix. The assigned vibrational spectra presented here provide a benchmark for the ability of theory to predict the spectrum of a helical peptide.
Introduction
The gas phase provides a medium for studying the intrinsic conformational preferences of peptides that is not dissimilar to the low dielectric constant environment of biological membranes. In the absence of solvent a peptide can adopt conformations driven by only intramolecular forces, providing the opportunity to study these interactions in detail using high-resolution spectroscopic techniques. A major goal of this work is to identify spectroscopic signatures associated with different types of secondary structures and compare them with the predictions of theory. One of the most common secondary structural elements in proteins is the helix. There are several different types of helices, which differ in their hydrogen-bonding patterns. The most common is the α-helix, which has a repeating pattern of hydrogen bonds between the amide carbonyl oxygen on the ith residue and the amide NH of the i + 4th residue, forming hydrogen-bonded rings of 13 atoms, denoted C13. The 310 helix is a tighter, less common structure formed by 10-membered rings (C10) of an i, i + 3 bonding pattern.Over the last few years, infrared-ultraviolet (IR-UV) double-resonance spectroscopy has become an important tool in the determination of the gas-phase structure of biological molecules, especially in combination with gentle vaporization techniques such as laser desorption and electrospray.1–3 Structural information comes from the pattern of spectral shifts of vibrational bands induced by internal hydrogen bonding, in comparison with theoretical predictions, since constraining the molecule at just a few points can be enough to fix its conformation. Such analyses have revealed a wealth of conformational structures in neutral peptides that are recognizable as the building blocks of the secondary structure in proteins, including extended conformations reminiscent of β-sheets and folded conformations including one or more turns.4–23 Many protonated gas-phase peptides have been studied using infrared multiphoton dissociation spectroscopy, which, while not conformation-specific, can give clearly identifiable spectral features attributed to different protonation sites and different conformations.24–34
Several groups have identified helical structures in gas-phase peptides using IR spectroscopy. The IRMPD spectrum of gas-phase potassiated cytochrome C was assigned mostly to features indicative of α-helices, in keeping with its known solution structure.25Infrared spectra in the hydride stretch region of gas-phase Ac-Ala-Phe-Ala-NH2 and Ac-Aib-Phe-Aib-NH2 measured by Mons and coworkers led them to assign each as having two rings of a 310-helix.11,16 De Vries and coworkers measured infrared spectra of the pentapeptide FDASV and several 15-residue gramicidin peptides in the hydride stretch region and suggest the presence of gas-phase helical structures, the former containing a single C13α-turn, and the latter each showing a large, unresolved band in the NH-stretch region attributed to hydrogen bonds involved in a helix.13,14 We have previously reported35 initial results of a spectroscopic study of cold, protonated, lysine-capped polyalanine helices for which some structural information is available from ion mobility studies.36 Jarrold and coworkers established that peptides consisting of seven or more alanines with a lysine at the C-terminus form extremely stable gas-phase helices, while those without the lysine or with the lysine at the N-terminus are globular.36,37 Indeed Vaden et al. have recently used IRMPD spectroscopy in the high-frequency stretch region to confirm the globular nature of small alanine peptides lacking the C-terminal lysine.33 The importance of the lysine in helix formation lies partially in its role as the protonation site, with the positive charge stabilizing the macro-dipole formed by alignment of the amide NH and CO groups, and partially in the ability of the lysine side-chain to hydrogen-bond to dangling carbonyl groups at the end of the helix. In our initial report35 we assigned the spectroscopic features associated with the major conformers of the 7-residue peptide Ac-Phe-(Ala)5-Lys-H+, and speculated as to the corresponding conformers observed in the 12-residue Ac-Phe-(Ala)10-Lys-H+.
In the present work we provide a more detailed account of the spectroscopy of these molecules and show evidence of additional stable conformations. Moreover, we use nitrogen-15 isotopic substitution in Ac-Phe-(Ala)5-Lys-H+ to provide a definitive confirmation of the spectroscopic assignments made previously on the basis of DFT calculations. Finally, in a quest to understand the spectroscopic signatures of helical peptides, we have investigated the spectroscopy of a presumably globular peptide, Ac-Lys(H+)-Phe-(Ala)10.
Experimental methods
Our experimental approach is based on ultraviolet photofragmentation spectroscopy of protonated, gas-phase biomolecules in a 6 K, 22-pole ion trap.38,39 We produce the protonated peptides in the gas phase by nanoelectrospray from a 0.2 mM solution in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50:0.2 methanol:water:acetic acid or pure methanol and collect them in an RF-only hexapole trap. Once released from the trap, the ion packet passes through a quadrupole mass filter to select ions of a particular m/z. A series of lenses and an octopole ion guide direct the ions into the 22-pole ion trap, where they cool by collisions with helium to ∼10 K.38 An ultraviolet laser pulse excites some fraction of these ions when the laser frequency is resonant with an electronic transition. After allowing several milliseconds for the excited molecules to fragment, we release all the ions from the trap and select fragmentation products of a particular m/z with a second quadrupole mass filter before detecting them with a channeltron electron multiplier.
