Recent development of reactions with α-diazocarbonyl compounds as nucleophiles
Yan
Zhang
ab and
Jianbo
Wang
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS), Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry, Peking University, Beijing 100871, China
bThe State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 354 Fenglin Lu, Shanghai 200032, China. E-mail: wangjb@pku.edu.cn; Fax: +8610-6275-1708
First published on 22nd July 2009
Abstract
The nucleophilic addition reaction of diazo-compound derived anions or enolates with electrophilic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds have been summarized. The subsequent reactions of the addition products demonstrate diverse reactivities of the diazocarbonyl compounds bearing other functional groups and their potential in organic synthesis. Asymmetric catalysis is also possible with diazocarbonyl compounds as nucleophiles .
O and CN bonds have been summarized. The subsequent reactions of the addition products demonstrate diverse reactivities of the diazocarbonyl compounds bearing other functional groups and their potential in organic synthesis. Asymmetric catalysis is also possible with diazocarbonyl compounds as nucleophiles .
 Yan Zhang | Yan Zhang received her PhD degree in 2002 from Lanzhou University. She continued her research as a postdoctoral fellow in Hong Kong University of Science and Technology (2002–2004), University of Innsbruck & Leibniz Institute of Surface Modification (IOM) e. V. (2004–2005), University of Missouri-St. Louis (2005–2006), and Auburn University (2006–2008). She is currently an associate professor in Peking University since 2008. Her research focuses on the application of transition metal complexes of N-heterocyclic carbenes and synthesis of small biological compounds. |
 Jianbo Wang | Jianbo Wang received his BS degree from Nanjing University of Science and Technology in 1983, and his PhD from Hokkaido University (under the supervision of Prof. H. Suginome) in 1990. He was a postdoctoral associate at the University of Geneva from 1990 to 1993 (with Prof. C. W. Jefford), and University of Wisconsin–Madison from 1993 to 1995 (with Prof. H. E. Zimmerman and F. A. Fahien). He began his academic career at Peking University in 1995. His research interests include catalytic metal carbene transformations. |
1. Introduction
α-Diazocarbonyl compounds have been extensively studied over many years. One of the most important applications of α-diazocarbonyl compounds is for the generation of metal carbene species. In the reaction of diazo compounds with transition metal catalysts the negatively polarized diazo carbon interacts with the empty orbital of the transition metal, followed by dinitrogen extrusion. The electrophilic metal carbene species thus generated have diverse reactivities, which can typically undergo cyclopropanations, X–H (X = C, O, S, N, etc.) bond insertions, 1,2-shift and ylide formations. Some of these reactions have been well established as standard synthetic methodologies.1
Diazo compounds are ambiphilic reagents. As shown in Scheme 1, from the resonance structure B, C, D of the diazo group it is obvious that the carbon, to which the diazo group is attached, has a partial negative charge and thus is nucleophilic .2Electrophiles usually attack at the carbon atom, while the terminal nitrogen of the diazo group is attacked by nucleophiles . Although in general diazo compounds decompose rapidly when treated with acids, they are relatively stable under basic conditions. Thus it is possible to generate the anion E which bears a diazo group through base-promoted deprotonation of acyl diazomethane. This type of anion is highly reactive and can add to CO or CN bond efficiently. In this nucleophilic addition, the diazo group remains unchanged, which allows further transformations.
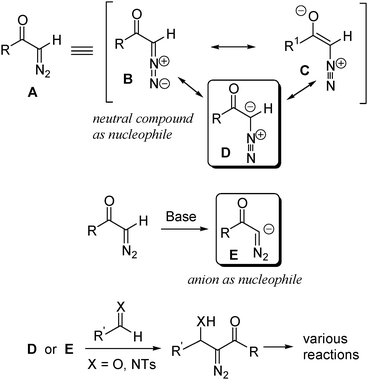 | ||
| Scheme 1 | ||
Compared with acyl diazomethanes, α-diazo β-ketoesters F are more stable. For these diazo compounds, transition metal catalysis usually requires high temperature and long reaction time. If α-diazo β-ketoesters are treated with strong base, deacylation may occur from G to afford the anion of diazoacetate H. On the other hand, it is possible to generate Ti or Si enolates I from α-diazo β-ketoesters. The enolates thus generated can undergo CO, CN additions, as well as Michael additions. Similarly, the addition product J retains the diazo functionality, which allows further transformations (Scheme 2).
 | ||
| Scheme 2 | ||
The nucleophilic addition with diazo compounds as nucleophiles has been known for a long time. Some of these reactions have been developed into useful methods in organic synthesis. For example, the homologation of ketones with diazoalkanes is the nucleophilic addition of diazoalkenes to a carbonyl group followed by 1,2-alkyl migration (Buchner–Curtius–Schlotterbeck reaction).3 This reaction is particularly useful in one-carbon ring expansion of cyclic ketones with diazoalkanes (eqn (1)). Another example is the Roskamp reaction in which the Lewis acid-catalyzed addition of α-acyl diazomethane is followed by a 1,2-H shift and extrusion of dinitrogen, affording β-keto carbonyl compounds (eqn (2)).4
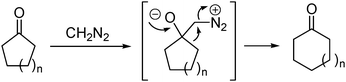 | (1) |
 | (2) |
More recently, asymmetric catalysis with diazo compounds as nucleophiles has made some progress. Particularly, high enantioselectivities have been achieved in several catalytic systems of CN addition with acyl diazomethanes. These new developments have aroused the interest in the chemistry of these “old” compounds. In this review, we will summarize the most recent developments in this area, which will include: (1) nucleophilic addition by acyl diazomethanes; (2) nucleophilic addition by α-diazo β-ketoester derived enolates ; (3) asymmetric nucleophilic addition with diazo compounds; (4) α-diazo carbonyl compounds as nucleophiles in Pd-catalyzed reactions.
