A new approach for measuring the redox state and redox capacity in milk
Tomer
Noyhouzer
ab,
Ron
Kohen
b and
Daniel
Mandler
*a
aInstitute of Chemistry, The Hebrew University of Jerusalem, Jerusalem, 91904, Israel. E-mail: mandler@vms.huji.ac.il; Fax: +972 2 658 5319; Tel: +972 2 658 5831
bDepartment of Pharmaceutics, School of Pharmacy, The Hebrew University of Jerusalem, Jerusalem, 91120, Israel
First published on 7th September 2009
Abstract
Milk is one of the most fundamental ingredients in our diet. It is a complex biological fluid, which contains numerous substances, ranging from metal ions to enzymes. There is a constant search for an improved way of monitoring its quality and freshness. These are highly affected by the redox state of milk, which is governed by different species. In this study, we investigated the redox state and capacity of milk. Specifically, milk was potentiometrically titrated using different redox mediators, which enabled facilitation of electron transfer between different oxidizable species and the electrode. We found that the iodine/iodide redox couple was superior for measuring the redox capacity of milk. These measurements revealed that milk is not a well-poised system due to the presence of at least two different oxidizable species, one of which is hydrophobic while the other is hydrophilic and therefore could be separated by phase separation of milk.
Introduction
Milk is the only element of our diet whose sole function in nature is to serve as food. It is an excellent source of energy, protein, and of minerals and vitamins.1,2 However, from a chemical point of view milk is a complex biological fluid, which contains numerous substances, ranging from metal ions to enzymes.3–5 Furthermore, milk has a wide biological activity, which persists even after pasteurization.3,6,7 The estimated worldwide milk consumption in 2007 was more than 40 billions liters, with prices ranging from 20 to 70 US cent per liter.8 Milk prices are still rising due to the global warming effects, search for renewable energy sources and the continuous increase in demand.9In accordance, the production, storage and distribution of milk and diary products are highly monitored.10–13 Monitoring of milk is complex and ranges from pH to biological and microbial activity. The current main procedures for testing milk quality consist of a variety of measurements. Milk filtration is carried out in order to determine the degree of impurities, whereas the freezing point is a measure of the water–milk ratio. Other tests include the degree of pasteurization, the amount of fat in milk and bacteria and germs counting.14–16
Nevertheless, there is no simple method to determine the freshness of milk.17–22 The freshness and quality of milk is undoubtedly affected and reflected by its redox state. It is documented23 that the redox potential has a significant effect on micro-organisms, which play an essential role in all fermented dairy products, such as cheese. These micro-organisms are involved in the manufacturing and ripening of cheese.24 Several studies showed a clear correlation between the degree of oxidation and the quality of cheese.14–16 Milk spoilage is also related to oxidation–reduction reactions.25,26 These reactions result in alteration of different compounds. Hence, knowledge of the actual redox conditions is important for interpretation of milk quality and freshness.
Previous studies27–31 pointed to the effect and control of the redox state of dairy products, especially in the cheese industry, on their flavor and taste. However, the effect of the redox state of milk has been almost completely neglected. Although the correlation between heat, pH and redox potential in milk was addressed32,33 measuring the redox potential as an indicator for milk quality was disregarded. Vuillemard et al.26 added a reducing agent, i.e., cysteine to milk in order to prolong its shelf life. De Coninck et al.24 applied a similar approach, however, using electric current for reduction. Brasca et al.34 tried to characterize the reduction potential of milk. But only Lundstrom et al.35–37 and Hitam et al.19 addressed the issue of characterizing the milk freshness or quality by a direct measure of the redox potential. Their approach was based on an electronic tongue.
Determination of the redox state of a system is still not a simple task and no universally accepted procedure exists.38 Methods to control the redox potential were evaluated in the food industry but they have the disadvantage of using chemical products such as addition of cystein or vitamin C.32 Measuring of the redox state involves a thermodynamic measurement, which is very similar to measuring pH. Nevertheless, redox measurements are by far less popular than pH measurements due to the irreversibility of most redox processes in nature.21,39 Yet, there is no doubt that determination of the redox state is likely to be an important factor in the future.40–44Oxidation–reduction processes are defined in terms of electron transfer between compounds. Redox or oxidation–reduction potential (ORP) which is an intrinsic indication of biological media,45 can be defined as the measure of the ability of a chemical/biochemical system to oxidize (lose electrons) or reduce (gain electrons). The redox potential is described by the Nernst equation, which assumes reversible electron transfer between all species.