:50:0.2 methanol:water:acetic acid or pure methanol and collect them in an RF-only hexapole trap. Once released from the trap, the ion packet passes through a quadrupole mass filter to select ions of a particular m/z. A series of lenses and an octopole ion guide direct the ions into the 22-pole ion trap, where they cool by collisions with helium to ∼10 K.38 An ultraviolet laser pulse excites some fraction of these ions when the laser frequency is resonant with an electronic transition. After allowing several milliseconds for the excited molecules to fragment, we release all the ions from the trap and select fragmentation products of a particular m/z with a second quadrupole mass filter before detecting them with a channeltron electron multiplier.
We record an ultraviolet photofragment excitation spectrum by monitoring the fragment ion signal as a function of UV laser wavelength. To collect as many fragment ions as possible and obtain the highest-quality spectrum, we operated the analyzing quadrupole at low resolution such that product ions of different m/z can be transmitted simultaneously.
We record the infrared spectrum associated with a particular UV transition by monitoring the fragment ion signal induced by that transition while introducing an IR laser 100 ns earlier. If the ions absorb an IR photon and if the UV absorption or fragmentation is less efficient for the vibrationally-excited ions, the UV-induced photofragmentation signal will be depleted. By measuring this depletion as a function of IR wavenumber, we obtain the conformation-specific infrared spectrum.
All peptides were synthesized using solid phase Fmoc chemistry on an Applied Biosystems 433 A Synthesizer.
Computational methods
A conformational search of Ac-Phe-(Ala)5-Lys-H+, using the AMBER force field40 in Macromodel,41 gives an initial collection of over 1000 structures under 50 kJ mol−1 in energy, nearly all of which are helical. We have observed in calculations of protonated amino acids39 that AMBER tends to overestimate the stabilizing effect of an NH⋯OH C5 interaction at the C-terminus by about 15 kJ mol−1, so we eliminated from further consideration all structures that had this type of interaction, retaining instead those with the more favorable NH⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C C5 structure. We calculated single-point energies using B3LYP/6-31G**42,43 in Gaussian 0344 for the remaining structures and performed a full geometry optimization and harmonic frequency analysis on a subset that included all major degrees of flexibility of the backbone, the phenylalanine side-chain, and other flexible coordinates as described below. In all, 45 structures were optimized, and vibrational frequencies were calculated for the 21 most stable of these. The energies we report are corrected for zero-point energies using the unscaled harmonic frequencies, while the hydride stretch frequencies are scaled by a factor of 0.952 for comparison to the measured infrared spectra.
C C5 structure. We calculated single-point energies using B3LYP/6-31G**42,43 in Gaussian 0344 for the remaining structures and performed a full geometry optimization and harmonic frequency analysis on a subset that included all major degrees of flexibility of the backbone, the phenylalanine side-chain, and other flexible coordinates as described below. In all, 45 structures were optimized, and vibrational frequencies were calculated for the 21 most stable of these. The energies we report are corrected for zero-point energies using the unscaled harmonic frequencies, while the hydride stretch frequencies are scaled by a factor of 0.952 for comparison to the measured infrared spectra.
With our current computational capabilities, we cannot perform a full set of calculations on the larger peptides Ac-Phe-(Ala)10-Lys-H+ and Ac-Lys(H+)-Phe-(Ala)10. Nevertheless, comparison with assigned spectra of the smaller peptide allows us to identify important structural features in these larger molecules.
Results
Ac-Phe-(Ala)5-Lys-H+
Fig. 1 shows the previously reported electronic excitation spectrum of Ac-Phe-(Ala)5-Lys-H+ obtained by collecting the UV-induced photofragments as a function of the laser wavenumber.35 We performed IR-UV double resonance spectroscopy to ascertain the parent ion conformation giving rise to each of the transitions labeled in the UV spectrum of Fig. 1. The two largest peaks, labeled A and B, have different IR spectra (Fig. 2) and thus belong to two different conformers. The next most intense peaks, 10 cm−1 and 38 cm−1 to the blue of peaks A and B, have the same IR spectra as A and B, so we assign them as vibronic bands of the same conformers, and label them in Fig. 1 with the corresponding lower-case letter. Just to the red of A and B are two transitions that have different infrared spectra (Fig. 2), and therefore represent the third and fourth conformers, C and D. Although the infrared laser usually causes depletions in the ultraviolet spectrum in IR-UV double resonance experiments, there are also some gains in the infrared spectra of conformers C and D. These gains arise because of statistical broadening45 in the UV spectrum of conformers A and B following IR excitation. This spreads a small amount of absorption intensity to the UV wavelengths at which conformers C and D are probed. Because the fragmentation signals from conformers A and B are significantly larger than those of C and D, this gain on the fragmentation signal is on the same scale as the IR-induced depletion of C and D. The populations of C and D are sufficiently small that the inverse is not perceptible.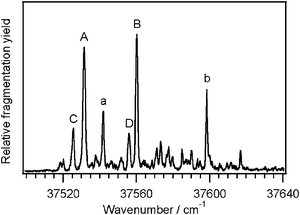 | ||
