Nucleoside H-boranophosphonates: a new class of boron-containing nucleotide analogues†
Renpei
Higashida
,
Natsuhisa
Oka
,
Toshihide
Kawanaka
and
Takeshi
Wada
*
Department of Medical Genome Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Bioscience Building 702, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8562, Japan. E-mail: wada@k.u-tokyo.ac.jp; Fax: +81 4 7136 3612; Tel: +81 4 7136 3612
First published on 19th February 2009
Abstract
A study on the synthesis of nucleosideH-boranophosphonates, a new class of nucleotide analogues having a P→BH3 and a P–H group, via condensation of the corresponding nucleosides with H-boranophosphonate derivatives is described.
Chemical modifications of natural nucleotides and oligonucleotides by replacing the non-bridging oxygen atoms of their phosphate groups with other elements or substituents (e.g., sulfur atom) have been widely used, especially for the development of nucleic acid-based drug candidates.1,2Nucleoside boranophosphates, in which a non-bridging oxygen atom of the phosphate group is replaced with a BH3 group, are one of the most promising candidates owing to their significant stability to nucleases and high lipophilicity, which may save the need for an elaborate delivery system.3 Their low cytotoxicity has also been suggested.4 In addition, oligonucleoside boranophosphates have shown promising RNA interference activity,5 and may also be useful as target-specific 10B carriers for boron-neutron capture therapy (BNCT).6
These studies on the nucleoside boranophosphates have generated interest in other nucleotide analogues containing a P→BH3 group. However, only a limited number of such analogues are available7,8 with the methods developed for the synthesis of nucleoside boranophosphates.9–11
To expand the availability of the boron-containing nucleotide analogues, we designed a novel nucleotide analogue having a P→BH3 and a P–H group (nucleosideH-boranophosphonate 1, Fig. 1) with the expectation that it would work as a versatile precursor of a variety of boron-containing P-modified nucleotide analogues via the activation of the P–H function by bases or transition metals followed by reactions with electrophiles.12 The P→BH3 moiety may also work as a modifiable site via deboronation and subsequent reaction with electrophiles, which would expand the availability of P-modified nucleotide analogues.12a,13
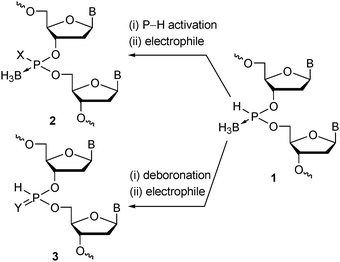 | ||
| Fig. 1 Strategy for synthesis of various P-modified oligonucleotide analogues 2 and 3 by substitution of P–H and P–B bonds of oligonucleoside H-boranophosphonates 1, respectively. B = nucleobase. | ||
To the best of our knowledge, there are only two reports on the synthesis of H-boranophosphonate derivatives.12c,14 Centofanti reported the synthesis of dimethyl H-boranophosphonate from dimethyl phosphonite decades ago.14 In addition, the synthesis of silylH-boranophosphonate derivatives and their applications as precursors of alkylboranophosphonates were reported by Montchamp et al. very recently.12c However, it is difficult to apply these methods to the synthesis of more functionalized molecules, such as nucleotide analogues.
We anticipated that an H-boranophosphonate derivative having at least one free P–O− function would undergo condensation reactions with the hydroxy group of nucleosides to give the nucleosideH-boranophosphonates under mild reaction conditions. Based on this idea, we synthesized inorganicH-boranophosphonate as a monopyridinium salt (Scheme 1, 7). Phosphinic acid4 was treated with N,O-bis(trimethylsilyl)benzamide15 to afford bis(trimethylsilyl) phosphonite 5, which was then boronated by treatment with BH3·THF. Montchamp et al. have also synthesized bis(trialkylsilyl) H-boranophosphonates and alkyl trialkylsilyl H-boranophosphoates via the corresponding phosphonite derivatives, though the bis(TMS) derivative 5 was not used for further applications due to its instability. In contrast, we used the silylation as a transient protection and the intermediate 6 was desilylated by treatment with MeOH and pyridine to synthesize 7. The resultant pyridinium H-boranophosphonate 7 was stable to air oxidation and water, and can be stored for at least several months at −30 °C without decomposition.
 | ||
| Scheme 1 Synthesis of pyridinium H-boranophosphonate 7. | ||
Pyridinium H-boranophosphonate 7 was applied to the synthesis of nucleosideH-boranophosphonate derivatives. Firstly, nucleoside 5′-H-boranophosphonates were synthesized by condensation of 7 with appropriately protected nucleosides having a 5′-hydroxy group (8a–e, Table 1). When the reaction was carried out under conditions similar to those used for the boranophosphorylation of nucleosides,11a the isolated yields of the desired thymidine 5′-H-boranophosphonate 9a were low mainly due to the formation of a 5′,5′-dinucleoside H-boranophosphonate diester (entries 1 and 2). In sharp contrast, condensations promoted by Bop-Cl in pyridine afforded the desired nucleoside 5′-H-boranophosphonates 9a–e in excellent yields without observable side-reactions (entries 3–7).

