The formation of a hydrated homochiral helix from an achiral zwitterionic salt, spontaneous chiral symmetry breaking and redox chromism of crystals†
Qing-Xia
Yao
a,
Wei-Min
Xuan
b,
Hui
Zhang
b,
Chao-Yang
Tu
a and
Jie
Zhang
*a
aState Key Laboratory of Structural Chemistry, Fujian Institute of Research on the Structure of Matter and Graduate School of the Chinese Academy of Sciences, Fuzhou, Fujian, 350002, China. E-mail: zhangjie@fjirsm.ac.cn; Fax: (+86) 591-83710051
bDepartment of Chemistry College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China
First published on 18th November 2008
Abstract
A unique supramolecular helix assisted by water tetramers is obtained by static evaporation of aqueous solution of a zwitterionic salt, 1,1′-bis(4-carboxylatebenzyl)-4,4′-bipyridinium; spontaneous chiral symmetry breaking occurs in crystallization; interestingly, redox chromism behavior of crystals upon exposure to alcohol atmosphere has been observed.
Chirality is an essential element of life, and plays key roles in functional materials for applications in optical devices, sensors, enantioselective separation and catalysts.1 Great efforts have been made to construct chiral molecular assemblies assisted by means of metal–ligand coordination interactions or other intermolecular non-covalent forces.2 Especially, hydrogen bonding is of special interest in this regard due to the strength, directionality, and specificity of the interaction, as well as its biological relevance.3 Besides enantioselective synthesis using enantiopure chiral species, which is generally tedious and expensive, one appealing approach towards chiral systems is based on the use of achiral precursors under spontaneous resolution.4 However, when assembling components are intrinsically achiral, right-handed and left-handed isomers usually condense as a racemate which contains equal amounts of both enantiomers, or spontaneous resolution into enantiomers (so-called conglomerates) occurs and the products are normally a racemic mixture of chiral crystals, although each crystal is a single enantiomer.5 Challenges remain in achieving homochirality or enantiomeric excess (ee) from an achiral or a racemic state through a chiral symmetry breaking transition. It is very important to understand the mutual recognition that exists between the achiral assembling components for the ultimate goal of controlling spontaneous resolution in crystals.5a In this paper, we report the formation of a hydrated homochiral helix from an achiral zwitterionic viologen, 1,1′-bis(4-carboxylatebenzyl)-4,4′-bipyridinium (Bpybc) based on “locked” chirality through non-covalent forces. Two types of hydrogen bonding interactions, O–H⋯O and C–H⋯O, are responsible for the helix formation and intracrystal chirality preservation, respectively. Different from some elegant examples that may achieve asymmetric amplification upon external stimuli, such as stirred crystallization,6 chemical manipulation of statistical fluctuation in crystallization,7 or an abrasion/grinding technique,8 spontaneous chiral symmetry breaking of the title compound occurs during static solvent evaporation without any chiral auxiliary component. Interestingly, due to the inherent redox activity of viologens,9 the crystalline solids could undergo significant color change from colorless to blue due to reduction in methanol or ethanol vapor, enabling visual detection of alcohol molecules.
Colorless crystals of Bpybc·5H2O for X-ray diffraction study were obtained from aqueous solution by slow evaporation.‡ The structure analysis reveals the formation of a hydrated supramolecular helix. The Bpybc undergoes conglomerate crystallization and the crystal chosen for X-ray analysis belongs to the chiral trigonal space groupP3221 (or its enantiomorph P3121). The asymmetric unit (Fig. S1, ESI†) contains one half of a Bpybc molecule and two and half water molecules. Each Bpybc adopts a twisted conformation with C2 symmetry (Fig. 1(a)) in which the C1–C8–C8#A–C1#A torsion angle is 84.3°. A careful examination of lattice water molecules reveals the formation of water tetramers by O1W, O2W and their symmetry related water molecules O1W#B and O2W#B, where the distances of O1W⋯O2W, O1W⋯O1W#B and O2W⋯O2W#B are 2.935(4), 2.811(6) and 2.817(6) Å respectively. The coordination environment of lattice water molecules is shown in Fig. 1(b). Each O1W and O2W in the tetramers are hydrogen-bonded to carboxylate oxygen atoms O1 and O2, respectively, and each O3W to two carboxylate oxygen atoms O1 from two different Bpybc molecules. The most remarkable structural feature is that these twisted organic molecules are hydrogen-bonded together by lattice water molecules to form a single left-handed (or right-handed) hydrated helix running along the crystallographic c axis (Fig. 1(c)). The helix, with a pitch of 22.674 Å winding around the threefold screw axis, has a C2 axis which accommodates symmetry of the molecules.