| Fig. 1 The UV photofragmentation spectrum of Ac-Phe-(Ala)5-Lys-H+. The transitions labeled in capital letters are band origins of the four conformers, while the lower-case letters are vibronic bands built upon the corresponding origins. | ||
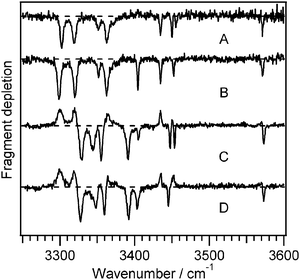 | ||
| Fig. 2 Infrared-ultraviolet double resonance spectra of Ac-Phe-(Ala)5-Lys-H+ recorded at the correspondingly-labeled UV transitions in Fig. 1. The positive-going signals in the spectra of C and D are gains are due to conformers A and B. | ||
The four infrared spectra of Fig. 2 all contain the C-terminal carboxylic acid OH stretch band around 3572 cm−1 and all but D show seven resolved amide NH stretches. In general, the amide NH stretches below 3400 cm−1 belong to the NH groups involved in stronger hydrogen bonds, whereas the higher-frequency NH stretch bands arise from those in weaker interactions. The substantial differences in the low-frequency region between the two pairs of conformers A/B and C/D suggest different hydrogen-bonding patterns for each pair. We also observed several broad transitions between 2900–3100 cm−1 (not shown), which we assign to the NH stretches of the ammonium group of the lysine side chain. In protonated amino acids, ammonium NH stretches appear around 3350 cm−1 in the absence of hydrogen bonding, and around 3000 cm−1 when hydrogen-bonded,39 implying that the ammonium NH groups in Ac-Phe-(Ala)5-Lys-H+ are involved in hydrogen bonds. While CH stretch bands will also occur in this region, they are generally much weaker.39
To help assign the amide NH bands, we also recorded infrared spectra of three isotopologues of Ac-Phe-(Ala)5-Lys-H+, each with an alanine amide nitrogen replaced by nitrogen-15: 15N-Ala2, 15N-Ala4, and 15N-Ala6, where the right superscript index of the residue indicates its position from the N-terminus. In a simple diatomic model for an NH oscillator, the difference in reduced mass due to the heavier isotope would shift the NH stretch frequency to lower energy by ∼8 cm−1.
Fig. 3 shows the IR spectra of all four conformers of isotopically labeled Ac-Phe-(Ala)5-Lys-H+. In each case, primarily a single transition is shifted by ∼8 cm−1, allowing us to assign that transition to the NH stretch of the labeled residue and suggesting that these amide NH stretches are localized. From these spectra we assign the NH stretch of Ala2 to the peak near 3450 cm−1, which is the highest-frequency amide NH stretch for all conformers except C. The NH stretch of Ala4 appears near 3320 cm−1 in conformers A and B, and near 3390 cm−1 in conformers C and D. The difference in the transition frequencies of the Ala4NH stretches between the pairs of conformers highlights our qualitative assessment that the hydrogen-bonding motifs of the pairs must be different. The Ala6NH stretch is associated primarily with the transition at 3340 cm−1 in all four conformers. We complete the assignment of the spectra and their attribution to particular conformations by comparing the experimental spectra to calculated spectra.
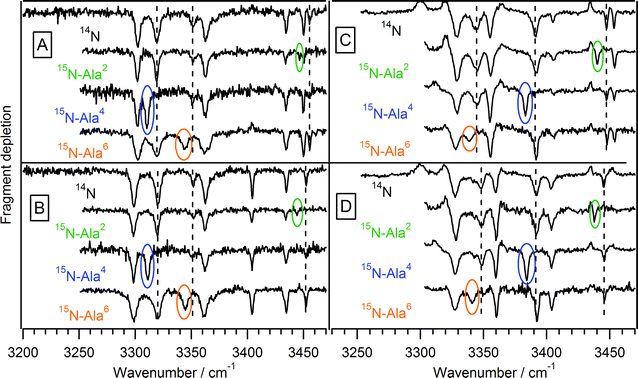 | ||
| Fig. 3 Infrared-ultraviolet double resonance spectra of isotopically substituted Ac-Phe-(Ala)5-Lys-H+. For each conformer, the unsubstituted peptide spectrum is shown at the top, with the singly-substituted peptides shown underneath. The shifted peaks are circled, while a dashed line shows the position of those transitions in the unsubstituted peptide. | ||
We classify the calculated structures of Ac-Phe-(Ala)5-Lys-H+ first according to the hydrogen-bonding scheme of the peptide backbone. Nearly all of the low-energy structures are helical, and the vast majority contain amide NH groups involved in only C10 and C13hydrogen-bonded rings. The global minimum at the B3LYP/6-31G** level of theory is a helix with two C10 and two C13 rings, as shown schematically in Fig. 4a, and denoted backbone I. The hydrogen-bonded rings involve the amide NH groups of Ala3, Ala4, Ala5, and Ala6 in such a way that the middle two share a carbonyl. The amide NH groups of Phe1, Ala2, and Lys7 are at the ends of the molecule and do not participate in C10 or C13NH⋯CO hydrogen bonds. The lysine amide NH is in a C5 arrangement with the C-terminus, and the phenylalanine NH can be in a π-hydrogen bond, depending on the orientation of the phenylalanine side chain. The second-lowest-energy hydrogen-bonding pattern, II, has three C10 rings and one C13 ring [Fig. 4b], with the Ala5 and Ala6NH groups hydrogen-bonded to the same carbonyl. The hydrogen bonding schemes for the end residues are the same as backbone I. We also considered backbones consisting of four C10 rings (III) and one C10 and three C13 rings (IV), which are 310- and α-helices, respectively. Each backbone structure locks in not only the amide NH groups of the participating alanines, but also the Lys7 ammonium group, as each backbone conformation provides a particular subset of the carbonyls as hydrogen-bond acceptors.