In a complex fluid such as milk, several oxidation–reduction systems are active simultaneously and their effect on the oxidation–reduction potential depends on several factors, such as the reversibility of the system, its E0 value, the ratio of oxidant to reductant, and the concentration of the active compounds of the system.46
We chose to approach the issue of the “problematic” potential measurements through the concept of redox or poising capacity (ρ). While buffer capacity is a well known factor, defined as the amount of strong acid or base needed to add to the system in order to change the pH by a unit. The poising capacity at any potential, E, is defined as the quantity of strong oxidant which must be added to one liter of the examined solution in order to change E by 1 V (eqn (1)).47
 | (1) |
The objective of this study is to measure the redox state of milk by a potentiometric method based on the introduction of different redox mediators that shuttle charge from a wide range of electroactive materials to a solid electrode.48 These mediators are used to overcome the fundamental problem in potetiometry, i.e., slow kinetics between the potential determining substances and the electrode surface.49 The redox couples: hexacyanoferrate(III/II) (E0 = 0.361 V), hexachloroiridate(IV/III)) (E0 = 0.867 V) and iodine/iodide (E0 = 0.536 V) (all potentials are versus NHE) have been employed in the redox titration mode.
Experimental section
Instrumentation
Potentiometric and cyclic voltammetry (CV) measurements were performed with an Autolab PGSTAT10 potentiostat (EcoChemie, Utrecht, The Netherlands) and 2000 multimeter (Keithley, OH, USA). Centrifugation was performed using a Sorvall RC 5c Plus (Themo Fisher, Waltham, MA, USA) with a GSA centrifuge head. pH measurements were carried out with a Cyberscan 510 pH meter and a pH electrode (Eutech instruments, Singapore).The electrochemical measurements were conducted with commercial Au and Pt disk electrodes (2 mm diameter, CH instruments, TX, USA) and a home-made glassy carbon electrode (GCE) which was assembled by sealing a glassy carbon rod (2 mm dia., Atomergic Chemetals, NY, USA) in a Teflon tube under pressure. A Pt wire was used as a counter electrode and an Ag/AgCl (KCl saturated) was used as a reference electrode.
Materials
Four different types of milk were used: 3% skimmed milk, UHT milk, 3% skimmed milk pre-pasteurized from Tnuva dairy manufactures (Jerusalem, Israel) and raw milk from a dairy farm (Ma'ale Hahamisha, Israel). Iodine 99.99%, potassium iodide 99%, potassium hexacyanoferrate(III) 99%, potassium hexacyanoferrate(II) 99%, potassium hexachloroiridate(IV) 99.99%, potassium hexachloroiridate(III) 99.99%, and 2,2-azobis(2-methylpropionamide) dihydrochloride (AAPH) were purchased from Aldrich. Potassium phosphate used for preparing buffer solutions was purchased from J.T.Baker (The Netherlands). All aqueous solutions were prepared from deionized water (Barnstead Easypure UV system).Procedures
The working electrodes were polished with alumina (1 and 0.05 µm, Buehler, IL, USA) and washed with deionized water before use. Potentiometric measurements were carried out under an anaerobic environment (using nitrogen or argon) with a two-electrode cell (working and reference electrodes). The milk was diluted (1 : 1 v/v) with a phosphate buffer solution (pH 6.7) in order to maintain a constant pH during the measurements. The CV measurements were conducted in a three-electrode cell similarly to the potentiometric measurements. The fat was separated from milk by centrifuging the milk for 30 min at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm at a temperature of 4 °C. Milk was artificially oxidized using 25 mM of AAPH for 30 min at 37 °C.
000 rpm at a temperature of 4 °C. Milk was artificially oxidized using 25 mM of AAPH for 30 min at 37 °C.