| Entry | 8 | BPRO | R2 | R3 | Reagents and conditions | Yield of 9a–e (%) |
|---|---|---|---|---|---|---|
| a TEAB = triethylammonium bicarbonate; BPRO = (protected) nucleobase; Thbz = N3-benzoylthymin-1-yl; DMTr = 4,4′-dimethoxytrityl; Piv-Cl = pivaloyl chloride; NT = 3-nitro-1H-1,2,4-triazole; Bop-Cl = bis(2-oxo-3-oxazolidinyl)phosphinic chloride; Pac = phenoxyacetyl. | ||||||
| 1 | a | Thbz | H | DMTr | 7 (1.5 equiv.), Piv-Cl (1.5 equiv.), iPr2NEt (3 equiv.), NT (1.5 equiv.), MeCN, rt, 3 h | 39 |
| 2 | a | Thbz | H | DMTr | 7 (2 equiv.), Bop-Cl (2 equiv.), iPr2NEt (4 equiv.), NT (2 equiv.), MeCN, rt, 2 h | 43 |
| 3 | a | Thbz | H | DMTr | 7 (1.2 equiv.), Bop-Cl (1.2 equiv.), Py, rt, 2 h | 95 |
| 4 | b | Th | H | DMTr | 7 (1.6 equiv.), Bop-Cl (1.6 equiv.), Py, rt, 1 h | 95 |
| 5 | c | Th | H | Bz | 7 (1.2 equiv.), Bop-Cl (1.2 equiv.), Py, rt, 30 min | 92 |
| 6 | d | Th | H | Pac | 7 (1.2 equiv.), Bop-Cl (1.2 equiv.), Py, rt, 40 min | 90 |
| 7 | e | Ur | OPac | Pac | 7 (2 equiv.), Bop-Cl (2 equiv.), Py, rt, 2 h | 98 |
Next, we attempted to synthesize fully-deprotected nucleoside 5′-H-boranophosphonates, which would be interesting as a new class of boron-containing nucleoside monophosphate analogues. Firstly, the detritylation of 3′-O-DMTr-thymidine 5′-H-boranophosphonate 9b was attempted by treatment with an 80% AcOH aqueous solution. However, decomposition of the H-boranophosphonate moiety (ca. 70%) was observed after 30 min (Table 2, entry 1). It is probably due to the reaction of the liberated DMTr+ with the BH3 group of the H-boranophosphonate moiety as observed in the case of boranophosphates.10b,11a,16 In contrast, the H-boranophosphonate monoesters were stable under conventional ammonolysis conditions, and the nucleoside 5′-H-boranophosphonates 10a,b were isolated in good yields (entries 2 and 3).

The reaction conditions optimized for the 5′-boranophosphonylation were also applicable to the synthesis of nucleoside 3′-H-boranophosphonates. The condensation of 5′-O-DMTr-nucleosides 11a,b with 7 in pyridine in the presence of Bop-Cl afforded the desired nucleoside 3′-H-boranophosphonates 12a,b in excellent yields (Table 3, entries 3 and 4), whereas the reactions promoted by Piv-Cl or Bop-Cl in MeCN in the presence of iPr2NEt and NT resulted in low yields of 12 mainly due to the generation of a 3′,3′-dithymidine H-boranophosphonate derivative (entries 1 and 2).