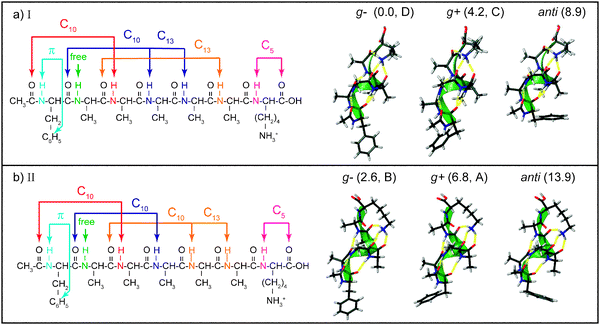 | ||
| Fig. 4 Schematic depictions of the structures of the lowest-energy conformers of Ac-Phe-(Ala)5-Lys-H+, with the hydrogen-bonding schemes of the helices on the left and calculated structures on the right. The calculated structures have the helix axes aligned to better show the orientation of the phenylalanine ring: g+, g−, or anti. The conformers are also labeled with their zero-point-corrected energy in kJ mol−1 and their assignment (A, B, C, or D). | ||
For each backbone conformation, there are other degrees of freedom that give rise to different conformers. First, the orientation of the phenylalanine side chain changes by rotation around the Cα–Cβ bond, such that the Phe1χ1 angle, measured along the N–Cα–Cβ–Cγ atoms, takes values of approximately 180, +60, or −60°, corresponding to labels of anti, gauche + (g +), and gauche− (g−). In all the structures we examined, the gauche structures were lower in energy because they allowed the Phe1amide NH to form a favorable π-hydrogen bond with the aromatic ring, which, based on the vibrational frequencies, seems to be especially strong in the g− structures (Fig. 4). Two other flexible coordinates involve the lysine. First, the C-terminal carboxylic acid group can be rotated away from the C5 interaction, but this costs 10–15 kJ mol−1 of energy and we did not consider structures without the C5 arrangement. Second, the butyl side chain of the lysine can be puckered in different ways without altering the hydrogen bonding of the ammonium group. These changes increase the energy by 8 kJ mol−1 or more, making these structures less likely to appear in our experiment, and we did not consider these further. Table 1 summarizes the twelve types of conformations that arise from the four types of helices and three orientations of the Phe side chain. The final entry in Table 1, (backbone V) is the only conformation that is fairly low in energy, but which does not belong to one of the main backbone families.
| Backbone | Phe χ1 | Energy | Assignment | |
|---|---|---|---|---|
| I | C10–C10–C13–C13 | g− | 0.0 | D |
| I | C10–C10–C13–C13 | g+ | 4.2 | C |
| I | C10–C10–C13–C13 | anti | 8.9 | |
| II | C10–C10–C10–C13 | g− | 2.6 | B |
| II | C10–C10–C10–C13 | g+ | 6.8 | A |
| II | C10–C10–C10–C13 | anti | 13.9 | |
| III | C10–C10–C10–C10 | g− | 8.0 | |
| III | C10–C10–C10–C10 | g+ | 12.2 | |
| III | C10–C10–C10–C10 | anti | 20.4 | |
| IV | C10–C13–C13–C13 | g− | 10.7 | |
| IV | C10–C13–C13–C13 | g+ | 15.3 | |
| IV | C10–C13–C13–C13 | anti | 20.3 | |
| V | C14–C7–C16–C16 | g+ | 6.0 |
Fig. 5 shows the experimental IR spectra of conformers A and B along with several calculated spectra. The best matches to A and B are shown directly under the experimental spectra, and these both have the C10–C10–C10–C13hydrogen-bonding pattern (backbone II). Fig. 5 also displays calculated spectra of the lowest-energy conformers of backbone conformations I and III–V. The region below ∼3400 cm−1 reflects the different interaction strengths and cooperative effects of these structures, and no other backbone matches the experimental spectra as well as II. The structures assigned to conformers A and B have the g+ and g− orientations of the Phe1 side chain, whereas the anti orientation of this helix is both higher in energy than its g+ and g− counterparts (Table 1) and has a calculated spectrum (Fig. 5, IIanti) that does not match the experimental spectra as well. The g− structure proposed for conformer B has the Phe1NH group in a stronger π-hydrogen bond than does conformer A, giving the NH stretch a lower frequency in conformer B, which is the only significant difference between the two spectra. It is important to note that these assignments are in complete agreement with those made on the basis of the isotopic substitution, although some of the calculated frequencies, particularly for Ala5 and Ala6, do not match the measured values especially well.