Results and discussion
As stated above there is considerable resemblance between pH and redox measurements. In both cases equilibrium is measured (either pH or redox potential) upon adding the titrant. Both titrations yield a sigmoid curve by plotting α, which is defined as the concentration ratio between the titrant and the total concentration of both species, i.e., acid and base or oxidized and reduced states, versus pH or potential, respectively.50Fig. 1A shows such a redox curve, which was obtained by adding known amounts of hexacyanoferrate(III) into a buffer solution consisting of hexacyanoferrate(II). The redox capacity can readily be extracted from a modified curve (Fig. 1B) which depicts the change of α with potential as a function of potential. The derivative of the sigmoid curve (Fig. 1A) equals the redox capacity, which attains its maximum at the deflection point (Fig. 1B). Clearly, when the solution contains only one redox couple, the maximum must be equal to E0. In the case that more than one redox couple is present in the solution, a Gaussian should be obtained for each couple providing that their redox potential is well-separated and there is no interaction between the redox couples. The potential measured by an electrode that is in equilibrium with all redox couples in solution will be in between the redox potentials.39 | ||
| Fig. 1 (A) Redox measurement of a solution consisting of 10 mM of K4Fe(CN)6 in phosphate buffer (pH 6.7) to which volumes of 1 mM K3Fe(CN)4 were added. (B) The derivative of the redox curve. The maximum of the curve is the redox or poising capacity where E = E0. | ||
Several studies discussed the difference between pH and pE† measurements.21,39 While in pH we measure the activity of protons, we cannot measure directly the activity of electrons but electron transfer to and from the electrode. The main advantage of pH measurements is that while proton transfer is usually diffusion-controlled, electron transfer very often exhibits sluggish kinetics on an electrode surface. The kinetics of redox reactions is a very complex process, which is often affected by the number of electrons transferred per species as well as by the specific interactions between the redox couple and the electrode surface. Typically, multiple electron transfer is associated with complex changes in the molecular configuration, while single electron transfer often involves much less change in the molecular structure and therefore exhibits faster kinetics.49
The measurement of the true redox capacity of a system relies upon the ability to measure the true redox potential. In real systems each couple independently exchanges electrons at the electrode surface and therefore the concentration ratios of each couple in the solution are defined by the measured potential. Hence, we aimed at measuring the redox capacity of a complex system such as milk by adding a redox mediator, which is expected to undergo reversible electron transfer with most of the redox species in milk. Specifically, we examined the following redox mediators: Fe(CN)63−/4−, IrCl62−/3− and I3−/I−.
Fig. 2 shows the redox potential measured by a Pt electrode in a solution consisting of 1 : 1 v/v milk and phosphate buffer (0.1 M pH 6.7) upon adding increasing amounts of redox mediators. Portions of 100 µL of the titrant were added into 10 mL of solution every 300 s, therefore resulting in a negligible dilution effect. Furthermore, the solution was continuously purged with nitrogen or argon in order to maintain an anaerobic environment. Commercial 3% milk was used.
 | ||
| Fig. 2 Open circuit potential measured by a Pt electrode in 1 : 1 v/v diluted milk (in phosphate buffer pH 6.7) as a function of time upon adding different mediators. (black) Fe(CN)63−, (red) I−/I3− and (blue) IrCl62−. Each addition was of 0.01 mM (final concentration) of mediator. Inset: A close inspection of one of the second additions. | ||
It is clearly seen that the addition of the iron complex caused an abrupt increase in the measured potential followed by a slow decay. Moreover, the potential is constantly shifted to more positive potentials as more Fe(CN)63− was added. Eventually, the potential attains a constant value, which is very close to the potential of the initial jump. These findings can be explained by assuming that the milk contains oxidizable species that react with the iron complex. Yet, it is evident that the rate of this reaction is sluggish as it takes a relatively long time to establish a new equilibrium. That is, the initial jump of potential is due to the addition of the oxidized iron species, while the slow decay is a result of the sluggish kinetics between the milk species and hexacyanoferrate(III). A sigmoidal curve should have been obtained if the reduced species in the solution had reacted immediately under Nerstian conditions with the added oxidant. Nevertheless, the outer shape of the curve (Fig. 2) does resemble a sigmoidal shape. It is also possible that the redox potential of the hexacyanoferrate is not sufficiently positive and therefore not all the oxidizable species reacted with Fe(CN)63−. Fig. 2 (inset) also demonstrates the difference in kinetics between the various redox mediators. While the iron complex reacts relatively slowly as stated above, the iodine and the Ir complex show substantially faster kinetics. After each addition a new equilibrium is established and the potential stabilizes.