2. Nucleophilic addition by acyl diazomethanes
The traditional method of nucleophilic addition of an acyl diazomethane to an aldehyde or ketone requires deprotonation of the acyl diazomethane to generate highly reactive anion species. The deprotonation is usually achieved by the treatment of the acyl diazomethane at low temperature with strong base, such as butyllithium, lithium diisopropyl amide (LDA), sodium hydride, potassium hexamethyl disilyl amide (KHMDS), or NaOH. Although the anion species thus generated can efficiently add to ketones or aldehydes to afford α-diazo-β-hydroxy carbonyl compounds, the strong basic reaction conditions make these reactions less attractive for synthetic organic chemists. The use of organometallic derivatives of diazocarbonyl compounds enables the same transformation under milder conditions.5 Moody and Morfitt report that ethyl diazoacetate (EDA) can be converted to zinc EDA derivatives, which subsequently react smoothly with aldehydes to afford α-diazo-β-hydroxy esters in reasonable yields that are comparable or better than those obtained from other methods.6 Sarabia and López-Herrera later utilized this diethylzinc approach to the reaction of EDA with aldehyde sugars, leading to the improvement of yields with shorter reaction times.7We have recently developed more straightforward reaction conditions for this nucleophilic addition. With a catalytic amount of 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU), the acyl diazomethanes can add to aldehydes at room temperature, giving the expected addition products 3 in moderate to good yields (eqn (3)).8 Furthermore, it is found that this reaction can be carried out in H2O, affording the addition products in comparable yields.9
 | (3) |
Under the same reaction with DBU catalysis, the addition of acyl diazomethanes to N-tosylimines 4 also works well to afford the corresponding β-tosylamino α-diazo carbonyl compounds 5 in good yields (eqn (4)).8,10 Since the basicity of DBU is not enough to deprotonate the acyl diazomethane, the reaction must occur through direct nucleophilic addition of diazo compound to CO or CN bonds, followed by proton transfer.
 | (4) |
Kantam et al. have recently reported that Mg/La mixed oxide could promote the aldol-type reaction of EDA with aldehyde and N-tosylimine (eqn (5)). The reaction can be carried out in water. It was suggested that the EDA anion was formed through deprotonation by the mixed oxide.11
 | (5) |
The same research group further developed polymer-supported imidazonium salt 9 as the catalyst in the nucleophilic addition of EDA to aldehydes (eqn (6)). The reaction could be carried out in water. The catalyst could be separated by simple filtration, recovered and reused several times without distinct loss of activity.12
 | (6) |
Contrary to the direct addition under basic conditions, a different approach is to activate the ketone or aldehyde with Lewis acid, as shown in Scheme 3. Under such conditions, the dinitrogen extrusion usually occurs with the nucleophilic addition affording 1,3-dicarbonyl compounds as the products. This type of reactions has been an efficient method to prepare 1,3-dicarbonyl compounds, as has been represented by the Roskamp reaction.4
 | ||
| Scheme 3 | ||
Under certain conditions, the Lewis acid or protic acid catalyzed addition of diazoester to CO or CN bonds may proceeds with retention of the diazo functionality. This makes possible the asymmetric catalysis of C–C bond formation with chiral acids (vide infra).
The β-hydroxyl α-diazo carbonyl compounds 10 obtained by the nucleophilic addition can further undergo a variety of transformations. For instance, catalysis with a transition metal complex such as Rh2(OAc)4 gives 1,2-hydride shift products 11, which rearrange to 1,3-dicarbonyl compounds 12 (eqn (7)). This is the same transformation as the Lewis acid-promoted reaction of diazo compound with aldehydes.
 | (7) |
Recently, we have found that CuSO4 can efficiently catalyze the reaction of β-hydroxy α-diazo carbonyl compounds 13 in water, giving the 1,3-dicarbonyl compounds 14 in high yields (eqn (8)). In combination of our DBU-catalyzed addition of acyl diazomethane to aldehydes, this two-step transformation from aldehyde and acyl diazomethane constitutes a ‘green’ and convenient preparative method for β-keto esters.13
 | (8) |
The β-hydroxyl group of the addition products can be further transformed into other groups with the diazo functionality kept intact. Subsequent transition metal catalyzed reaction may result in a different reaction pattern of the diazo compounds. For example, when the β-hydroxy is converted to β-acetoxy group, the Rh(II)-catalyzed reaction of the resultant β-acetoxy α-diazo carbonyl compounds 16 results in 1,2-acetoxy group migration, rather than 1,2-hydride migration (Scheme 4).14
 | ||
| Scheme 4 | ||
We have systematically studied the effects of β-substituents on the 1,2-migratory aptitude of Rh(II) carbenes. It is revealed that although 1,2-H migration is predominant in general, the β-substituent may override this migratory preference.10,15 This property could be useful in organic synthesis, as demonstrated by the Rh(II)-catalyzed reaction of β-trimethylsiloxy α-diazo esters 18 (Scheme 5).9 The predominant 1,2-aryl group migration provides a useful approach to prepare β-hydroxy α-arylacrylates 19. The preference for 1,2-aryl migration in these cases can be reasonably interpreted by steric effects. The bulky siloxy group promotes the large aryl group to migrate, instead of small hydrogen.
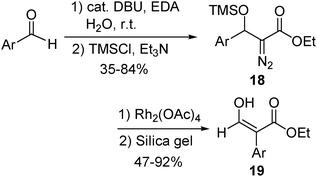 | ||
| Scheme 5 | ||
TfOH-catalyzed reaction of aryl diazoacetate with aldehydes has been studied by Maruoka and co-workers.16 The reaction proceeds through nucleophilic addition of diazo carbon to aldehyde, followed by 1,2-aryl migration rather than 1,2-H migration. The reaction provides a new way to generate all-carbon quaternary centers. Moreover, with aryl diazoacetates bearing chiral auxiliary as substrate, the reaction gives products 21 with high diastereoselectivity (eqn (9)).