| Entry | 11 | BPRO | Reagents and conditions | Yield of 12 (%) |
|---|---|---|---|---|
| 1 | a | Thbz | 7 (1.2 equiv.), Piv-Cl (1.5 equiv.), iPr2NEt (3 equiv.), NT (1.5 equiv.), MeCN, rt, 4 h | 41 |
| 2 | a | Thbz | 7 (1.2 equiv.), Bop-Cl (3 equiv.), iPr2NEt (6 equiv.), NT (3 equiv.), MeCN, rt, 1.5 h | 50 |
| 3 | a | Thbz | 7 (1.2 equiv.), Bop-Cl (1.2 equiv.), Py, rt, 1 h | 95 |
| 4 | b | Th | 7 (2 equiv.), Bop-Cl (2 equiv.), Py, rt, 3 h | 82 |
The 5′-O-protected nucleoside 3′-H-boranophosphonates are potentially useful as monomer units to synthesize oligonucleotide analogues having an H-boranophosphonate diester backbone. As we mentioned above, such oligonucleotide analogues would work as precursors of a variety of backbone-modified oligonucleotide analogues through the P–H or P–B modifications. To investigate the potential of the H-boranophosphonates, a dithymidine H-boranophosphonate was synthesized from the 5′-O-DMTr-nucleoside 3′-H-boranophosphonate 12a. The 3′-H-boranophosphonate 12a was allowed to condense with 3′-O-DMTr-N3-benzoyl-thymidine 8a in MeCN in the presence of Bop-Cl and 2,2,6,6-tetramethylpiperidine (Scheme 2, reaction i). The reaction proceeded smoothly at rt. Although a partial decomposition during silica gelcolumn chromatography was observed, the dithymidine H-boranophosphonate diester13a was isolated in a modest yield.
 | ||
| Scheme 2 (i) Synthesis of dithymidine H-boranophosphonates 13a,b, (ii) 5′-O-detritylation of 13b and (iii) conversion of 13a into dithymidine boranophosphorothioate 15 by P–H modification. Reagents and conditions: (i) N3-benzoyl-3′-O-R3-thymidine (0.75 equiv.), Bop-Cl (2.5 equiv.), 2,2,6,6-tetramethylpiperidine (6 equiv.), MeCN, rt, 1 h; (ii) 3% dichloroacetic acid in CH2Cl2–Et3SiH (3 : 1, v/v), rt, 1 min; (iii) S8 (3 equiv.), Et3N (3 equiv.), MeCN, rt, 3 h. | ||
To explore the potential of the nucleosideH-boranophosphonates, we carried out the following two experiments. Firstly, a 3′-O-benzoyl-dithymidine H-boranophosphonate 13b was synthesized as a crude product in a similar manner and treated with 3% dichloroacetic acid in CDCl3–Et3SiH (1 : 1, v/v)11b,16 for 5′-O-detritylation. In contrast to the detritylation of the H-boranophosphonate monoester 9b by treatment with 80% AcOH (Table 2, entry 1), which caused the decomposition of the H-boranophosphonate moiety, the 5′-O-DMTr group of 13b was quantitatively removed without decomposition of the product (Scheme 2, reaction ii). It indicates that a solid-phase synthesis of oligonucleoside H-boranophosphonates via condensation and 5′-O-detritylation is feasible.
Secondly, the possibility of the P–H modification of the H-boranophosphonate diester linkage was investigated by treating the dithymidine H-boranophosphonate 13a with S8 in the presence of Et3N under anhydrous conditions. A 31P NMR analysis of the reaction showed that 13a (δP 135.1, 133.7) was quantitatively converted into the corresponding dithymidine boranophosphorothioate 15 (δP 162.5, 161.0)8a,b within 3 h at rt, and 15 was isolated in good yield (Scheme 2, reaction iii).
In conclusion, nucleosideH-boranophosphonates were synthesized via condensation of the corresponding nucleosides and H-boranophosphonate derivatives. The mild reaction conditions and high efficiency of this method are attractive for further applications to the synthesis of a broad spectrum of H-boranophosphonate derivatives. In addition, conversion of the P–H group of a dinucleoside H-boranophosphonate into a P–S– group demonstrated that the H-boranophosphonate derivatives are potential precursors of a variety of boron-containing oligonucleotide analogues. Further studies on the synthesis of nucleosideH-boranophosphonates and their applications are in progress.
We thank Professor Kazuhiko Saigo (University of Tokyo) for helpful suggestions. This research was supported by a Grant-in-Aid for Young Scientists (B) (No. 20750127) from MEXT Japan (N.O.).
Notes and references
- C. Wilson and A. D. Keefe, Curr. Opin. Chem. Biol., 2006, 10, 607 CrossRef CAS.
- T. Calogeropoulou, A. Detsi, E. Lekkas and M. Koufaki, Curr. Top. Med. Chem., 2003, 3, 1467 CrossRef CAS.
- P. Li, Z. A. Sergueeva, M. Dobrikov and B. R. Shaw, Chem. Rev., 2007, 107, 4746 CrossRef CAS.