Similar titration experiments can be conducted in a slightly different way. Instead of adding the oxidizing agent into milk, it is possible to add milk into a solution containing the reduced and oxidized form of hexacyanoferrate. The advantage of this approach lies in the fact that the initial solution has a well-defined potential and the addition of milk does not cause a sudden change in the measured potential. The latter is because of the slow electron exchange rate between the oxidizable species in milk and the platinum electrode. Fig. 3 shows the results of such an experiment. The initial potential is 0.238 V vs. Ag/AgCl which is equal to the redox potential of the hexacyanoferrate species. Two different concentrations (1.0 and 0.1 mM) of the redox species were used. It is evident that the addition of milk into the solution caused a shift of the potential to more negative values. The change is more significant as the concentration of the redox species decreased. This suggests that milk behaves, indeed, as a reducing agent, which reacts with Fe(CN)63− thus increasing the ratio between the reduced to oxidized hexacyanoferrate species and therefore shifting the potential to more negative values. It can be seen that the shift in the potential ceases (for 0.1 mM of the hexacyanoferrate) after 90 mV. This corresponds to a change of 95% of the concentration of the reduced and oxidized species. The reason for the relatively significant shift of the potential is due to the high portions of milk that were added (1 ml). In fact, this is a titration of Fe(CN)63− by milk.
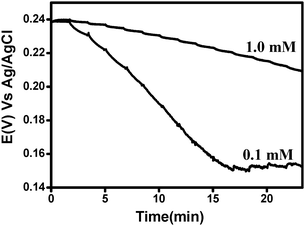 | ||
| Fig. 3 The change of the open circuit potential measured by a Pt electrode in a solution consisting of 1 : 1 Fe(III)(CN)63− : Fe (II)(CN)64− (in two different concentrations: 1.0 and 0.1 mM) and phosphate buffer pH 6.7 as a function of time upon adding milk. 1 ml of milk was added every 100 s. | ||
As a result, we concluded that hexacyanoferrate(III) is not the best mediator for milk due to its sluggish kinetics with the different oxidizable components in this medium.
Hence, we examined additional and stronger oxidants. Specifically, we employed iodine, hexachloroiridate(IV) and Ce(IV).
Fig. 4A–B shows the potentiometric titrations of 3% milk in a phosphate buffer (1 : 1 v/v), upon adding constant volumes of I−/I3− (concentration ratio 10 : 1, Fig. 4A), and IrCl63− (Fig. 4B). Three different redox concentrations were used and additions were made after every 100 s. Both mediators result in a sigmoidal curve (as one would expect from a titration experiment) suggesting that milk is oxidized. There are a few differences between the two oxidants. Iodine attains equilibrium and therefore a final potential much faster than hexachloroiridate implying that charge transfer between iodine and the various components in milk is faster than that of IrCl62−. Furthermore, the final potential attained upon adding IrCl62− is more positive than that of iodine due to its more positive redox potential (eqn (2)).
| E0IrCl63−/4− = 0.867 V, E0I3−/I2 = 0.536 V vs. NHE. | (2) |
 | ||
| Fig. 4 The open circuit potential of a Pt electrode in a solution of 3% milk (diluted 1 : 1 v/v with phosphate buffer pH 6.7) that was titrated with: (A) I−/I2 (ratio 10 : 1) and (B) IrCl63−. In both cases the concentrations were: 5 (black), 10 (red) and 20 (blue) µM. | ||
Fig. 4A–B also shows the effect of decreasing the concentration of the added oxidants. The effect is more evident for iodine addition. As expected the curve is shifted to longer times which represent a larger number of additions that are required as a result of the low concentration of the oxidant. This will be shown in Fig. 5. We also conducted a set of experiments with Ce(IV), which is a very strong oxidant (E0Ce4+/3+ = 1.72 V vs. NHE), however, this species requires a very acidic environment (pH < 3) to prevent its hydrolysis. The low pH caused an immediate spoilage of milk, which made it impossible to use this mediator.
In order to obtain a better understanding of the titration curves and study the potential changes in the course of the initial additions, we diluted our oxidizing agents by a factor of ten and repeated the experiments. No significant change was observed for hexacyanoferrate(III), and the results of the hexachloroiridate(IV) were not obsolete either. Yet, decreasing the concentration of iodine (0.5, 1 and 2.5 µM) had a noticeable effect on the titration curves as seen in Fig. 5. Lucid double sigmoidal curves were obtained for both types of milk, i.e., fresh and 3% milk. This double sigmoidal shape alludes to the presence of two major oxidizable components that are sequentially oxidized by iodine. Lowering the concentration of iodine separates these two processes.
