First total syntheses of (−)-macrosphelides J and K and elucidation of their absolute configuration†
Hwayoung
Yun
,
Seung-Mann
Paek
,
Jong-Wha
Jung
,
Nam-Jung
Kim
,
Seok-Ho
Kim
and
Young-Ger
Suh
*
College of Pharmacy, Seoul National University, San 56-1, Sillim-Dong, Gwanak-Gu, Seoul, 151-742, Korea. E-mail: ygsuh@snu.ac.kr; Fax: +82-2-888-0649; Tel: +82-2-880-7875
First published on 15th January 2009
Abstract
First total syntheses of (–)-macrosphelides J and K and their structural elucidation are described.
All thirteen macrosphelides including macrosphelide M, isolated from a strain of Periconia byssoides separated from the sea hare Aplysia kurodai in 2007,1 have been reported during the past two decades.2 These natural macrolides are of great interest both for their excellent biological activities such as potent cell–cell adhesion inhibition2a,b,e and for their unique structural features. In particular, some of these macrocyclic polyketides are considered potential leads for the treatment of various cancers, although their structures have not yet been completely elucidated.3 These all prompted us to initiate studies on the synthesis of macrosphelides, including macrosphelides J and K (Fig. 1).4
 | ||
| Fig. 1 Structures of macrosphelides B, J and K. | ||
(−)-Macrosphelides J and K are structurally unique members of their class, and they were first isolated from the Microsphaeropsis sp. FO-5050 in 1999 by Ōmura et al.5 They consist of three core fragments, including a γ-keto-trans-α,β-unsaturated acid, which is commonly connected via an ester linkage throughout the series. These macrolides also contain five stereogenic centers, of which stereochemistries have yet to be elucidated. Generally, the stereochemistries of the three stereogenic centers corresponding to C-8, C-9 and C-15 of (−)-macrosphelides J and K are known to be identical. Thus, we assumed that the configurations of C-8, C-9 and C-15 of (−)-macrosphelides J and K were also identical to those of other macrosphelides. In addition, we anticipated the configuration of C-3 to be as illustrated in Scheme 1. This configuration was suspected because it has been proposed that macrosphelides J and K are derived from macrosphelide B,3e probably via incorporation of an alkoxy unit to the γ-keto-trans-α,β-unsaturated acid moiety of macrosphelide B,6 which exhibits strong immunosuppressant activity equal to that of rapamycin.7 Regarding the stereochemistry of C-12, we assumed its configuration as shown in the structure of 1 (Scheme 1) on the basis of our preliminary molecular modeling study. The molecular modeling study revealed a favorable addition of the alkoxy unit to the γ-keto-trans-α,β-unsaturated acid moiety from the α-face. Thus, we have executed syntheses of (−)-macrosphelides J (1) and K (2). We herein report the first total syntheses of (−)-macrosphelides J and K and their structural elucidation.
 | ||
| Scheme 1 Retrosynthesis. | ||
Our retrosynthetic approach to the proposed structures of macrosphelides J and K is outlined in Scheme 1. For the syntheses of macrosphelides J and K, we initially applied the strategy previously employed for the synthesis of macrosphelides A and B.4n However, our preliminary studies on the synthesis of macrosphelides J and K revealed that our previous intramolecular nitrile oxide–olefin cycloaddition strategy could not provide the requisite stereochemistry for the C-12 stereogenic center. Thus, we turned our attention to the asymmetric intermolecular nitrile oxide–olefin cycloaddition to install the correct stereochemistry of C-12, as well as a masked form of the labile γ-keto-α-hydroxy acid moiety.