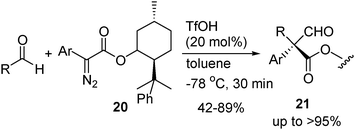 | (9) |
Aggarwal and co-workers studied the reaction of lithium (trimethylsilyl)diazomethane22 with aldehydes (Scheme 6). Under ordinary conditions terminal alkynes are expected as the product. However, if the nucleophilic reaction at −78 °C was followed by addition of MeOH and catalytic Rh2(OAc)4 and then warmed to 0 °C, terminal silyl enol ether 26 could be obtained in good yields. The reaction was suggested to involve Brook rearrangement of the addition intermediate 23 and Rh(II) carbene 1,2-hydride migration.17 In the following paper, a switch of 1,2 C–H migration to C–C migration was observed when Rh2(O2CCF3)4, rather then Rh2(OAc)4, was added as catalyst in the same reaction system. Normally, Rh(II) carbene 1,2 C–C migration is not competitive as compared with 1,2 C–H insertion. This study demonstrates remarkable effect of ligands in controlling the reaction pathway of the Rh(II) carbene.18
 | ||
| Scheme 6 | ||
Recently, Draghici and Brewer have reported a SnCl4-promoted ring fragmentation of γ-siloxy-β-hydroxy-α-diazoesters 28, which can be a useful method to prepare tethered aldehyde ynoates 29 (Scheme 7). The reaction is suggested to proceed through elimination of the Lewis acid-activated β-hydroxy group.19
 | ||
| Scheme 7 | ||
Nenoto investigated the nucleophilic addition of lithiated diazoacetate to benzocyclobutenone (Scheme 8). The initial addition product alkoxide 31 could be transformed into the corresponding alcohol 32, which could be isolated in high yield when the reaction was quenched at −78 °C. When the alcohol 32 was further treated with LDA at −78 °C and then warmed to room temperature, diazepine product 35 was obtained in good yield. The formation of diazepine is attributed to oxy-anion accelerated electrocyclic ring-opening, which is followed by 8π electrocyclization. Diazepine could be obtained directly in the reaction of lithiated diazoacetate to benzocyclobutenone by warming the reaction to room temperature upon the completion of nucleophilic addition. The overall transformation is a net insertion of diazomethane unit into cyclobutenone.20
 | ||
| Scheme 8 | ||
The homologation of cyclic ketones with diazo alkanes is a widely utilized approach in organic synthesis. A recent development of this reaction is the realization of a Sc(III)-catalyzed process with substituted diazomethanes. Unlike the acyl-substituted diazomethanes, alkyl or aryl-substituted diazomethane derivatives are highly unstable. Consequently, their application in organic synthesis is rather limited as compared with their acyl substituted counterparts. With the recent development of several mild protocols to prepare non-stabilized diazoalkanes,21 their reactions as diazonucleophiles are revisited. Moebius and Kingsbury have reported that with Sc(III) catalysts, the ring expansion of cyclic ketones with a variety of alkyl- or aryl-substituted diazomethanes such as 37 proceed well at room temperature (eqn (10)).22
 | (10) |
Very recently, Maruoka and co-workers reported a BF3·OEt2-catalyzed six-membered ring expansion with an α-alkyl diazoacetate.23 The reaction generates seven-membered rings having an all-carbon quaternary center in highly stereoselective manner, as demonstrated by the formation of 41 from 39 and 40 (eqn (11)).
 | (11) |
Nucleophilic addition of deprotonated EDA to N-tert-butylsulfonyl cycloketimines occured to afford addition products, which can be converted into cyclic enamines viaRh2(OAc)4-catalyzed ring expansion (Scheme 9).24
The acid-catalyzed reaction of acyl diazomethane with imines led to the formation of aziridine products (aza-Darzens reaction). This is an alternative method for nitrogen transfer to alkenes as compared to the common metal nitronoid process. Johnston and co-workers discovered that TsOH was a highly efficient Brønsted acidcatalyst that catalyzed the direct aza-Darzens reaction, affording aziridines with high cis selectivity (Scheme 10).25
 | ||
| Scheme 9 | ||
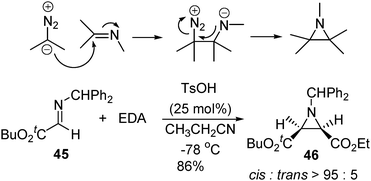 | ||
| Scheme 10 | ||
The nucleophilic addition of diazo phosphonate 47 to aldehydes under basic conditions is a valuable way to synthesize terminal and internal alkynes (Seyforth–Gilbert reaction).26 The reaction is driven by the interaction of the oxygen anion and the PO double bond of the nucleophilic addition intermediate48 (Scheme 11). This reaction has been widely used in organic synthesis. More recently, milder reaction conditions have been developed by using dimethyl-2-oxopylphosphonate, from which the diazophosphonate anion was generated by base-promoted deacetylation.27
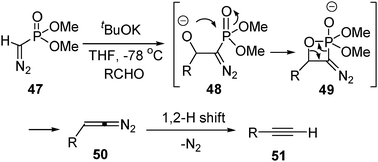 | ||
| Scheme 11 | ||
The nucleophilic addition of α-diazophosphonate 47 to aryl N-tosylimine 4 in the presence of catalytic DBU has been studied in our group (Scheme 12). The reaction afforded α-aryl β-(N-tosyl)amino phosphonate derivatives 52 in good yields as stable products. These addition products were further subjected to TsOH-catalyzed diazodecomposition to yield α-aryl β-(N-tosyl)enamino phosphonates 53 through 1,2-aryl migration. The Rh(II)-catalyzed reaction of 52 afforded a mixture of 1,2-aryl and 1,2-H migration products.28
 | ||
| Scheme 12 | ||
3. Nucleophilic addition by α-diazo β-ketoester derived enolates