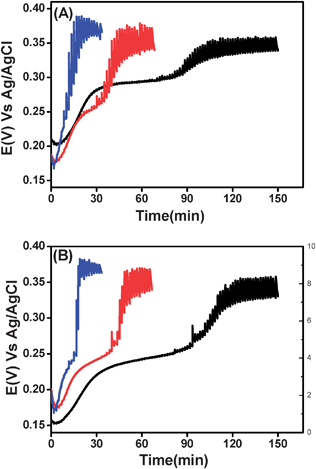 | ||
| Fig. 5 The same as Fig. 4, however, milk samples were titrated with iodine at a lower concentration: 0.5 (black), 1 (red) and 2.5 (blue) µM. (A) 3% milk, (B) raw milk. | ||
From the curves it was possible to extract the poising capacity of milk (Fig. 6). Plotting the derivative of the oxidant concentration with potential as a function of potential, results in a double-peak curve. This is in accordance with the discussion above (Fig. 1).
 | ||
| Fig. 6 Redox capacity as a function of potential measured for: (A) 3% milk and (B) fresh milk. | ||
The redox (poising) capacity calculated from the experimental results is basically the value of dOx/dE at the peaks. The potential of the redox capacity corresponds to the redox potential of the species in milk, which is ca. 0.21 and 0.30 V for 3% milk and 0.25 and 0.31 V for fresh milk, respectively. These potentials are believed to correspond to the same species. This relatively large difference in potential implies that the system is not well poised39,47 namely, there is no interaction between the different electroactive species in milk and therefore there is no true equilibrium. The fact that the system is not well poised can explain the relatively large range of redox potential of milk reported in the literature (150–300 mV).26,51,52 It is evident that the measured potential is likely to be significantly affected by the kinetics of this species with the electrode used. Hence, our mediator approach is clearly a better method for evaluating the redox potential and capacity of milk.
To confirm that the redox phenomena we observed are related specifically to the milk and not to the buffer solution we used (to maintain a steady pH) we carried out an identical experiment with adding water instead of milk. Instead of observing a positive shift of the potential upon adding the iodine/iodide, we noticed a negative shift of potential, which was due to a dilution effect. Namely, diluting the I3−/I− solution shifts the equilibrium towards the formation of I− and I2 in order to maintain the equilibrium constant, thus shifting the potential towards negative values (eqn (3)).
 | (3) |
Fig. 7 shows the effect of decreasing the amounts of milk that were titrated. It is evident that as the amount of milk decreases, the concentration of the titrant that is required to affect the potential decreases as well. Moreover, a linear dependence can be seen (R2 = 0.989, Fig. 7B) by plotting the concentration of I2 that caused the initial change of the second titration curve (see Fig. 7A) vs. the percentage of milk in the solution. This confirms also that the sigmoidal curves are indeed due to an oxidation–reduction process between the titrant and milk and that the species being oxidized originate from the milk.
 | ||
| Fig. 7 The open circuit potential of a Pt electrode measured in different dilutions of milk: 20% (black), 40% (red), 60% (green) and 80% (blue) in phosphate buffer pH 6.7 upon adding (every 100 s) 1 mM (final concentration) of I−/I2 (ratio 10 : 1). (B) The correlation (R2 = 0.991) between the potential at the foot of the second titration curve as a function of the percentage of milk in the sample. | ||
Previous works53–57 suggested that the oxidation–reduction reactions are primarily a result of lipid oxidation. Jensen et al.56 states that phospholipids are the main fat molecules sensitive to oxidative stress because of their position on the milk fat globule membrane and the fact that they are composed of polyunsaturated fatty acids. On the other hand, the aqueous phase of milk has not been thoroughly investigated as the fatty phase with respect to redox processes. Evidently, there are several known electroactive species in the aqueous phase, such as citric and ascorbic acid as well as trace amounts of metals, e.g., iron, which will affect the redox potential. A number of enzymes that catalyze electron transfer reactions can be found either in the fatty or in the aqueous phase.58–60 In the quest to find the origin of the reducing species which were observed in milk, a few approaches were examined. In order to eliminate the possibility that one or more species is related to the casein in milk, we separated the casein from the milk using the Rowland method.61 The casein-free milk was mixed with phosphate buffer pH 6.7 (1 : 1 v/v) while a highly-diluted iodine solution (1 nM) was used for titrating this solution. A diluted iodine solution was applied because the milk was highly diluted in the course of this process. Fig. 8 shows that a double-sigmoidal titration curve was still obtained in spite of removing the casein. This implies that the oxidizable species are not casein associated.