The revised strategy envisioned that both (−)-macrosphelides J and K could be effectively synthesized from the common advanced intermediate 34q by a reductive N–O cleavage of isoxazoline followed by O-alkylation. The key macrocyclic isoxazoline 3 would be synthesized via a modular assembly of the three fragments 4, 6 and 7. The 16-membered macrocyclic skeleton of 3 can be conveniently constructed by Yamaguchi macrolactonization of the corresponding ω-hydroxy acid. A stereoselective nitrile oxide cycloaddition of 8 and 9 would allow to effectively prepare the labile γ-keto-α-hydroxy-ester moiety of 3 in a stable isoxazoline form.8a The optically active γ,δ-dihydroxy-trans-α,β-unsaturated acid moiety 4 could be provided via trans-vinylogous OBO ester anion addition to the appropriate carbonyl system, which was previously developed in our laboratory.9
The synthetic procedure for the isoxazoline fragment 12 from the (S)-2-(4-methoxybenzyloxy)propanal (10)10 is described in Scheme 2. Exposure of the aldehyde10 to hydroxylamine hydrochloride in the presence of sodium acetate afforded the aldoxime8 as a [3 + 2] cycloaddition precursor in 96% yield.11 The pivotal diastereoselective nitrile oxide–olefin cycloaddition of aldoxime8 was performed with the acrylate 9,12 which was prepared on a multi-gram scale from (1R)-(+)-2,10-camphorsultam. The cycloaddition proceeded smoothly to produce the isoxazoline 7 in 78% yield and with high diastereoselectivity (>10 : 1).8b Ti-mediated transesterification of the isoxazoline 7 with allyl alcohol gave the ester 11 in 84% yield and it gave the recovered auxiliary in 90% yield.13 The alcohol12 was finally obtained by an oxidative cleavage of the PMB protected intermediate 11 in 90% yield.
 | ||
| Scheme 2 Stereoselective synthesis of isoxazoline moiety. | ||
Next, the alcohol12 was coupled with (S)-3-(4-methoxybenzyloxy)butanoic acid (6)14 in the presence of the EDCI, providing the ester 13 (Scheme 3). This ester was then deprotected with DDQ to yield the free alcohol5. Similarly, coupling of the alcohol5 and the optically active γ,δ-dihydroxy-trans-α,β-unsaturated acid 44n in the presence of EDCI provided the diester14, which was then deprotected by DDQ treatment to give the free alcohol15. Finally, sequential deallylation of 15 and macrolactonization of the resulting ω-hydroxy acid in a one-pot process by Pd(PPh3)4 and Yamaguchi lactonization, respectively, afforded the key isoxazoline macrolide 3 in 60% yield.15
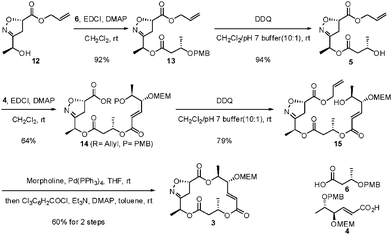 | ||
| Scheme 3 Preparation of common isoxazoline intermediate. | ||
At the stage of conversion of the isoxazoline 3 to the β-hydroxy ketone 16 in the presence the labile hydroxyacrolate moiety, we encountered serious problems upon attempt of N–O bond cleavage of the isoxazoline. We could not obtain the β-hydroxy ketone 16, though we tried a variety of procedures for the reductive N–O bond cleavage such as catalytic Raney-Ni hydrogenation,16a Zn in AcOH reduction,16bSmI2 reduction,16cCuSO4 reduction,16d Mo(CO)6/H2O reduction16e and Mo(CO)3(CH3CN)3/SiO2 reduction.16f Hydrogenolysis of 3 in the presence of Pd/C gave the olefin saturated product in 80% yield. However, we finally obtained the desired β-hydroxy ketone 16 in 64% yield by using Ti(Oi-Pr)4/EtMgBr (Scheme 4).16g
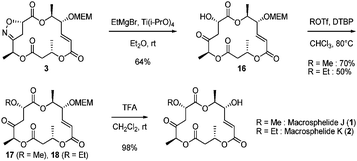 | ||
| Scheme 4 Completion of the synthesis. | ||
