
Recent development of reactions with α-diazocarbonyl compounds as nucleophiles
Abstract
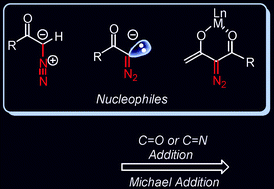
The ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds have been summarized. The subsequent reactions of the addition products demonstrate diverse reactivities of the diazocarbonyl compounds bearing other functional groups and their potential in organic synthesis. Asymmetric catalysis is also possible with diazocarbonyl compounds as
O and CN bonds have been summarized. The subsequent reactions of the addition products demonstrate diverse reactivities of the diazocarbonyl compounds bearing other functional groups and their potential in organic synthesis. Asymmetric catalysis is also possible with diazocarbonyl compounds as
 Please wait while we load your content...
Please wait while we load your content...

