DNA functionalized gold nanoparticles for bioanalysis
Yang-Wei
Lin
,
Chi-Wei
Liu
and
Huan-Tsung
Chang
*
Department of Chemistry, National Taiwan University, 1, Section 4, Roosevelt Road, Taipei, Taiwan. E-mail: changht@ntu.edu.tw; Fax: +011-886-2-33661171; Tel: +011-886-2-33661171
First published on 7th September 2009
Abstract
Gold nanoparticles (Au NPs) have become one of the most interesting sensing materials because of their unique size- and shape-dependent optical properties, high extinction coefficients, and super-quenching capability. Au NPs that are bioconjugated with DNA (DNA-Au NPs) have been demonstrated for selective and sensitive detection of analytes such as mercury(II) ions, platelet-derived growth factor (PDGF), and adenosine triphosphate (ATP). This review focuses on approaches using DNA-Au NPs for colorimetric, fluorescent, and scattering detection of biopolymers and small solutes. We highlight the important roles that the size and concentration of Au NPs, the length and sequence of DNA, the nature of the capping agents, and the ionic strength and pH of solution play in determining the specificity and sensitivity of the nanosensors for the analytes. The advantages and disadvantages of different detection methods for sensing of interesting analytes using DNA-Au NPs will be discussed.
 Yang-Wei Lin | Dr Yang-Wei Lin has worked on the development of bioseparation and biosensing techniques. He is employed currently as a postdoctoral fellow, focusing on the synthesis of functional nanomaterials and fabrication of (micro) nanoparticle arrays for high-sensitivity bioassays, in the Department of Chemistry, National Taiwan University with Professor Huan-Tsung Chang. |
 Chi-Wei Liu | Dr Chi-Wei Liu has worked on the preparation and applications of functional nanomaterials for highly selective and sensitive detection of heavy metal ions, proteins, and DNA. He obtained his PhD degree from the Department of Chemistry, National Taiwan University in July 2009. |
 Huan-Tsung Chang | Huan-Tsung Chang is Professor of Chemistry at National Taiwan University. The interests of his research group (http://www.ch.ntu.edu.tw/%7Ehtchang/) are broadly within the areas of bioanalytical chemistry and nanoscience focussing on the preparation and application of nanomaterials and the development of capillary electrophoresis-based separation systems. |
Introduction
Nanomaterials are materials of size 1–100 nm and have different chemical and physical properties from their bulk materials because of quantum effects.1–3 Nanomaterials are popular in many fields due to their unique optical, electronic, magnetic, and catalytic properties, which make them ideal candidates for signal generation and transduction in the detection of analytes of interest, including DNA,4–11proteins,12–20 and small solutes,21–35 and for sensing of cells such as cancer cells and neurons.36 Some interesting sensing nanomaterials include gold nanoparticles (Au NPs), quantum dots such as CdSe and CdTe,37–40TiO2 NPs,41SiO2 NPs,42,43 and Fe3O4 NPs.44,45 Among them, Au NPs are the most commonly used optical sensing nanomaterials because of their interesting size- and shape-dependent optical properties. This tunable optical property makes Au NPs ideal nanomaterials for sensing of analytes. With their great extinction coefficients, Au NPs are usually used in colorimetric-based detection systems.4–10,12–17,20–33 Recently, many Au NPs sensing systems based on fluorescence detection have been developed by taking advantage of their super quenching capability.11,18,19,34,35 In addition, fluorescent Au NPs, so-called gold nanodots, have become useful sensing materials because of their low toxicity and bio-compatible properties.19Au NPs are usually prepared from the reduction of Au ions (HAuCl4) by reducing agents such as citrate.46Citrate ions also act as capping agents to stabilize the as-prepared Au NPs. Au NPs by themselves do not have target recognition abilities necessary for selective binding of analytes of interest. In order to selectively detect analytes of interest, antibodies, proteins, short-chain organic acids, aminothiols, and DNA have been used to modify the surface properties of Au NPs to provide target recognition sites.1–3 These molecules can be introduced to Au NPs through electrostatic attractions or covalent bonding.4–35 Although the former is easier and more straightforward, they are not as stable as those prepared by the latter.
Because of their high sensitivity and selectivity, many Au NP-based sensing systems have been demonstrated for various purposes such as sensing of trace analytes and cell imaging. It is thus beyond our scope to review all of the papers. In this study, we focus on DNA functionalized Au NPs (DNA-Au NPs). Readers that are interested in different functionalized Au NPs should refer to other review papers.1–3DNA is one of the most important biomolecules for sensing analytes including DNA, proteins, small solutes, and metal ions.4–35 Since Mirkin's work on the detection of DNA through hybridization, DNA functionalized Au NPs have become popular sensing materials for many studies like single-nucleotide polymorphisms (SNPs). In addition to those DNA, aptamers14–16,18–22,27–29 and DNAzymes (acting as protein enzymes for catalytic reactions)25,26,32 that both contain short single stranded DNA or RNA are used to prepare bioconjugated Au NPs, mainly because of their specific binding ability for proteins and small solutes. Bioconjugation of Au NPs with DNA can be easily performed through covalent bonding or electrostatic attraction. The main advantages of covalent bonding through thiol or amino groups of DNA fragments are that the ratio of DNA to Au NPs is easily controlled and the DNA molecules are more stable on the surface. However, DNA modified with a thiol or amine moiety is relatively higher cost-wise. In addition, Au NPs that are bioconjugated with unmodified and modified DNA may coexist, which affects the sensitivity and reproducibility of the assays . On the other hand, modification of Au NPs with DNA through electrostatic attractions is quite simple and less expensive. However, in high-ionic-strength solution they are not as stable as covalently bonded DNA-Au NPs. In addition, the difficulty of controlling the conformation of DNA on the Au NP surface may be problematic.