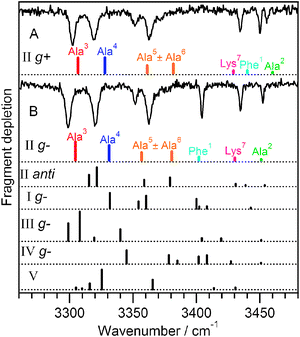 | ||
| Fig. 5 Comparison between experimental and calculated spectra for conformers A and B of Ac-Phe-(Ala)5-Lys-H+. The best-matching calculated spectra are presented directly under each experimental spectrum and both have backbone II, C10–C10–C10–C13. They differ by the Phe ring orientation. Calculated spectra belonging to other conformers are shown below for comparison. | ||
Fig. 6 shows the IR spectra of the minor conformers, C and D, together with several calculated spectra. The experimental data are the same as those shown in Fig. 2, but the gains have been removed by subtracting the summed spectra of conformers A and B. Because the subtraction process is not perfect, it occasionally gives rise to small artificial peaks, such as the one at 3440 cm−1 in conformer D, which we ignore. The calculated spectra that give the best match to the experimental spectra both correspond to conformers with backbone I, C10–C10–C13–C13; the calculated spectra for backbones II–V clearly do not reproduce the experimental transitions in the region below 3400 cm−1. We make the assignment of the Phe ring orientation on the basis of the Phe1NH stretch, which is present above 3430 cm−1 in conformer C and the g+ structure. The anti conformer (Fig. 6, Ianti) is a reasonable match to conformer C, but as Table 1 shows, it is higher in energy, so we presume that the g+ conformer is more likely to appear in the low-temperature conditions of our experiment. In conformer D, the Phe1NH stretch is apparently shifted from that of Ala2 (which was assigned by isotopic substitution), and the calculated spectrum for the g− structure suggests that it may be buried underneath that of Ala4 or Lys7. In the spectrum of 15N-Ala4 conformer D, the Ala4 transition shifts to the red but reveals no underlying Phe1NH stretch, so perhaps it lies underneath that of the Lys7 amide NH transition at 3404 cm−1, which appears broader in conformer D than in conformer C (Fig. 6).
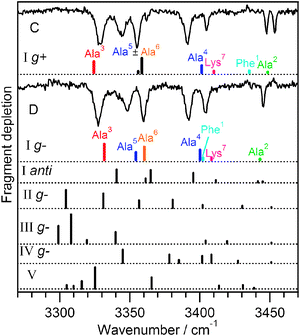 | ||
| Fig. 6 Comparison between experimental and calculated spectra for conformers C and D of Ac-Phe-(Ala)5-Lys-H+, from which the gains were removed by subtracting spectra of conformers A and B. The best-matching calculated spectra are presented directly under each experimental spectrum and both have backbone I, C10–C10–C13–C13. They differ by the phenylalanine ring orientation. Calculated spectra belonging to other conformers are shown below for comparison. | ||
Some of the assignments made by isotopic substitution disagree with the calculated transition assignments for conformers C and D. One instance is the assignments of the NH stretches of Ala5 with Ala6 in both conformers, which are reversed in the calculation. The other deficiency is in the underestimation of the Phe1NH stretch frequency in conformer C.
Helical Ac-Phe-(Ala)10-Lys-H+ and globular Ac-Lys(H+)-Phe-(Ala)10
We previously35 reported the infrared spectrum of Ac-Phe-(Ala)10-Lys-H+, which we assume to be helical on the basis of ion mobility measurements of the similar peptide Ac-(Ala)10-Lys-H+.36 We now compare Ac-Phe-(Ala)10-Lys-H+ with a peptide that we expect to be non-helical, Ac-Lys(H+)-Phe-(Ala)10, since the positively charged lysine at the N-terminus destabilizes the helix macro-dipole.36Fig. 7 shows the UV photofragment excitation spectra of these two peptides. We have already assigned the most intense UV transitions of the helical peptide in Fig. 7a to the band origins of two distinct conformers on the basis of their IR spectra.35 The smaller peaks between the two are a Franck–Condon progression in a 9 cm−1 vibrational mode, with an associated hot band just to the red of the band origin. Two similar progressions appear built off of the largest transition in the UV spectrum in Fig. 7b, at spacings of 12 and 15 cm−1.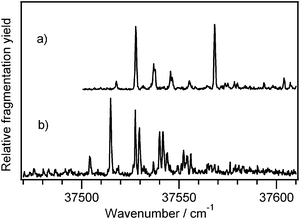 | ||
| Fig. 7 Ultraviolet photofragmentation spectrum of (a) Ac-Phe-(Ala)10-Lys-H+ and (b) Ac-Lys(H+)-Phe-(Ala)10. | ||
In Fig. 8 we show IR spectra of Ac-Phe-(Ala)10-Lys-H+ and Ac-Lys(H+)-Phe-(Ala)10, where (a) and (b) are associated with the two UV band origins of the former and (c) shows the spectrum recorded using the largest transition of the latter. The vibronic bands in the UV spectra of Fig. 7 each have the same IR spectrum as their respective band origins, confirming that they arise from the same conformer. A few smaller peaks to the red of the band origin in Fig. 7b (not shown) have IR spectra different from that of the major conformer, indicating the presence of at least one other conformation with a relatively small population.