For the O-alkylation of the alcohol16, we also examined a variety of alkylation conditions. Our initial attempts included O-methylation under the conditions such as Me2SO4, Ag2O or NaH or KOt-Bu or KHCO3/MeI, TMSCHN2 and O-ethylation under the conditions of Ag2O or NaH or KOt-Bu or KHCO3/EtI. However, each of these resulted in hydrolysis of the ester linkages. Fortunately, the best result was achieved upon treatment of 16 with alkyl trifluoromethanesulfonate (ROTf) in the presence of 2,6-di-tert-butylpyridine (DTBP), which gave the corresponding ethers17 (R = Me) and 18 (R = Et) without the problematic isomerization or epimerization.17 Finally, MEM-deprotection of 17 and 18 by TFA treatment of the resulting alcohol afforded macrosphelides J (1) and K (2). The synthetic macrosphelides J and K exhibited all spectral data identical to those of the authentic natural products.5,18
In summary, we have accomplished the first syntheses of (−)-macrosphelides J (1) and K (2) from ethyl (S)-(−)-lactate in 15 steps. We also confirmed their structures, particularly the stereochemistries of their five stereogenic centers. Considering that the γ-keto-trans-α,β-unsaturated acid moiety of macrosphelide B possibly functions as a Michael acceptor of O-nucleophiles for their biological activity as well as the structural similarity of macrosphelides B and J or K, our syntheses of the proposed structures on the basis of the mechanistic hypothesis are quite important in terms of further studies on the macrosphelides. The total synthesis of other macrosphelides employing this strategy, as well as their biological evaluations, are in progress and these will be published in due course.
This work was supported by the Center for Bioactive Molecular Hybrids, Yonsei University and Seoul R&BD Program.
Notes and references
- T. Yamada, K. Minoura, R. Tanaka and A. Numata, J. Antibiot., 2007, 60, 370–375 CrossRef CAS.
- (a) M. Hayashi, Y.-P. Kim, H. Hiraoka, M. Natori, S. Takamatsu, T. Kawakubo, R. Masuma, K. Komiyama and S. Ōmura, J. Antibiot., 1995, 48, 1435–1439 CAS; (b) S. Takamatsu, Y.-P. Kim, M. Hayashi, H. Hiraoka, M. Natori, K. Komiyama and S. Ōmura, J. Antibiot., 1996, 49, 95–98 CAS; (c) S. Takamatsu, H. Hiraoka, Y.-P. Kim, M. Hayashi, M. Natori, K. Komiyama and S. Ōmura, J. Antibiot., 1997, 50, 878–880 CAS; (d) A. Numata, M. Iritani, T. Yamada, K. Minoura, E. Matsumura, T. Yamori and T. Tsuruo, Tetrahedron Lett., 1997, 38, 8215–8218 CrossRef CAS; (e) S. Ōmura, M. Hayashi and H. Tomoda, Pure Appl. Chem., 1999, 71, 1673–1681 CrossRef CAS; (f) T. Yamada, M. Iritani, M. Doi, K. Minoura, T. Ito and A. Numata, J. Chem. Soc., Perkin Trans. 1, 2001, 3046–3053 RSC; (g) T. Yamada, M. Iritani, K. Minoura, A. Numata, Y. Kobayashi and Y.-G. Wang, J. Antibiot., 2002, 55, 147–154 CAS.
- For medicinal chemistry of macrosphelides, see: (a) Y. Matsuya, T. Kawaguchi, H. Nemoto, H. Nozaki and H. Hamada, Heterocycles, 2003, 59, 481–484 CAS; (b) Y. Matsuya, T. Kawaguchi and H. Nemoto, Heterocycles, 2003, 61, 39–43 CAS; (c) Y. Matsuya, K. Ishihara, N. Funamori, T. Kawaguchi and H. Nemoto, Heterocycles, 2003, 61, 59–63 CAS; (d) K. Ishihara, T. Kawaguchi, Y. Matsuya, H. Sakurai, I. Saiki and H. Nemoto, Eur. J. Org. Chem., 2004, 3973–3978 CrossRef CAS; (e) Y. Matsuya and H. Nemoto, Heterocycles, 2005, 65, 1741–1749 CAS; (f) Y. Matsuya, T. Kawaguchi, K. Ishihara, K. Ahmed, Q.-L. Zhao, T. Kondo and H. Nemoto, Org. Lett., 2006, 8, 4609–4612 CrossRef CAS.