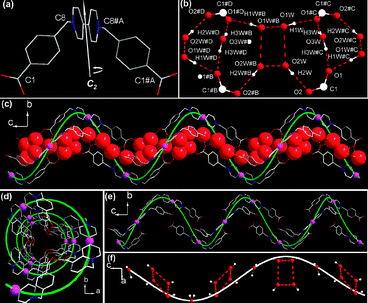 | ||
| Fig. 1 Crystal structure of Bpybc·5H2O for (a) twisted Bpybc with C2 symmetry; (b) hydrogen bonds between carboxylate groups of Bpybc and water molecules (including water tetramer and single water molecule O3W); (c) view of the left-handed hydrated helix running along the crystallographic c axis, water oxygen atoms are shown in space-filling mode; (d) view down the c axis showing a central projection of the helical alignment of Bpybc with all the carboxylate groups pointing towards the inner wall of the channel; (e) view of the helical alignment of Bpybc molecules running along the c axis; (f) the helical arrangement of water O3W and water tetramers running along the c axis. Hydrogen bonds are shown as dotted lines. An imaginary 32 helix (colored green) runs through the pink dummy atoms which are inserted between C13 and C13#A. | ||
Some water-mediated supramolecular helical architectures have been reported in which water molecules are intervened between two consecutive organic molecules.10 However, the helix described here is quite different for it can be viewed as a helical supramolecular host with aqua pores anchoring helical water chains which comprise alternate single water molecules and tetrameric water clusters. The helical arrangement of Bpybc molecules results in channels oriented along the crystallographic c axis (Fig. 1(d), (e)). Due to this packing arrangement, all the carboxylate groups point towards the inner wall of the channels and thus make the channels remarkably hydrophilic. Single water molecule O3W and water tetramer are alternately entrapped in the helical channels (Fig. 1(c), (f)). It is interesting to note that both helical alignments of Bpybc molecules and water molecules are only stabilized by hydrogen bonds between them, with all carboxylate oxygen atoms as acceptors and water molecules as donors. Therefore, the mutual recognition between the organic and water molecules through hydrogen bonds is responsible for the formation of the helix. The structural analysis indicates that methylene C8 atom (with H6) forms a hydrogen bond with O1W from the neighboring helix, in which the C8⋯O1W distance and C–H⋯O1W angle are 3.532 Å and 148.88°, respectively.11 Adjacent helices are linked via such C–H⋯O hydrogen bonds and hence the chiral information will be transmitted along the [100], [010] and [110] directions, respectively, resulting in the formation of homochiral crystals (Fig. 2). Therefore, interhelical C–H⋯O hydrogen bonds play a vital role in chirality transfer within the crystal. In fact, this water-assisted non-covalent force not only contributes to the propagation of helical organization, but also furnishes an origin to chirality in the present system since the prochiral methylene carbon atom can be converted into chiral center by attaching one water molecule to an enantiotopic hydrogen atom of the methylene group through this C–H⋯O interaction (Fig. S3, ESI†). The adoption of a chiral space group by the compound prompts an investigation of the optical activity with SHG (second harmonic generation) response about 2.1 times that of KDP (potassium dihydrogen phosphate).