β-Keto α-diazocarbonyl compounds are more stable than acyl diazomethanes and enolates can be generated from them. Calter and co-workers reported the nucleophilic addition to aldehydes of Ti(IV) enolate 55 that was derived from β-keto α-diazocarbonyl compounds.29 However, the reaction of the same Ti(IV) enolate with less reactive ketones was found to be very sluggish. To enhance the reactivity of the carbonyl group, one equivalent of Ti(OiPr)4 was introduced to the reaction system, and under such conditions the nucleophilic addition occurred smoothly and afforded the expected alcohol in reasonable yields.30 The addition product could be dehydrated with (CF3CO)2O–Et3N at room temperature, affording α,β-unsaturated diazocarbonyl compounds 56. Subsequent Rh2(acam)4-catalyzed intramolecular C–H insertion yielded bicyclic fused cyclopentenone derivatives 57 in moderate yields (Scheme 13). This three-step operation constitutes an efficient approach to bicyclic fused cyclopentenone derivatives, which are important structural units of many natural products. | ||
| Scheme 13 | ||
The nucleophilic addition of Ti(IV) enolates to α,β-unsaturated carbonyl compounds such as 58 inevitably meets selectivity problems, namely 1,2- vs. 1,4-addition. An interesting Lewis-acid additive effect was observed in this type of nucleophilic addition. In the reaction of enone 58 with Ti(IV) enolate 55, a mixture of 1,2- and 1,4- addition products 59 and 60 was obtained in a ratio of 67 : 33 when there was no additive added. Adding one equivalent of Ti(OiPr)4 increased the ratio to 99 : 1. Surprisingly, with TiCl4 as additive under otherwise identical conditions the selectivity was completely reversed to 1 : 99 (Scheme 14). This remarkable Lewis acid-controlled switch of selectivity can be rationalized by a dimeric structure that involves bridging chlorine atoms (Fig. 1).31
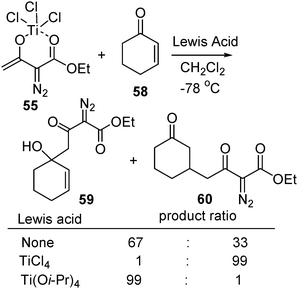 | ||
| Scheme 14 | ||
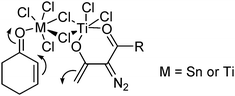 | ||
| Fig. 1 | ||
The addition products 63 of Ti(IV)-enolate 62, derived from β-keto α-diazo carbonyl compounds, to ketones can be further converted into 3-keto tetrahydrofuran derivatives 64 by Rh2(OAc)4-catalyzed diazodecomposition. Alternatively, the same addition products undergo photo-induced Wolff rearrangement/intramolecular nucleophilic addition of keteneintermediate65, affording γ-butyrolactone derivatives 66 (Scheme 15).32
 | ||
| Scheme 15 | ||
Deng and co-workers reported the nucleophilic addition of EDA to alkynyl aldehydes (Scheme 16).33 The hydroxyl group of the addition products 68 could be further converted to β-keto α-diazo carbonyl compounds69 through oxidation and hydrogenation, in which the triple bond was hydrogenated to a cisdouble bond. Finally, Rh2(OAc)4-catalyzed intramolecular C–H insertion gives cyclopentenone derivatives 70 in excellent yields.
 | ||
| Scheme 16 | ||
Silicon enolates derived from β-keto α-diazoesters such as 71 have been successfully used in Mukaiyama-aldol type reaction with aromatic and aliphatic aldehydes. Doyle and co-workers reported that scandium(III) triflate was an efficient catalyst in this reaction for the aldehydes with strong electron withdrawing groups. Rh2(OAc)4-catalyzed reaction of the addition products 70 affords cyclobutanone derivatives 73 and 74 (Scheme 17). The predominant 1,4 C–H insertion is attributed to the activation effect of the OTBS group. Furthermore, with the strategy of using Lewis-acid activation as the case of addition to ketones, the nucleophilic addition of β-keto α-diazoester-derived enolates can be extended to the reaction with imines.34
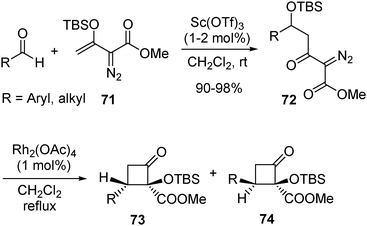 | ||
| Scheme 17 | ||
Catalytic Mukaiyama–Michael type addition of methyl 3-(trialkylsilyoxy)-2-diazo-3-butenoate 71 to α,β-unsaturated enones 75 has been reported by Doyle and co-workers. In this reaction, zinc(II) triflate was found to be a suitable catalyst, and under the optimized reaction conditions the Mukaiyama–Michael addition products 76 were obtained in excellent yields for a wide range of enone substrates under the optimized reaction conditions (Scheme 18).35
 | ||
| Scheme 18 | ||
The reaction of Ti(IV)-enolates derived from β-keto α-diazo carbonyl compounds with N-tosylimine was found very sluggish. Similar to the reaction of Ti(IV)-enolates with ketones, it was found that through activation of N-tosylimine with one equivalent of TiCl4, the reaction with Ti(IV)-enolates occurred smoothly, affording δ-(N-tosyl)amino α-diazocarbonyl compounds 78 in good yields (Scheme 19). Subsequent Rh2(OAc)4-catalyzed reaction of the addition products led to the formation of pyrrole derivatives 80, presumably through elimination of the initially formed intramolecular N–H insertion products 79. Similarly, irradiation of the addition products 78 with a high-pressure Hg lamp resulted in Wolff rearrangement, affording ketene intermediates 81, which were trapped by an internal N-tosyl-protected amino group to finally give γ-lactam derivatives 82.36
 | ||
| Scheme 19 | ||
4. Asymmetric nucleophilic addition with diazo compounds