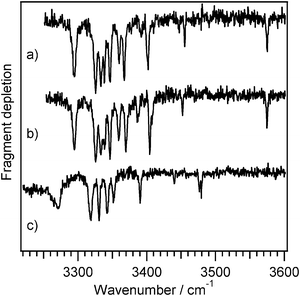 | ||
| Fig. 8
IR-UV double resonance spectra of (a) Ac-Phe-(Ala)10-Lys-H+ recorded at the first band origin at 37258 cm−1, (b) Ac-Phe-(Ala)10-Lys-H+ recorded at the second band origin at 37568 cm−1, and (c) Ac-Lys(H+)-Phe-(Ala)10 recorded at the largest transition at 37515 cm−1. | ||
Thus far we have focused on the amide NH stretch region because in the smaller peptide it provides detailed information on the hydrogen-bonding patterns of the helix. The exceptional feature of the IR spectrum of the globule, however, is the carboxylic acid OH stretch, which rather than occurring as a sharp peak around 3570 cm−1, appears to be broadened and shifted to 3270 cm−1. Its position is similar to that observed in conformers of neutral amino acids that contain a hydrogen bond between the COOH and amino groups,46–48 although it is likely here that the hydrogen-bond acceptor is a carbonyl group. The fact that the OH group is hydrogen-bonded is consistent with the peptide being non-helical, since the rigid structure of the helix would prevent the C-terminus from wrapping around to form any hydrogen bonds.
Even though Ac-Phe-(Ala)10-Lys-H+ and Ac-Lys(H+)-Phe-(Ala)10 are too large to carry out a full set of calculations, we can categorize their amide NH transitions by comparison with the smaller peptide. The transitions above ∼3380 cm−1 should correspond to free NH groups or those involved in π-hydrogen bonds or C5 interactions, while transitions below 3380 cm−1 reflect stronger hydrogen bonding. We have previously suggested that vibrations of NH groups in α-helical C13 rings appear in the region 3320-3350 cm−1.35 However, the globular peptide also shows at least four transitions in this region, indicating that they are not specifically diagnostic of an α-helical peptide. For Ac-Lys(H+)-Phe-(Ala)10 they may represent a short helical turn, spanning a few amino acids, or they may simply represent medium-strength hydrogen bonded NH groups that happen to fall in the same region.
Discussion and conclusions
Our conformational assignments reflect a trend that has been reported recently by several groups, that even relatively large peptides appear as single conformations or a small number of conformations of similar structure.13,14 In our case, the larger helical peptide Ac-Phe-(Ala)10-Lys-H+ exhibits fewer observed conformations (two) than the smaller Ac-Phe-(Ala)5-Lys-H+ which has four. This may be a result of the extreme stability of α-helical structures37 such that the only degree of freedom in Ac-Phe-(Ala)10-Lys-H+ that is not completely locked in at low temperature is that of the phenyl ring orientation. It is perhaps more surprising that the globular structure also has a single major conformer, implying that it has as well-defined of a structure as the helix, although we do not know what that structure is.The IR spectra of the helical peptides provide the opportunity to examine the relationship between peptide structure and amide NH frequency. Chin et al. recently carried out such an analysis for neutral peptides, relating the amide NH frequency to the type of hydrogen bonding interaction (C5, C10, etc).2 However, the frequencies of our larger, charged peptides do not coincide with that scheme, as the NH stretches in C10 interactions in the neutral peptides appeared exclusively above 3360 cm−1, while we observe several such bands as low as 3300 cm−1 in the protonated peptides. The factors leading to these differences are likely a combination of local geometry of the hydrogen bonding interaction itself, such as the distance and angle between the NH and CO groups, as well as the presence of charge and effects of cooperativity in chains of hydrogen bonds. Several computational studies have investigated the effects of hydrogen bond cooperativity on amide frequencies.49–52 One such study examined the NH vibrational frequencies of a neutral glycine pentapeptide in a 310-helical conformation, finding that the furthest red-shifted transition belonged to the first amide NH participating in the hydrogen bonding network of the helix, which belongs to the third residue from the N-terminus,51 in agreement with our assigned spectra of Ac-Phe-(Ala)5-Lys-H+. However, the Ala3NH stretch we observe is 30 cm−1 lower in conformers A and B than in C and D, despite similar geometries of the hydrogen bond and the same C10 interaction. This might imply that helices with different backbones and hence different chains of hydrogen bonds exhibit different degrees of cooperativity.
While the level of theory used here was sufficient to help assign the infrared spectra, it was not particularly accurate for predicting precise vibrational frequencies or the relative energies of the various conformers (as determined from the observed relative intensities of the band origins in the UV spectra). Although the DFT methods and modest basis set used here performed surprisingly well, Hobza and coworkers had previously noted unsatisfactory results in other peptides because of the lack of inclusion of dispersive interactions.12 Dispersion may also be the reason that Phe1NH frequencies are predicted rather poorly in the g+ conformers A and C of Ac-Phe-(Ala)5-Lys-H+. The infrared spectra of helical peptides are likely to involve contributions from local geometry, longer-range cooperative effects, and charge, and the high resolution infrared spectra of Ac-Phe-(Ala)5-Lys-H+ that we have assigned with the help of isotopic substitution provide an unprecedented benchmark for testing the accuracy of theoretical predictions. If a particular theoretical approach cannot accurately predict the structure and the spectrum of this well defined system, one should question its applicability for larger systems in more complicated environments.