- For total syntheses of macrosphelides, see: (a) T. Sunazuka, T. Hirose, Y. Harigaya, S. Takamatsu, M. Hayashi, K. Komiyama, S. Ōmura, P. A. Sprengeler and A. B. Smith III, J. Am. Chem. Soc., 1997, 119, 10247–10248 CrossRef CAS; (b) Y. Kobayashi, B. G. Kumar and T. Kurachi, Tetrahedron Lett., 2000, 41, 1559–1563 CrossRef CAS; (c) M. Ono, H. Nakamura, F. Konno and H. Akita, Tetrahedron: Asymmetry, 2000, 11, 2753–2764 CrossRef CAS; (d) Y. Kobayashi, G. B. Kumar, T. Kurachi, H. P. Acharya, T. Yamazaki and T. Kitazume, J. Org. Chem., 2001, 66, 2011–2018 CrossRef CAS; (e) H. Nakamura, M. Ono, Y. Shida and H. Akita, Tetrahedron: Asymmetry, 2002, 13, 705–713 CrossRef CAS; (f) Y. Kobayashi and Y.-G. Wang, Tetrahedron Lett., 2002, 43, 4381–4384 CrossRef CAS; (g) H. Nakamura, M. Ono, M. Makino and H. Akita, Heterocycles, 2002, 57, 327–336 CAS; (h) G. V. M. Sharma and C. C. Mouli, Tetrahedron Lett., 2002, 43, 9159–9161 CrossRef CAS; (i) T. Takahashi, S.-i. Kusaka, T. Doi, T. Sunazuka and S. Ōmura, Angew. Chem., Int. Ed., 2003, 42, 5230–5234 CrossRef CAS; (j) Y. Matsuya, T. Kawaguchi and H. Nemoto, Org. Lett., 2003, 5, 2939–2941 CrossRef CAS; (k) S.-i. Kusaka, S. Dohi, T. Doi and T. Takahashi, Tetrahedron Lett., 2003, 44, 8857–8859 CrossRef CAS; (l) T. Kawaguchi, N. Funamori, Y. Matsuya and H. Nemoto, J. Org. Chem., 2004, 69, 505–509 CrossRef CAS; (m) T. Sunazuka, T. Hirose, N. Chikaraishi, Y. Harigaya, M. Hayashi, K. Komiyama, P. A. Sprengeler, A. B. Smith III and S. Ōmura, Tetrahedron, 2005, 61, 3789–3803 CrossRef CAS; (n) S.-M. Paek, S.-Y. Seo, S.-H. Kim, J.-W. Jung, Y.-S. Lee, J.-K. Jung and Y.-G. Suh, Org. Lett., 2005, 7, 3159–3162 CrossRef CAS; (o) G. V. M. Sharma and K. V. Babu, Tetrahedron: Asymmetry, 2007, 18, 2175–2184 CrossRef CAS; (p) B.-L. Wang, Z.-X. Jiang, Z.-W. You and F.-L. Qing, Tetrahedron, 2007, 63, 12671–12680 CrossRef CAS; (q) S.-M. Paek, H. Yun, N.-J. Kim, J.-W. Jung, D.-J. Chang, S. Lee, J. Yoo, H.-J. Park and Y.-G. Suh, J. Org. Chem., 2009, 74, 554–561 CrossRef CAS.