- (a) I. H. Hall, B. S. Burnham, K. G. Rajendran, S. Y. Chen, A. Sood, B. F. Spielvogel and B. R. Shaw, Biomed. Pharmacother., 1993, 47, 79 CrossRef CAS; (b) H. Li, C. Hardin and B. R. Shaw, J. Am. Chem. Soc., 1996, 118, 6606 CrossRef CAS.
- (a) A. H. S. Hall, J. Wan, E. E. Shaughnessy, B. R. Shaw and K. A. Alexander, Nucleic Acids Res., 2004, 32, 5991 CrossRef CAS; (b) A. H. S. Hall, J. Wan, A. Spesock, Z. Sergueeva, B. R. Shaw and K. A. Alexander, Nucleic Acids Res., 2006, 34, 2773 CrossRef CAS.
- B. S. Burnham, Curr. Med. Chem., 2005, 12, 1995 CrossRef CAS.
- (a) J. Lin and B. R. Shaw, Chem. Commun., 2000, 2115 RSC; (b) P. Li and B. R. Shaw, Org. Lett., 2002, 4, 2009 CrossRef CAS; (c) P. Li and B. R. Shaw, Chem. Commun., 2002, 2890 RSC; (d) J. Baraniak, R. Kaczmarek and E. Wasilewska, Tetrahedron Lett., 2004, 45, 671 CrossRef CAS; (e) P. Li and B. R. Shaw, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 675 CrossRef CAS; (f) P. Li and B. R. Shaw, J. Org. Chem., 2005, 70, 2171 CrossRef CAS.
- (a) J. Lin and B. R. Shaw, Chem. Commun., 1999, 1517 RSC; (b) J. Lin and B. R. Shaw, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 587 CrossRef CAS; (c) J. Lin and B. R. Shaw, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 1325 CrossRef CAS.
- (a) A. Sood, B. R. Shaw and B. F. Spielvogel, J. Am. Chem. Soc., 1990, 112, 9000 CrossRef CAS; (b) H. A. Brummel and M. H. Caruthers, Tetrahedron Lett., 2002, 43, 749 CrossRef CAS; (c) H. B. McCuen, M. S. Noé, A. B. Sierzchala, A. P. Higson and M. H. Caruthers, J. Am. Chem. Soc., 2006, 128, 8138 CrossRef CAS.
- (a) J. Zhang, T. Terhorst and M. D. Matteucci, Tetrahedron Lett., 1997, 38, 4957 CrossRef CAS; (b) A. P. Higson, A. Sierzchala, H. Brummel, Z. Zhao and M. H. Caruthers, Tetrahedron Lett., 1998, 39, 3899 CrossRef CAS; (c) D. S. Sergueev and B. R. Shaw, J. Am. Chem. Soc., 1998, 120, 9417 CrossRef CAS.
- (a) T. Wada, M. Shimizu, N. Oka and K. Saigo, Tetrahedron Lett., 2002, 43, 4137 CrossRef CAS; (b) M. Shimizu, T. Wada, N. Oka and K. Saigo, J. Org. Chem., 2004, 69, 5261 CrossRef CAS; (c) M. Shimizu, K. Saigo and T. Wada, J. Org. Chem., 2006, 71, 4262 CrossRef CAS.
- (a) T. Imamoto, T. Kusumoto, N. Suzuki and K. Sato, J. Am. Chem. Soc., 1985, 107, 5301 CrossRef CAS; (b) T. Oshiki and T. Imamoto, J. Am. Chem. Soc., 1992, 114, 3975 CrossRef CAS; (c) Y. Belabassi, M. I. Antczak, J. Tellez and J.-L. Montchamp, Tetrahedron, 2008, 64, 9181 CrossRef CAS.
- (a) H. Brisset, Y. Gourdel, P. Pellon and M. Le Corre, Tetrahedron Lett., 1993, 34, 4523 CrossRef CAS; (b) M. Shimizu, K. Tamura, T. Wada and K. Saigo, Tetrahedron Lett., 2004, 45, 371 CrossRef CAS; (c) T. Kawanaka, M. Shimizu and T. Wada, Tetrahedron Lett., 2007, 48, 1973 CrossRef CAS.
- L. F. Centofanti, Inorg. Chem., 1973, 12, 1131 CrossRef CAS.
- T. Wada, A. Mochizuki, Y. Sato and M. Sekine, Tetrahedron Lett., 1998, 39, 7123 CrossRef CAS.
- Z. A. Sergueeva, D. S. Sergueev and B. R. Shaw, Nucleosides, Nucleotides Nucleic Acids, 2001, 20, 941 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra. See DOI: 10.1039/b901045a |
| This journal is © The Royal Society of Chemistry 2009 |