Aptamers act as antibodies to recognize the target molecules, depending on their sequences and conformation. Aptamers can be isolated by a process called “SELEX”, an acronym for “systematic evolution of ligands by exponential enrichment”. Aptamers can fold into complex three-dimensional shapes, forming binding pockets and clefts for the specific recognition and tight binding of any given molecular target, from metal ions and small solutes to large proteins and higher order protein complexes, whole cells, viruses, or parasites.47–49Aptamers are advantageous over antibodies, including a high stability of their structures, simplicity of their synthesis, ease of structural modification, and high affinity.
This review focuses on DNA-Au NPs for the detection of biopolymers and small solutes through colorimetric, fluorescent, and scattering approaches. We discuss important factors such as the size and concentration of Au NPs, the length and sequence of DNA, the nature of capping agents, and the ionic strength and pH of solution that play important roles in determining the specificity and sensitivity of the nanosensors for the analytes. The advantages and disadvantages of the DNA-Au NPs for bioanalysis will be highlighted. We also briefly address the future trend of DNA functionalized nanomaterials.
1. Colorimetric detection
Bulk gold has a familiar golden color, due to a reduction in reflectivity for light at the end of the spectrum in the visible region.50 When a particle of gold is small enough, its color is ruby red, due to its strong absorption of green light at about 520 nm, corresponding to the frequency at which a plasmon resonance occurs within the gold metal. Kreibig and Vollmer have shown that plasmon excitation is present when the radius of a particle is large compared with the wavelength of light.51 Freely mobile electrons are trapped in such metal boxes and show a characteristic collective oscillation frequency of the plasma resonance, giving rise to the so-called surface plasmon resonance (SPR) band observed at near 520–530 nm for the Au NPs having sizes ranging 5–20 nm in diameter.3 For such small sizes of Au NPs, there is a gap between the valence band and the conduction band, unlike in bulk gold materials.It's well known that the intrinsic properties of Au NPs are mainly governed by their size, shape, crystallinity, and structure.3 For example, the SPR band of spherical Au NPs undergoes red-shifts when their size becomes larger. Unlike spherical Au NPs, there are two SPR bands (transverse and longitudinal) in rod-like nanomaterials, such as gold and gold–silver nanorods.52–54 By using such interesting optical characteristics, many sensing systems have been invoked based on colorimetric changes due to alternation in their absorption spectra upon aggregation. Aggregation can be through cross-linking and non-cross-linking events. Cross-linking aggregation among Au NPs is through specific binding such as DNA–DNA hybridization, antibody–antigen interaction, and metal–ligand coordination. Non-cross-linking aggregation usually occurs through analyte-induced changes in the surface charge density on the surfaces of Au NPs. Electrolyte,55 ligand,56–58 and acid-induced aggregations59–61 are the three most common phenomena occurring in the Au NP sensing systems. They are all about screening the surface charge (non-cross-linking aggregation) and inducing some cross-linking aggregation. Thus, factors affecting the surface properties of Au NPs have been suggested as important in determining aggregation of Au NPs, including nanoparticle size, species and concentration of ligands, pH, ionic strength, solvent composition, and temperature.62
1.1 Cross-linking assays
In 1996, Mirkin et al. first reported DNA-Au NPs for the detection of target DNA based on the aggregation of Au NPs through hybridization of two complementary DNA strands on Au NPs.63 Interestingly, the melting profile of the DNA-Au NP aggregates was extraordinarily sharp, occurring over a temperature range much narrower than that of the original DNA. By controlling temperature, this approach provides a limit of detection (LOD) down to nanomolar levels for DNA, with a single base mismatch resolution. In order to further improve the sensitivity, bio-barcode amplification approaches using two different particles including Au NPs and magnetic microparticles have been demonstrated.5,12,13 Magnetic microparticles that have a magnetic iron oxide core with an amine-modified silane coating were functionalized with alkanethiol-capped oligonucleotides. The oligonucleotides are complementary to the 12-mer portion (5′-ATTGATAAGGAT-3′) of a target sequence using a sulfosuccinimidyl 4-N-maleimidomethyl cyclohexane-1-carboxylate (sulfo-SMCC) linker. The second