Asymmetric C–C bond formation has been one of the most important topics in organic synthesis over many years. Aldol reaction and Michael addition are the two powerful and efficient C–C bond-forming reactions and their asymmetric catalysis has attracted great attentions. Since the α-diazocarbonyl compounds can serve as nucleophiles in aldol-type and Michael-type reactions, it would be a natural extension to further develop asymmetric reactions with these diazo group-containing nucleophiles .Thus we proceeded to investigate the reaction of EDA with aldehydes under chiral Lewis acid catalysis. After screening a series of catalysts, including B(III), Ti(IV) and Al(III) complexes, and various BINOL derived chiral ligands, it was concluded that a Zr(IV) catalyst, prepared from Zr(OtBu)4 and 2.2 equivalents of (S)-6,6-Br2-BINOL 84 was effective for the addition reaction, giving the products 83 with moderate to good enantioselectivity (Scheme 20). Doubleactivation of the aldehyde substrate and the EDA by the chiral Zr(IV) complex has been proposed to account for the enantioselectivity.37
 | ||
| Scheme 20 | ||
Nishida and co-workers then developed a reaction system with a chiral phase-transfer catalyst for the aldol-type reaction of aldehydes with tert-butyl diazoacetate. By using a cinchonidinium salt as a chiral catalyst, a similar level of enantioselectivity has been achieved (Schemes 21).38
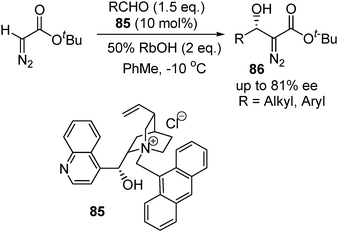 | ||
| Scheme 21 | ||
More recently, Trost and co-workers reported a Mg-catalyzed system for the direct aldol addition of EDA to aldehydes. With their chiral prophenol ligand 87 and a diol additive, the reaction affords the addition products with enantioselectivity up to 98% ee (Scheme 22).39
 | ||
| Scheme 22 | ||
The direct aldol addition product can be further converted into synthetically useful compounds. The diazo group can be oxidized with dimethyl dioxirane (DMD) to a keto group, which is subsequently reduced with NaBH4 to a hydroxyl group with high anti selectivity.40 Thus the overall transformation from EDA and aldehydes is a three-step synthesis of anti α,β-dihydroxy esters.
The corresponding asymmetric addition of diazo group-containing nucleophiles to CN bonds was initially attempted by the chiral auxiliary approach. α-Diazocarbonyl compounds 89 tethered with chiral auxiliaries were synthesized and their reaction with N-tosylbenzaldimine was examined. Diazo amides tethered with Evans’ chiral oxazolidinone auxiliary afforded addition products 90 with good diastereoselectivity (Scheme 23). The nucleophilic addition proceeded with high diastereoselectivity over a wide range of aromaticN-tosylimines. It was suggested that the rigidity of the amide bond in the deprotonated diazoamide was responsible for the observed high diastereoselectivities, as shown in Fig. 2. The diazo group of the addition products 90 were oxidized with dimethyldioxirane (DMD) after removal of the chiral auxiliary. Subsequent reduction of the oxo group with NaBH4 produced anti-α-hydroxy-β-amino esters 91 and 92 with high diastereoselectivities, while Pd/C-catalyzed hydrogenation afforded syn-α-hydroxy-β-amino esters.41
 | ||
| Scheme 23 | ||
 | ||
| Fig. 2 | ||
The next challenge for this reaction would be asymmetric catalytic CN bond addition. This has been recently reported by Terada et al. with chiral binaphthol monophosphoric acid 93 (Scheme 24),42 and by Maruoka with chiral dicarboxylic acid 94 (Scheme 25). In both cases, high enantioselectivities have been achieved.43
 | ||
| Scheme 24 | ||
 | ||
| Scheme 25 | ||
Catalytic enantioselective aza-Darzens reactions have recently repeated by Maruoka44a and Akiyama.44b In Maruoka’s report, an axially chiral dicarboxylic acidcatalyst is found to be highly efficient in initiating the reaction of N-Boc imines with diazoacetamides 96, affording aziridines with high enantioselectivies (Scheme 26).
 | ||
| Scheme 26 | ||
Asymmetric catalysis in Mukaiyama aldol type reaction with β-keto α-diazoester-derived silicon enolate 98 has been developed by Doyle and co-workers. With AgOTf and (S)-BINAP ligand 99, high enantioselectivity has been achieved (up to 92% ee) (Scheme 27).45
 | ||
| Scheme 27 | ||
The corresponding asymmetric CN bond addition was studied with the chiral auxiliary approach. Mukaiyama type addition with the silyl enol of α-diazoacetate and chiral N-(benzylidene)-p-toluene 100 was first investigated under catalysis with Lewis acids, such as Et2O·BF3, TiCl4, Cu(OTf)2, La(OTf)3 and TBSOTf. However, only TBSOTf showed reaction and provided a low yield of the expected addition product. It was then found that straightforward generation of the enolate with a strong base [lithium bis(trimethylsilyl)amide] LHMDS gave the addition product in good yield with excellent diastereoselectivity. The addition products 102, after converting the N-sulfinyl group to N-butyloxycarbonyl group, could be transferred to 2-oxopyrrolidines 103 and 3-oxopyrrolidines 104 with photo-induced Wolff rearrangement or Rh(II)-catalyzed intramolecular N–H insertion (Scheme 28). The overall transformation constitutes a new route to 5-substituted 2-oxo- and 3-oxopyrrolidines of high enantiomeric purity.46
 | ||
| Scheme 28 | ||
The stereochemical outcome of the nucleophilic addition is consistent with the previous reports of the addition of enolates derived from esters or ketones to N-sulfinyl imines. We could thus conclude that the sense of induction can be similarly predicted by invoking a six-membered Zimmerman–Traxler-type transition state as previously suggested for the similar addition of acetateenolates to N-sulfinyl imines (Fig. 3), in which the chelation of Li+ with the sulfinyl oxygen plays a vital role in controlling the attack of the enolate to the Re face of the imine.