- A. Fukami, Y. Taniguchi, T. Nakamura, M.-C. Rho, K. Kawaguchi, M. Hayashi, K. Komiyama and S. Ōmura, J. Antibiot., 1999, 52, 501–504 CAS.
- For recent examples of Michael addition in natural product, see: (a) M. Groll, B. Schellenberg, A. S. Bachmann, C. R. Archer, R. Huber, T. K. Powell, S. Lindow, M. Kaiser and R. Dudler, Nature, 2008, 452, 755–759 CrossRef CAS; (b) E.-H. Kim and Y.-J. Surh, Biochem. Pharmacol., 2006, 72, 1516–1528 CrossRef CAS.
- S. Ōmura and K. Komiyama, PCT Int. Appl., WO 0147516 A1.
- (a) D. P. Curran, J. Am. Chem. Soc., 1983, 105, 5826–5833 CrossRef CAS; (b) D. P. Curran, B. H. Kim, J. Daugherty and T. A. Heffner, Tetrahedron Lett., 1988, 29, 3555–3558 CrossRef CAS.
- Y.-G. Suh, S.-Y. Seo, J.-K. Jung, O.-H. Park and R.-O. Jeon, Tetrahedron Lett., 2001, 42, 1691–1694 CrossRef CAS.
- W. Yu, Y. Zhang and Z. Jin, Org. Lett., 2001, 3, 1447–1450 CrossRef CAS.
- N.-K. Huang, Y. Chern, J.-M. Fang, C.-I. Lin, W.-P. Chen and Y.-L. Lin, J. Nat. Prod., 2007, 70, 571–574 CrossRef CAS.
- (a) M. Vandewalle, J. Van der Eycken, W. Oppolzer and C. Vullioud, Tetrahedron, 1986, 42, 4035–4043 CrossRef CAS; (b) J. Y. Lee, Y. J. Chung and B. H. Kim, Synlett, 1994, 197–198 CrossRef.
- W. Oppolzer and P. Lienard, Helv. Chim. Acta, 1992, 75, 2572–2582 CrossRef CAS.
- M. Eh, D. Schomburg, K. Schicht and M. Kalesse, Tetrahedron, 1995, 51, 8983–8992 CrossRef CAS.
- L. D. Julian, J. S. Newcom and W. R. Roush, J. Am. Chem. Soc., 2005, 127, 6186–6187 CrossRef CAS.
- (a) J. W. Bode, N. Fraefel, D. Muri and E. M. Carreira, Angew. Chem., Int. Ed., 2001, 40, 2082–2085 CrossRef CAS; (b) G. S. King, P. D. Magnus and H. S. Rzepa, J. Chem. Soc., Perkin Trans. 1, 1972, 437–443 RSC; (c) J. W. Bode and E. M. Carreira, Org. Lett., 2001, 3, 1587–1590 CrossRef CAS; (d) N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2003, 125, 6038–6039 CrossRef CAS; (e) P. G. Baraldi, A. Barco, S. Benetti, S. Manfredini and D. Simoni, Synthesis, 1987, 276–278 CrossRef CAS; (f) A. Guarna, A. Guidi, A. Goti, A. Brandi and F. De Sarlo, Synthesis, 1989, 175–178 CrossRef CAS; (g) D. H. Churykau, V. G. Zinovich and O. G. Kulinkovich, Synlett, 2004, 1949–1952 CAS.
- J. Carpenter, A. B. Northrup, D. M. Chung, J. J. M. Wiener, S.-G. Kim and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2008, 47, 3568–3572 CrossRef CAS.
- Optical rotation of the synthetic (−)-1: [α]D20 −40 (c 0.10, MeOH), Optical rotation of the synthetic (−)-2: [α]D20 −55 (c 0.17, MeOH). The spectral data of the final product synthesized from the enantiomer of 9 was not identical to those of the authentic natural products.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds along with copies of 1H and 13CNMR spectra. See DOI: 10.1039/b817693k |
| This journal is © The Royal Society of Chemistry 2009 |