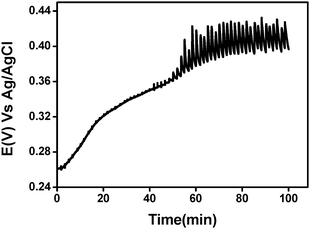 | ||
| Fig. 8 Potentiometric titration curve of casein free-milk sample using I−/I2 (ratio 10 : 1) 1nM (final concentration). | ||
We also targeted the milk fat and therefore used a rapid lipidseparation method described by Garnsworthy et al.62 which separated the milk into an aqueous and a fatty phase. The viscosity of the fat is too high to allow a proper potentiometric measurement. Hence, we concentrated on the aqueous phase. The latter was titrated as described above with iodine/iodide solution. Fig. 9 shows that only one sigmoidal curve is detected implying that one of the oxidizable species is dissolved in the aqueous phase, while the other species is probably more soluble in the fatty phase.
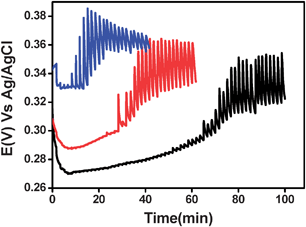 | ||
| Fig. 9 The open circuit potential of a Pt electrode measured in fat-free milk, diluted in phosphate buffer (1 : 1 v/v) pH 6.7 upon adding (every 100 s) 0.5 µM (black), 1 µM (red) and 2.5 µM (blue) of I−/I2 (ratio 10 : 1). | ||
Other evidence for supporting the presence of the oxidizable species in the aqueous phase stems from the titration curves measured by hexacyanoferrate(III)/hexacyanoferrate(II) (Fig. 3). This redox couple is highly charged and insoluble in an organic (fat) phase and yet, a titration curve (with one sigmoidal curve) is observed. Finally, we also applied the ferric reducing ability of a plasma (FRAP) assay .63 Milk samples were diluted with the FRAP solution and doubly distilled water (DDW). The maximum dilution ratio between milk and the solution was 1 : 18, any higher a ratio led to saturation between the FRAP reagent and the milk. A clear linear dependence (R2 = 0.93) between the amount of milk and absorbance due to the formation of an iron(II) complex is evident (Fig. 10). This also indicates the presence of oxidizable species in the aqueous phase of milk.
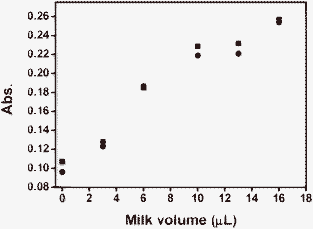 | ||
| Fig. 10 Absorption of milk samples (duplicates) with different dilutions assayed with FRAP. | ||
Conclusions
The redox capacity of milk was determined using different redox mediators. We found that the redox titration of milk exhibited two clear and separated redox processes associated with at least two oxidizable species. From separating the phases of milk it was concluded that at least one of these species is hydrophobic and tends to stay in the fatty phase. On the other hand and as opposed to previous reports, one of the redox species is highly hydrophilic and could be titrated in the aqueous phase after phase separation.This study clearly shows that milk, indeed, contains oxidizable species, which are not in equilibrium and therefore milk is not a well-poised system. This finding was made possible due to the application of a mediator, which was found to exchange charge with the different species present in milk and the electrode surface. The mediator approach is therefore suitable for determining the redox capacity of complex systems, such as milk. Moreover, we believe that this procedure could be highly relevant to the measurement of the freshness of milk and presumably also other biological fluids.
Acknowledgements
This study was partially supported by the Ring Foundation (at the Hebrew University). RK is the incumbent of the Richard and Jean Zarbin Chair in Medical Studies at the Hebrew University of Jerusalem. Ya. I. Tur'yan is warmly acknowledged for his valuable notes.References
- H. C. Sherman, Chemistry of Food and Nutrition, The Macmillan Company, New York, 1947 Search PubMed.
- D. Goff, Diary Science and Technology, http://www.foodsci.uoguelph.ca/dairyedu/home.html Search PubMed.