 | ||
| Fig. 3 | ||
5. α-Diazocarbonyl compounds as nucleophiles in Pd-catalyzed reactions
Palladium-catalyzed cross-coupling reactions are among the most powerful methods for the formation of C–C bonds. In general, the Pd-catalyzed cross-coupling involves (1) oxidative addition; (2) transmetalation/nucleophilic substitution/ligand exchange; (3) reductive elimination. In the second step, various carbon nucleophiles derived from active methylene compounds, such as ketones, esters, amides and nitriles, have been applied. However, diazo compounds have not been used as cross-coupling partners in Pd-catalyzed reactions. A recent study from our group has indicated that EDA undergoes efficient Pd-catalyzed cross-coupling with aryl or vinyl iodides (Scheme 29). Remarkably, the diazo functionality remains unchanged in this reaction. Moreover, carbonylation occurs prior to the coupling when the reaction is carried out under an atmosphere of CO, affording β-keto α-diazo carbonyl compounds105 in moderate yields. This reaction represents a novel approach to introduce diazo functionality to organic compounds.47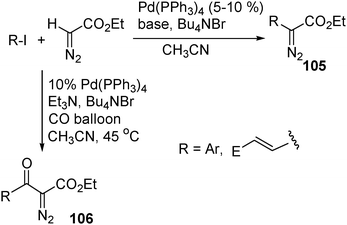 | ||
| Scheme 29 | ||
The proposed mechanism for this reaction involves oxidative addition of Pd(0)Ln with aryl or vinyliodide affording the Pd(II) intermediate 107 followed by EDA coordination to Pd(II) intermediate108 (Scheme 30). Deprotonation followed by reductive elimination afforded the final coupling product. Remarkably, nitrogen extrusion, which is a very facile process for transition metal-complexed diazo compounds, does not occur in this process. This indicates the electron back donation from Pd in intermediate 108 is not as strong as from Rh(II) or Cu(I) in similar complexes, which are known to generate metal carbenes efficiently from diazo compounds.1
 | ||
| Scheme 30 | ||
π-Allylic palladium complexes are important intermediates in palladium-catalyzed reactions. The π-allylic palladium complexes are electrophilic in character and react with various nucleophiles such as amines, alcohols and carbon nucleophiles . In particular, the reactions with carbon nucleophiles have been developed into a valuable method for the formation of C–C bonds. The common carbon nucleophiles used in Pd-catalyzed allylation include anions or enolates derived from carbonyl compounds such as ketones, esters and 1,3-dicarbonyl compounds. We have conceived that it may be possible to employ α-diazocarbonyl compounds as nucleophiles in the reaction of π-allylic palladium complexes. The investigation based on this idea led to the development of a palladium-catalyzed reaction of allylic halides 110 with aryl diazoacetates 111. The reaction resulted in dinitrogen extrusion and generation of carbon–carbon double bonds, affording 1,3-diene derivatives 112 as the products (eqn (12)). The reaction with aryl diazoacetates proceeds well for a variety of allylic bromides and chlorides and also with high stereoselectivity.48
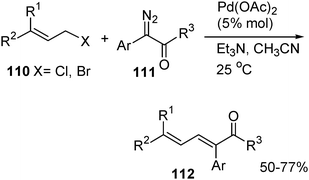 | (12) |
A plausible reaction pathway to account for the formation of dienes is considered as shown in Scheme 29. The reaction is initiated by the reduction of Pd(II) to Pd(0) by the diazo compound, followed by the formation of π-allylpalladium complex 115. For the diazo substrate to interact with the π-allylpalladium complex, there are two possibilities. One is direct attack of the allyl ligand, and the other is the attack of palladium. In the chemistry of π-allylpalladium complexes, it has been suggested that “soft” carbon centered nucleophiles , defined as those derived from conjugate acids whose pKa < 25, usually attack the allyl ligand. On the contrary, “hard” carbon-centered nucleophiles , defined as those derived from conjugate acids whose pKa > 25, attack the metal center of the π-allyl palladium complex, and the substitution occurs through subsequent transmetallation and reductive elimination. The pKa values of conjugate acids of diazoesters and diazoketones have been estimated to be between −5 and −2, respectively.1a Therefore, the α-diazocarbonyl compounds are categorized as “soft” nucleophiles , which attack the allyl ligand. Subsequent deprotonation and dinitrogen extrusion from intermediate 114 lead to the formation of 1,3-diene product 115 (Scheme 31). However, other pathways involving coordination of the diazo substrate to palladium, and formation of a Pd carbene can not be strictly ruled out.
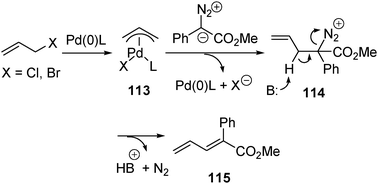 | ||
| Scheme 31 | ||
Conclusions
The transformations shown in this article demonstrates that α-diazocarbonyl compounds can function as nucleophiles under various conditions without decomposition of diazo functionality. This makes possible a variety of transformations other than the traditional process that directly decomposes the diazo functionality with generation of the metal carbeneintermediate. Although in general α-diazo carbonyl compounds are prepared by diazo transfer reactions of the corresponding carbonyl compounds, the addition reactions of nucleophiles containing diazo groups provide alternative and complementary methods to prepare highly functionalized diazo compounds. Further transition metal catalyzed reactions of the addition products demonstrate novel reactivity of these functionalized diazo compounds. Some of these reactions may find synthetic utilities. In particular, the condensation of diazo compounds with CO or CN is very useful because it can introduce a diazo group as well as a hydroxyl or an amino group in a single reaction step. Although further efforts are needed to improve both reactivity and selectivity, the stereocontrol that has been achieved in this type of nucleophilic addition demonstrates the potential of this novel approach to generate C–C bonds with chiral centers.