The difficulty of assigning amide NH stretches without the extensive use of calculations is demonstrated by the comparison between the helical and globular peptides. Given the likelihood that Ac-Phe-(Ala)10-Lys-H+ is helical (based on ion mobility studies), we have suggested that the clustered region between 3320–3350 cm−1 belongs to the NH groups in C13 interactions in the center of the helix,35 but this same region shows several transitions in a peptide that is very likely globular, Ac-Lys(H+)-Phe-(Ala)10. A more decisive measure of whether the peptide is helical comes from the hydrogen-bonded carboxylic acid OH, which is red-shifted in the globular peptide by 300 cm−1 relative to its free position in the helical peptides.
Acknowledgements
Funding was provided by the École Polytechnique Fédérale de Lausanne and the Fonds National Suisse through grant no. 200020-112071. We are grateful to Catherine Servis and the University of Lausanne Protein and Peptide Chemistry Facility for synthesis of the peptides.References
- M. Gerhards, in Principles of Mass Spectrometry Applied to Biomolecules, ed. J. Laskin and C. Lifshitz, John Wiley & Sons, Inc, Hoboken, NJ, 2006, pp. 3–61 Search PubMed.
- W. Chin, F. Piuzzi, I. Dimicoli and M. Mons, Phys. Chem. Chem. Phys., 2006, 8, 1033–1048 RSC.
- M. S. de Vries and P. Hobza, Annu. Rev. Phys. Chem., 2007, 58, 585–612 CrossRef CAS.
- B. C. Dian, A. Longarte, S. Mercier, D. Evans, D. J. Wales and T. S. Zwier, J. Chem. Phys., 2002, 117, 10688–10702 CrossRef CAS.
- H. Fricke, A. Gerlach, C. Unterberg, P. Rzepecki, T. Schrader and M. Gerhards, Phys. Chem. Chem. Phys., 2004, 6, 4636–4641 RSC.
- I. Huenig and K. Kleinermanns, Phys. Chem. Chem. Phys., 2004, 6, 2650–2658 RSC.
- J. M. Bakker, C. Pluetzer, I. Huenig, T. Haeber, I. Compagnon, G. von Helden, G. Meijer and K. Kleinermanns, ChemPhysChem, 2005, 6, 120–128 CrossRef CAS.
- W. Chin, J.-P. Dognon, C. Canuel, F. Piuzzi, I. Dimicoli, M. Mons, I. Compagnon, G. von Helden and G. Meijer, J. Chem. Phys., 2005, 122 Search PubMed , 054317/054311–054317/054318.
- W. Chin, M. Mons, J.-P. Dognon, R. Mirasol, G. Chass, I. Dimicoli, F. Piuzzi, P. Butz, B. Tardivel, I. Compagnon, G. Von Helden and G. Meijer, J. Phys. Chem. A, 2005, 109, 5281–5288 CrossRef CAS.
- W. Chin, F. Piuzzi, J.-P. Dognon, I. Dimicoli and M. Mons, J. Chem. Phys., 2005, 123 Search PubMed , 084301/084301–084301/084311.
- W. Chin, F. Piuzzi, J. P. Dognon, I. Dimicoli, B. Tardivel and M. Mons, J. Am. Chem. Soc., 2005, 127, 11900–11901 CrossRef CAS.
- D. Reha, H. Valdes, J. Vondrasek, P. Hobza, A. Abu-Riziq, B. Crews and M. S. de Vries, Chem.–Eur. J., 2005, 11, 6803–6817 CrossRef CAS.
- A. Abo-Riziq, J. E. Bushnell, B. Crews, M. Callahan, L. Grace and M. S. de Vries, Chem. Phys. Lett., 2006, 431, 227–230 CrossRef CAS.
- A. Abo-Riziq, B. O. Crews, M. P. Callahan, L. Grace and M. S. d. Vries, Angew. Chem., Int. Ed., 2006, 45, 5166–5169 CrossRef CAS.
- I. Compagnon, J. Oomens, G. Meijer and G. Von Helden, J. Am. Chem. Soc., 2006, 128, 3592–3597 CrossRef CAS.
- V. Brenner, F. Piuzzi, I. Dimicoli, B. Tardivel and M. Mons, J. Phys. Chem. A, 2007, 111, 7347–7354 CrossRef CAS.
- H. Fricke, G. Schafer, T. Schrader and M. Gerhards, Phys. Chem. Chem. Phys., 2007, 9, 4592–4597 RSC.
- E. Gloaguen, F. Pagliarulo, V. Brenner, W. Chin, F. Piuzzi, B. Tardivel and M. Mons, Phys. Chem. Chem. Phys., 2007, 9, 4491–4497 RSC.
- T. Haber, K. Seefeld and K. Kleinermanns, J. Phys. Chem. A, 2007, 111, 3038–3046 CrossRef.