- P. F. Fox and P. L. H. McSweeney, Dairy Chemistry and Biochemistry, Chapmna & Hall, London, 1998 Search PubMed.
- F. J. Francis, Encyclopedia of Food Science and Technology, Wiley, New York, 2000 Search PubMed.
- Cornell University, Milk Facts, http://www.milkfacts.info Search PubMed.
- U.S. Dept. of Health and Human Services, Public Health Service and Food and Drug Admin, 2005 Search PubMed.
- J. B. Enright, W. W. Sadler and R. C. Thomas, Am. J. Public Health Nations Health, 1957, 47 CrossRef.
- United Nations, Food and Agriculture Organization, http://www.fao.org.
- I. Flamenbaum, Cattle Milk Econ., 2007, 7 Search PubMed.
- M. Borkova and J. Snaselova, Czech J. Food Sci., 2005, 23 CAS.
- J. Van Oostdam, A. Gilman, E. Dewailly, P. Usher, B. Wheatley, H. Kuhnlein, S. Neve, J. Walker, B. Tracy, M. Feeley, V. Jerome and B. Kwavnick, Sci. Total Environ., 1999, 230 CrossRef CAS.
- A. R. Frost, C. P. Schofield, S. A. Beaulah, T. T. Mottram, J. A. Lines and C. M. Wathes, Comput. Electron. Agric., 1997, 17 CrossRef.
- D. J. Mcclements, Crit. Rev. Food Sci. Nutr., 1997, 37 CrossRef CAS PubMed.
- H. S. Adams, Milk and Food Sanitation Practice, Oxford University Press, London, 1947 Search PubMed.
- F. J. Francis, Encyclopedia of Food Science and Technology, Wiley, New York, 2000 Search PubMed.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, Wiley, New York, 1996 Search PubMed.
- T. M. P. Cattaneo, C. Giardina, N. Sinelli, M. Riva and R. Giangiacomo, Int. Dairy J., 2005, 15 CrossRef CAS.
- S. Labreche, S. Bazzo, S. Cade and E. Chanie, Sens. Actuators, B, 2005, 106 CrossRef CAS.
- M. Y. M. Sim, T. J. Shya, M. N. Ahmad, A. Y. M. Shakaff, A. R. Othman and M. S. Hitam, Sensors, 2003, 3 Search PubMed.
- Y. Karagul-Yuceer, M. Drake and K. R. Cadwallader, Freshness and Shelf Life of Foods, ACS Symposium Series, 2003, vol. 836, pp. 108–123, ed. K. R. Cadwallader and H. Weenen Search PubMed.
- T. H. Christensen, P. L. Bjerg, S. A. Banwart, R. Jakobsen, G. Heron and H. J. Albrechtsen, J. Contam. Hydrol., 2000, 45 CrossRef CAS.
- Am. Dairy Rev., 1980, 42 Search PubMed.
- L. Gram, L. Ravn, M. Rasch, J. B. Bruhn, A. B. Christensen and M. Givskov, Int. J. Food Microbiol., 2002, 78 CrossRef.
- S. Abraham, R. Cachon, B. Colas, G. Feron and J. De Coninck, Int. Dairy J., 2007, 17 CrossRef CAS.
- M. P. Bolduc, Y. Raymond, P. Fustier, C. P. Champagne and J. C. Vuillemard, Int. Dairy J., 2006, 16 CrossRef CAS.
- M. P. Bolduc, L. Bazinet, J. Lessard, J. M. Chapuzet and J. C. Vuillemard, J. Agric. Food Chem., 2006, 54 CrossRef CAS PubMed.
- A. Schreyer, M. Britten, J. M. Chapuzet, J. Lessard and L. Bazinet, Innovative Food Sci. Emerg. Technol., 2008, 9 CAS.
- A. Topcu, I. Mckinnon and P. L. H. Mcsweeney, J. Food Sci., 2008, 73 CrossRef CAS PubMed.
- H. J. Giroux, J. B. St-Amant, P. Fustier, J. M. Chapuzet and M. Britten, Food Res. Int., 2008, 41 CrossRef CAS.
- A. Kieronczyk, R. Cachon, G. Feron and M. Yvon, J. Appl. Microbiol., 2006, 101 CrossRef CAS PubMed.
- M. Balestrieri, M. S. Spagnuolo, L. Cigliano, G. Storti, L. Ferrara, P. Abrescia and E. Fedele, Food Chem., 2002, 77 CrossRef CAS.