![(a) Chirality transfer along the [100] direction through methylene C–H⋯O hydrogen bonds between adjacent helices; (b) chirality transfer along the [100], [010] and [110] directions. C–H⋯O hydrogen bonds are shown as white dotted lines.](/image/article/2009/CC/b815456b/b815456b-f2.gif) | ||
| Fig. 2 (a) Chirality transfer along the [100] direction through methylene C–H⋯O hydrogen bonds between adjacent helices; (b) chirality transfer along the [100], [010] and [110] directions. C–H⋯O hydrogen bonds are shown as white dotted lines. | ||
Upon conglomerate crystallization both types of enantiomorphic crystals should be formed in a single batch. In order to further evaluate the spontaneous resolution process, single crystals of sufficient size and quantity in the same crystallization are chosen to determine the solid-state CD spectra in a KCl matrix. As seen in Fig. 3, the crystals give the expected CD spectra which are mirror images of each other, revealing the formation of either M or P enantiomer in single crystals. A crystal with M helicates, whose structure is solved in the space groupP3221, exhibits a positive CD signal. Interestingly, based on the CD measurement of 30 single crystals chosen from one crystallization randomly, the result with 21 positive and 9 negative CD signals (Fig. S5, ESI†) suggests that the enantiomeric excess (ee) is 40%. Another strong support for an ee arises from the CD measurements of 12 batches of powdered bulk samples (Fig. S6, ESI†). All of them show a significant CD signal (10 positive and 2 negative). Such results undoubtedly confirm spontaneous chiral symmetry breaking in crystallization. The usual explanation for chiral symmetry breaking is secondary nucleation by which a randomly generated single mother crystal clones a large number of secondary crystals that are enantiomerically identical to itself. In this case, the handedness preference is random, each enantiomer can dominate with equal probability in different solutions. However, the present result with 10 positive and 2 negative signals for powdered bulk samples shows a nonstochastic distribution of handedness. This may be due to the sporadic presence of chiral crypto-impurities that may either favor the formation of one enantiomeric crystal or disfavor the growth of the other one by selective surface adsorption.12 The mechanism that a cryptochiral environment induces selective chiral symmetry breaking was first proposed by Viedma to explain the directed asymmetric amplification for sodium chlorate and sodium bromate.8a,12b
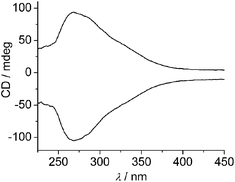 | ||
| Fig. 3 Solid-state circular dichroism spectra of Bpybc·5H2O | ||
Viologen derivatives may undergo one-electron reduction upon using strong reducing agents, or light irradiation in the presence of electron donors, to produce free radicals, accompanied by a blue coloration.9 Bpybc·5H2O is photosensitive and turns blue upon irradiation by a 500 W Xe lamp or sunlight within several minutes under air. The structural analysis suggests that the adjacent carboxylate oxygen atom is approximately perpendicular to the nitrogen atom of each pyridinium ring with the O2–N1–C13 angle of 103.00° and the O⋯N distance of 3.38 Å, and two central pyridinium rings are nearly coplanar with an interplanar angle of 7.42°. Such geometry and distance is favorable to the interaction between the donor unit and the bipyridinium acceptor,9f thus an electron transfer from the carboxylate group to bipyridinium core can be expected. Most viologen derivatives exist as dication salts, the photochromism of neutral zwitterionic viologen is rarely exploited. Until now, only the photochromic behavior and crystal structure of a zwitterionic viologen bearing sulfonate groups have been reported,9d,e in which the sulfonate oxygen atom lies almost perpendicularly above (or below) the nitrogen atom of the pyridinium ring with the O–N–C angle of 94.47° and the O⋯N distance of 2.97 Å.9e Different from some observations that viologen cations only show weak sensitivity to methanol or ethanol under irradiation,9b,c the solid Bpybc·5H2O is quite sensitive to them. Even in a dark room and under air, blue coloration is readily observed when Bpybc·5H2O is exposed to methanol or ethanol vapor. The diffuse reflectance spectra of Bpybc·5H2O obtained upon exposure to methanol show a characteristic absorption band centered at 618 nm (Fig. 4). This spectral feature resembles those of viologen radicals observed in crystalline samples or polymer matrices,9 suggesting that the color change arises from the reduction of colorless zwitterionic Bpybc to blue [Bpybc]˙− radicals. Upon removal of alcohol vapor, the fading occurs in air and under dark conditions, which is a typical behavior of viologen radical resulting from oxidation by molecular O2 in the absence of effective electron donors or photoexcitation.9 The discolored compound undergoes a color change to blue again upon exposure to alcohol vapor. This cycling and color change may enable Bpybc·5H2O to serve as a visual indicator of alcohol molecules.
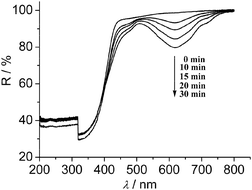 | ||
| Fig. 4 The diffuse reflectance spectra of Bpybc·5H2O upon exposure to methanol vapor. | ||
In summary, an interesting supramolecular helix assisted by water molecules is obtained. Two types of hydrogen bonding interactions Oaqua–H⋯O and methylene C–H⋯Oaqua, are responsible for the formation of helix and intracrystal chirality preservation, respectively. Notably, spontaneous chiral symmetry breaking occurs in each crystallization and the resulting enantiomeric excess of bulk samples are further confirmed by the solid-state CD spectra.
We thank the financial support from the NSFC (No. 20671090, 50372069), Key Project from CAS (KJCX2-YW-H01) and the NSF of Xiamen University (Series B, No. XDKJCX20061027).
Notes and references
- (a) Chirality in Industry II, eds. A. N. Collins, G. N. Sheldrake and J. Crosby, Wiley, New York, 1998 Search PubMed; (b) W. B. Lin, MRS Bull., 2007, 32, 544 CAS.