Acknowledgements
The project is generously supported by NSFC (Grant No. 20832002, 20772003, 20821062), the Ministry of Education of China, and National Basic Research Program of China (973 Program, No. 2009CB825300).Notes and references
- (a) Comprehensive reviews on α-diazocarbonyl compounds, see: T. Ye and M. A. McKervey, Chem. Rev., 1994, 94, 1091 Search PubMed; (b) A. Padwa and D. J. Austin, Angew. Chem., Int. Ed., 1994, 33, 1797 CrossRef; (c) A. Padwa and M. D. Weingarten, Chem. Rev., 1996, 96, 223 CrossRef CAS; (d) M. P. Doyle, M. A. McKervey and T. Ye, in Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, Wiley, New York, 1998 Search PubMed; (e) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS; (f) A. Padwa, J. Organomet. Chem., 2001, 617–618, 3 CrossRef CAS; (g) D. J. Timmons and M. P. Doyle, J. Organomet. Chem., 2001, 617–618, 98 CrossRef CAS; (h) D. M. Hodgson, F. Y. T. M. Pierard and P. A. Stupple, Chem. Soc. Rev., 2001, 30, 50 RSC; (i) H. M. L. Davies and E. G. Antoulinakis, J. Organomet. Chem., 2001, 617–618, 47 CrossRef CAS; (j) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS; (k) G. S. Singh and L. K. Mdee, Curr. Org. Chem., 2003, 7, 1821 CrossRef CAS; (l) H. Heydt, Sci. Synth., 2004, 27, 843 Search PubMed; (m) G. S. Singh, Curr. Org. Synth., 2005, 2, 377 Search PubMed; (n) M. M. Díaz-Requejo and P. J. Pérez, J. Organomet. Chem., 2005, 690, 5441 CrossRef CAS; (o) A. G. H. Wee, Curr. Org. Synth., 2006, 3, 499 Search PubMed; (p) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS.
- For a discussion on nucleophilicity of diazo compounds, see: T. Bug, M. Hartnagel, C. Schlierf and H. Mayr, Chem.–Eur. J., 2003, 9, 4068 Search PubMed.
- (a) E. Buchner and T. Curtius, Chem. Ber., 1989, 18, 2371; (b) C. D. Gutsche, Org. React., 1954, 8, 364.
- (a) C. R. Holmquist and E. J. Roskamp, J. Org. Chem., 1989, 54, 3258 CrossRef CAS; (b) C. R. Holmquist and E. J. Roskamp, Tetrahedron Lett., 1992, 33, 1131 CrossRef CAS; (c) A. Padwa, S. F. Hornbuckle, Z. Zhang and L. Zhi, J. Org. Chem., 1990, 55, 5297 CrossRef CAS; (d) K. Normura, T. Iida, K. Hori and E. Yoshii, J. Org. Chem., 1994, 59, 488 CrossRef CAS.
- J. Fink and M. Regitz, Synthesis, 1985, 569 CrossRef CAS.
- C. J. Moody and C. N. Morfitt, Synthesis, 1998, 1039 CrossRef CAS.
- F. Sarabia and F. J. López-Herrera, Tetrahedron Lett., 2001, 42, 8801 CrossRef CAS.
- N. Jiang and J. Wang, Tetrahedron Lett., 2002, 43, 1285 CrossRef CAS.
- F. Xiao, Y. Liu and J. Wang, Tetrahedron Lett., 2007, 48, 1147 CrossRef CAS.
- N. Jiang, Z. Ma, Z. Qu, X. Xing, L. Xie and J. Wang, J. Org. Chem., 2003, 68, 893 CrossRef CAS.
- M. L. Kantam, V. Balasubrahmanyam, K. B. S. Kumar, G. T. Venkanna and F. Figuerasb, Adv. Synth. Catal., 2007, 349, 1887 CrossRef CAS.
- P. R. Likhar, S. Roy, M. Roy, M. S. Subhas and M. L. Kantam, Catal. Commun., 2009, 10, 728 CrossRef CAS.
- M. Liao and J. Wang, Tetrahedron Lett., 2006, 47, 8859 CrossRef CAS.
- (a) N. Ikota, N. Takamura, S. D. Young and B. Ganem, Tetrahedron Lett., 1981, 22, 4163 CrossRef CAS; (b) F. J. Lopez-Herrera and F. Sarabia-Garcia, Tetrahedron Lett., 1994, 35, 6705 CrossRef; (c) F. J. Lopez-Herrera and F. Sarabia-Garcia, Tetrahedron, 1997, 53, 3325 CrossRef CAS.
- (a) N. Jiang, Z. Qu and J. Wang, Org. Lett., 2001, 3, 2989 CrossRef CAS; (b) W. Shi, N. Jiang, S. Zhang, W. Wu, D. Du and J. Wang, Org. Lett., 2003, 5, 2243 CrossRef CAS; (c) W. Shi, F. Xiao and J. Wang, J. Org. Chem., 2005, 70, 4318 CrossRef CAS; (d) F. Xu, W. Shi and J. Wang, J. Org. Chem., 2005, 70, 4191 CrossRef CAS; (e) F. Xiao and J. Wang, J. Org. Chem., 2006, 71, 5789 CrossRef CAS.