- H. J. Giroux, J. B. St-Amant, P. Fustier, J. M. Chapuzet and M. Britten, Food Res. Int., 2008, 41 CrossRef CAS.
- T. Mattilasandholm, T. Alivehmas, G. Wirtanen, U. Ronner and M. Sandholm, Int. J. Food Sci. Technol., 1991, 26 Search PubMed.
- M. Brasca, S. Morandi, R. Lodi and A. Tamburini, J. Appl. Microbiol., 2007, 103 CrossRef CAS PubMed.
- F. Winquist, R. Bjorklund, C. Krantz-Rulcker, I. Lundstrom, K. Ostergren and T. Skoglund, Sens. Actuators, B, 2005, 111 Search PubMed.
- F. Winquist, C. Krantz-Rulcker, P. Wide and I. Lundstrom, Meas. Sci. Technol., 1998, 9 Search PubMed.
- F. Winquist, P. Wide and I. Lundstrom, Anal. Chim. Acta, 1997, 357 CrossRef CAS.
- M. Whitfiel, Limnol. Oceanogr., 1969, 14 Search PubMed.
- T. Grundl, Chemosphere, 1994, 28 CrossRef CAS.
- M. J. Barcelona and T. R. Holm, Environ. Sci. Technol., 1991, 25 CrossRef CAS.
- T. Frevert, Swiss J. Hydrol., 1984, 46 Search PubMed.
- R. D. Lindberg and D. D. Runnells, Science, 1984, 225 CAS.
- R. Kohen, E. Moor and M. Oron, in Redox–Genome Interaction, ed. J. Fuchs, M. Podda and L. Packer, Marcel Dekker Inc., New York, 2004, pp. 13–42 Search PubMed.
- I. Grenthe, W. Stumm, M. Laaksuharju, A. C. Nilsson and P. Wikberg, Chem. Geol., 1992, 98 CrossRef CAS.
- A. Nyska and R. Kohen, Toxicol. Pathol., 2002, 30 CAS.
- J. M. Sherbon, Fundamentals of Dairy Chemistry, Aspen Publishers, Gaithersburg, MD, 1999 Search PubMed.
- E. R. Nightingale, Anal. Chem., 1958, 30 CrossRef CAS.
- A. J. Bergren and M. D. Porter, J. Electroanal. Chem., 2006, 591 CrossRef CAS.
- J. A. Bard and R. L. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, New York, 2000 Search PubMed.
- R. Delevie, J. Electroanal. Chem., 1992, 323 CAS.
- T. P. Beresford, N. A. Fitzsimons, N. L. Brennan and T. M. Cogan, Int. Dairy J., 2001, 11 CrossRef CAS.
- V. L. Crow, T. Coolbear, P. K. Gopal, F. G. Martley, L. L. Mckay and H. Riepe, Int. Dairy J., 1995, 5 Search PubMed.
- R. T. Marsili, J. Chromatogr. Sci., 1999, 37 CAS.
- H. Stapelfeldt, B. R. Nielsen and L. H. Skibsted, Int. Dairy J., 1997, 7 CrossRef CAS.
- J. N. Coupland and D. J. Mcclements, Trends Food Sci. Technol., 1996, 7 CrossRef CAS.
- R. G. Jensen, A. M. Ferris and C. J. Lammikeefe, J. Dairy Sci., 1991, 74 CAS.
- J. Hegenauer, P. Saltman, D. Ludwig, L. Ripley and P. Bajo, J. Agric. Food Chem., 1979, 27 Search PubMed.
- C. M. Harris, S. K. Sanders and V. Massey, J. Biol. Chem., 1999, 274 Search PubMed.
- J. Hunt, V. Massey, W. R. Dunham and R. H. Sands, J. Biol. Chem., 1993, 268 CAS.
- E. D. Jarasch, G. Bruder, T. W. Keenan and W. W. Franke, J. Cell Biol., 1977, 73 CrossRef CAS.
- J. S. Rowland, J. Diary Res., 1938, 9 Search PubMed.
- S. Feng, A. L. Lock and P. C. Garnsworthy, J. Dairy Sci., 2004, 87 CrossRef CAS.
- I. F. F. Benzie and J. J. Strain, Anal. Biochem., 1996, 239 CrossRef CAS PubMed.
Footnote |
†  |
| This journal is © The Royal Society of Chemistry 2009 |