- (a) C. Nuckolls, T. J. Katz, G. Katz, P. J. Collings and L. Castellanos, J. Am. Chem. Soc., 1999, 121, 79 CrossRef CAS; (b) P. Gangopadhyay and T. P. Radhakrishnan, Angew. Chem., Int. Ed., 2001, 40, 2451 CrossRef CAS; (c) Y. Cui, H. L. Ngo and W. B. Lin, Chem. Commun., 2003, 1388 RSC.
- (a) G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press, Oxford, 1999 Search PubMed; (b) J. E. Field and D. Venkataraman, Chem. Commun., 2002, 306 RSC; (c) Y. Imai, K. Kawaguchi, N. Tajima, T. Sato, R. Kuroda and Y. Matsubara, Chem. Commun., 2008, 362 RSC.
- (a) A. M. Berg and D. L. Patrick, Angew. Chem., Int. Ed., 2005, 44, 1821 CrossRef CAS; (b) Q. Z. Sun, Y. Bai, G. J. He, C. Y. Duan, Z. H. Lin and Q. J. Meng, Chem. Commun., 2006, 2777 RSC.
- (a) L. Pérez-García and D. B. Amabilino, Chem. Soc. Rev., 2002, 31, 342 RSC; (b) S. Khatua, H. Stoeckli-Evans, T. Harada, R. Kuroda and M. Bhattacharjee, Inorg. Chem., 2006, 45, 9619 CrossRef CAS; (c) P. L. Caradoc-Davies and L. R. Hanton, Chem. Commun., 2001, 1098 RSC.
- D. K. Kondepudi and K. Asakura, Acc. Chem. Res., 2001, 34, 946 CrossRef CAS.
- S. T. Wu, Y. R. Wu, Q. Q. Kang, H. Zhang, L. S. Long, Z. P. Zheng, R. B. Huang and L. S. Zheng, Angew. Chem., Int. Ed., 2007, 46, 8475 CrossRef CAS.
- (a) C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef; (b) P. S. M. Cheung, J. Gagnon, J. Surprenant, Y. Tao, H. Xu and L. A. Cuccia, Chem. Commun., 2008, 987 RSC.
- (a) K. B. Yoon and J. K. Kochi, J. Am. Chem. Soc., 1988, 110, 6586 CrossRef CAS; (b) H. Yoshikawa and S.-i. Nishikiori, Chem. Lett., 2000, 142 CrossRef CAS; (c) H. Yoshikawa, S.-i. Nishikiori, T. Watanabe, T. Ishida, G. Watanabe, M. Murakami, K. Suwinska, R. Luboradzki and J. Lipkowski, J. Chem. Soc., Dalton Trans., 2002, 1907 RSC; (d) H. Kamogawa and T. Suzuki, Bull. Chem. Soc. Jpn., 1987, 60, 794 CrossRef CAS; (e) L. A. Vermeulen and P. D. Robinson, Acta Crystallogr., Sect. C, 1996, 52, 984 CrossRef; (f) P. M. S. Monk, The Viologens: Physicochemical Properties Synthesis and Applications of the Salts of 4,4′-Bipyridine, John Wiley & Sons Ltd, Chichester, 1998 Search PubMed.
- (a) R. Parthasarathy, S. Chaturvedi and K. Go, Proc. Natl. Acad. Sci. U.S.A, 1990, 87, 871 CAS; (b) N. Ramasubbu and R. Parthasarathy, Biopolymers, 1989, 28, 1259 CrossRef CAS; (c) S. Guha, M. G. B. Drew and A. Banerjee, Tetrahedron Lett., 2006, 47, 7951 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS.
- (a) I. Weissbuch, M. Lahav and L. Leiserowitz, Cryst. Growth Des., 2003, 3, 125 CrossRef CAS; (b) C. Viedma, Cryst. Growth Des., 2007, 7, 553 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, characterization and additional structural data. CCDC 700752. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b815456b |
| ‡ Crystal data: C26H30N2O9, Mr = 514.53, trigonal, P3221, a = 9.6710(4), b = 9.6710(4), c = 22.6740(17) Å, γ=120°, V = 1836.54(17) Å3, T = 293 K, Z = 3, Dc = 1.385 g cm−3, μ = 0.106 mm−1, F(000) = 804, 13804 reflections, 1629 unique (Rint = 0.0246), 1518 observed I > 2σ(I), R1 (wR2) = 0.0431 (0.1176) and GOF = 1.075 for 180 parameters. |
| This journal is © The Royal Society of Chemistry 2009 |