- E. E. Baquero, W. H. James, S. H. Choi, S. H. Gellman and T. S. Zwier, J. Am. Chem. Soc., 2008, 130, 4784–4794 CrossRef CAS.
- E. E. Baquero, W. H. James, S. H. Choi, S. H. Gellman and T. S. Zwier, J. Am. Chem. Soc., 2008, 130, 4795–4807 CrossRef CAS.
- H. Fricke, A. Funk, T. Schrader and M. Gerhards, J. Am. Chem. Soc., 2008, 130, 4692–4698 CrossRef CAS.
- T. Haeber, K. Seefeld, G. Engler, S. Grimme and K. Kleinermanns, Phys. Chem. Chem. Phys., 2008, 10, 2844–2851 RSC.
- J. M. Bakker, L. M. Aleese, G. Meijer and G. von Helden, Phys. Rev. Lett., 2003, 91, 203003–203004 CrossRef.
- J. Oomens, N. Polfer, D. T. Moore, L. van der Meer, A. G. Marshall, J. R. Eyler, G. Meijer and G. von Helden, Phys. Chem. Chem. Phys., 2005, 7, 1345–1348 RSC.
- J. Oomens, B. G. Sartakov, G. Meijer and G. Von Helden, Int. J. Mass Spectrosc., 2006, 254, 1–19 CrossRef CAS.
- G. Gregoire, M. P. Gaigeot, D. C. Marinica, J. Lemaire, J. P. Schermann and C. Desfrancois, Phys. Chem. Chem. Phys., 2007, 9, 3082–3097 RSC.
- R. Wu and T. B. McMahon, J. Am. Chem. Soc., 2007, 129, 4864–4865 CrossRef CAS.
- R. Wu and T. B. McMahon, J. Am. Chem. Soc., 2007, 129, 11312–11313 CrossRef CAS.
- N. C. Polfer, J. Oomens, S. Suhai and B. Paizs, J. Am. Chem. Soc., 2007, 129, 5887–5897 CrossRef CAS.
- C. F. Correia, P. O. Balaj, D. Scuderi, P. Maitre and G. Ohanessian, J. Am. Chem. Soc., 2008, 130, 3359–3370 CrossRef CAS.
- T. D. Vaden, T. S. J. A. d. Boer, J. P. Simons and L. C. Snoek, Phys. Chem. Chem. Phys., 2008, 10, 1443–1447 RSC.
- T. D. Vaden, T. S. J. A. de Boer, J. P. Simons, L. C. Snoek, S. Suhai and B. Paizs, J. Phys. Chem. A, 2008, 112, 4608–4616 CrossRef CAS.
- A. Fujihara, H. Matsumoto, Y. Shibata, H. Ishikawa and K. Fuke, J. Phys. Chem. A, 2008, 112, 1457–1463 CrossRef CAS.
- J. A. Stearns, O. V. Boyarkin and T. R. Rizzo, J. Am. Chem. Soc., 2007, 129, 13820–13821 CrossRef CAS.
- R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1999, 121, 3494–3501 CrossRef CAS.
- M. Kohtani, T. C. Jones, J. E. Schneider and M. F. Jarrold, J. Am. Chem. Soc., 2004, 126, 7420–7421 CrossRef CAS.
- O. V. Boyarkin, S. R. Mercier, A. Kamariotis and T. R. Rizzo, J. Am. Chem. Soc., 2006, 128, 2816–2817 CrossRef CAS.
- J. A. Stearns, S. Mercier, C. Seaiby, M. Guidi, O. V. Boyarkin and T. R. Rizzo, J. Am. Chem. Soc., 2007, 129, 11814–11820 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. W. M. Jr, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- MacroModel, version 9.1, Schrödinger, LLC, New York, 2005.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. J. A. Montgomery, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision D.01), Gaussian, Inc., Pittsburgh, PA, 2004 Search PubMed.
- R. Bossart, O. V. Boyarkin, A. A. Makarov and T. R. Rizzo, J. Chem. Phys., 2007, 126, 054302–054307 CrossRef CAS.
- L. C. Snoek, E. G. Robertson, R. T. Kroemer and J. P. Simons, Chem. Phys. Lett., 2000, 321, 49–56 CrossRef CAS.
- L. C. Snoek, R. T. Kroemer, M. R. Hockridge and J. P. Simons, Phys. Chem. Chem. Phys., 2001, 3, 1819–1826 RSC.
- Y. Inokuchi, Y. Kobayashi, T. Ito and T. Ebata, J. Phys. Chem. A, 2007, 111, 3209–3215 CrossRef CAS.
- N. Kobko and J. J. Dannenberg, J. Phys. Chem. A, 2003, 107, 6688–6697 CrossRef CAS.
- N. Kobko and J. J. Dannenberg, J. Phys. Chem. A, 2003, 107, 10389–10395 CrossRef CAS.
- R. Wieczorek and J. J. Dannenberg, J. Am. Chem. Soc., 2003, 125, 14065–14071 CrossRef CAS.
- N. S. Myshakina and S. A. Asher, J. Phys. Chem. B, 2007, 111, 4271–4279 CrossRef CAS.
| This journal is © the Owner Societies 2009 |