- T. Hashimoto, Y. Naganawa and K. Maruoka, J. Am. Chem. Soc., 2008, 130, 2434 CrossRef CAS.
- V. K. Aggarwal, C. G. Sheldon, G. J. Macdonald and W. P. Martin, J. Am. Chem. Soc., 2002, 124, 10300 CrossRef CAS.
- M. Vitale, T. Lecourt, C. G. Sheldon and V. K. Aggarwal, J. Am. Chem. Soc., 2006, 128, 2524 CrossRef CAS.
- C. Draghici and M. Brewer, J. Am. Chem. Soc., 2008, 130, 3766 CrossRef CAS.
- Y. Matsuya, N. Ohsawa and H. Nemoto, J. Am. Chem. Soc., 2006, 128, 13072 CrossRef CAS.
- (a) M. E. Furrow and A. G. Myers, J. Am. Chem. Soc., 2004, 126, 12222 CrossRef CAS; (b) M. I. Javed and M. Brewer, Org. Lett., 2007, 9, 1789 CrossRef; (c) T. Toma, J. Shimokawa and T. Fukuyama, Org. Lett., 2007, 9, 3195 CrossRef CAS.
- D. C. Moebius and J. S. Kingsbury, J. Am. Chem. Soc., 2009, 131, 878 CrossRef CAS.
- T. Hashimoto, Y. Naganawa and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 6614 CrossRef CAS.
- S. Chen, Y. Zhao and J. Wang, Synthesis, 2006, 1905.
- A. L. Williams and J. N. Johnston, J. Am. Chem. Soc., 2004, 124, 1612 CrossRef.
- (a) D. Seyferth, P. Hilbert and R. S. Marmor, J. Am. Chem. Soc., 1967, 89, 4811 CrossRef CAS; (b) E. W. Colvin and B. J. Hamill, J. Chem. Soc., Chem. Commun., 1973, 151 RSC; (c) J. C. Gilbert and U. Weerasooriya, J. Org. Chem., 1979, 44, 4997 CrossRef CAS.
- (a) G. J. Roth, L. Bernd, S. G. Müller and H. J. Bestmann, Synthesis, 2004, 59 CrossRef CAS; (b) A. G. M. Barrett, B. T. Hopkins, A. C. Love and L. Tedeschi, Org. Lett., 2004, 6, 835 CrossRef CAS.
- Y. Zhao, N. Jiang and J. Wang, Tetrahedron Lett., 2003, 44, 8339 CrossRef CAS.
- (a) M. A. Calter, P. M. Sugathapala and C. Zhu, Tetrahedron Lett., 1997, 38, 3837 CrossRef CAS; (b) M. A. Calter and P. M. Sugathapala, Tetrahedron Lett., 1998, 39, 8813 CrossRef CAS; (c) M. A. Calter and C. Zhu, J. Org. Chem., 1999, 64, 1415 CrossRef CAS.
- G. Deng, X. Tian and J. Wang, Tetrahedron Lett., 2003, 44, 587 CrossRef CAS.
- G. Deng, X. Tian, Z. Qu and J. Wang, Angew. Chem., Int. Ed., 2002, 41, 2773 CrossRef CAS.
- M. Liao, S. Dong, G. Deng and J. Wang, Tetrahedron Lett., 2006, 47, 3927.
- G. Deng, B. Xu and J. Wang, Tetrahedron, 2005, 61, 10811 CrossRef CAS.
- M. P. Doyle, K. Kundu and A. E. Russell, Org. Lett., 2005, 7, 5171 CrossRef CAS.
- Y. Liu, Y. Zhang, N. Jee and M. P. Doyle, Org. Lett., 2008, 10, 1605 CrossRef CAS.
- (a) G. Deng, N. Jiang, Z. Ma and J. Wang, Synlett, 2002, 1913 CAS; (b) C. Dong, G. Deng and J. Wang, J. Org. Chem., 2006, 71, 5560 CrossRef CAS.
- W. Yao and J. Wang, Org. Lett., 2003, 5, 1527 CrossRef CAS.
- (a) S. Arai, K. Hasegawa and A. Nishida, Tetrahedron Lett., 2004, 45, 1023 CrossRef CAS; (b) K. Hesegawa, S. Arai and A. Nishida, Tetrahedron, 2006, 62, 1390 CrossRef CAS.
- B. M. Trost, S. Malhotra and B. A. Fried, J. Am. Chem. Soc., 2009, 131, 1674 CrossRef CAS.
- M. Liao, W. Yao and J. Wang, Synthesis, 2004, 16, 2633.
- Y. Zhao, Z. Ma, X. Zhang, Y. Zou, X. Jin and J. Wang, Angew. Chem., Int. Ed., 2004, 43, 5977 CrossRef CAS.
- D. Uraguchi, K. Sorimachi and M. Terada, J. Am. Chem. Soc., 2005, 127, 9360 CrossRef CAS.
- T. Hashimoto and K. Maruoka, J. Am. Chem. Soc., 2007, 129, 10054 CrossRef CAS.
- (a) T. Hashimoto, N. Uchiyama and K. Maruoka, J. Am. Chem. Soc., 2008, 130, 14380 CrossRef CAS; (b) T. Akiyama, T. Suzuki and K. Mori, Org. Lett., 2009, 11, 2445 CrossRef CAS.
- K. Kundu and M. P. Doyle, Tetrahedron: Asymmetry, 2006, 17, 574 CrossRef CAS.
- C. Dong, F. Mo and J. Wang, J. Org. Chem., 2008, 73, 1971 CrossRef CAS.
- C. Peng, J. Cheng and J. Wang, J. Am. Chem. Soc., 2007, 129, 8708 CrossRef CAS.
- S. Chen and J. Wang, Chem. Commun., 2008, 4198 RSC.
| This journal is © The Royal Society of Chemistry 2009 |