probe is Au NPs (30 nm) modified with two types of oligonucleotides; one is complementary to the target sequence of interest (5′-GGATTATTGTTAAAT-3′) and the other is complementary to a barcode sequence that is a unique identification tag for the target sequence. In this approach, the signal after silver enhancement was detected by a scanometric detection system, which provided an LOD down to 500 zM that is comparable to those obtained by PCR-based approaches. An isothermal amplification method with visual, colorimetric readout based on aggregation of DNA-Au NPs was developed for the detection of short sequences of DNA.6 The aggregation of DNA-Au NPs is initiated by a trigger oligonucleotide that is capable of bridging two sets of DNA in the DNA-Au NPs. The resulting color change from red to dark purple or blue is enhanced through spotting the solution onto a C18 reversed-phase thin-layer chromatography plate, leading to a detection of as low as 100 fM trigger oligonucleotides.A highly specific sensing system using aptamer-modified Au NPs (Apt-Au NPs) was developed for the detection of platelet-derived growth factors (PDGF).14PDGF is a growth factorprotein found in human platelets; it has growth-promoting activity toward fibroblasts, smooth muscle cells, and glial cells.64–68 The color of Apt-Au NPs changes from red to purple at low concentration (<400 nM) as the result of aggregation with cross-linking between the Au NPs. Interestingly, the color is reversed to red at very high PDGF concentrations (>400 nM) due to the repulsion and steric effects because the surface of the Apt-Au NPs quickly becomes saturated with PDGF molecules through aptamer-PDGF binding (Fig. 1). By plotting the ratios of the extinction coefficients of the Apt-Au NPs at 650 and 530 nm against the concentrations of PDGF-AA, the linear ranges of the increases and decreases in this extinction ratio are 25–75 and 75–200 nM, respectively. Furthermore, a homogeneous assay was developed to detect the PDGF receptor-β (PDGFR-β) at a concentration as low as 3.2 nM, on the basis of the competition between the Apt-Au NPs and PDGFR-β for PDGF-BB.
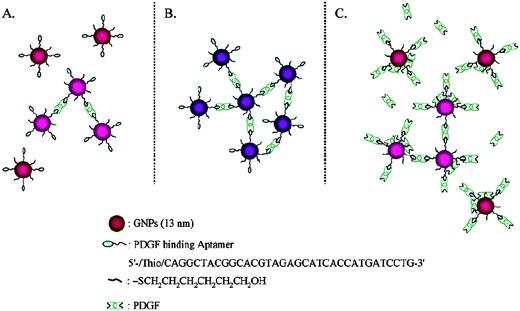 | ||
| Fig. 1 Schematic representation of the aggregation of Apt-Au NPs in the presence of PDGFs at (A) low, (B) medium, and (C) high concentrations. Reprinted with permission from ref. 14. | ||
DNA-Au NPs have proved sensitive and specific for the determination of heavy metal ions such as Hg2+. One example is the colorimetric detection of Hg2+ using two different DNA functionalized Au NPs, each functionalized with different thiolated-DNA sequences (5′-thiol-C10A10TA10 and 5′-thiol-C10T10TT10), which are complementary except for a single thymine–thymine (T–T) mismatch. It is known that Hg2+ ions coordinate selectively to a T–T pair.69–72 The Au NPs maintains aggregation at higher temperature (T < Tm) in the presence of Hg2+ ions. However, Au NP aggregates are re-dispersed in the presence of other metal ions. Because each increase in concentration of 1 µM Hg2+ results in an increase in Tm by about 5 °C, measurement of the changes in Tm is an easy way for determining Hg2+ concentration. This sensing system provides an LOD of 100 nM for Hg2+ ions. Alternatively, detection of DNA has been demonstrated using two DNA-functionalized Au NPs (3′-thiol-ATGCTCAACTCT and 5′-thiol-CGCATTCAGGAT) and an appropriate oligonucleotide linker. The linker has a sequence that recognizes the two particle probes and forms stable DNA duplexes (or Au NP aggregated networks) only in the presence of Hg2+ at a given operating temperature (room temperature). In the absence of Hg2+, these three DNA fragments do not form DNA-Au NP aggregates due to mismatches formed in the DNA duplexes. This approach provides LODs of 1 µM of Hg2+ by the naked eye. When compared to others using thiol-functionalized Au NPs, the DNA-Au NP sensor provides comparable sensitivity. Although the cost of DNA-Au NPs is higher, their selectivity is greater. When compared with MPA-Au NPs, the DNA (thymine)-Au NPs provide superior selectivity for mercury.73,74
When some Au NPs were bound to white microspheres, they were easily seen by the naked eye. Detection of target DNA by sandwiching it between white DNA-microspheres as separator and red DNA-Au NPs as reporter has been demonstrated.75 When compared to the above mentioned approaches, by the naked eye this approach is more sensitive because the color change is from white to red. A similar sensing system was developed for detecting proteins using biotinylated pyridyldithio propionate dextran microspheres and streptavidin–Au NPs through biotin–streptavidin specific binding.76 In order to further improve the sensitivity of microsphere assays for DNA, an approach based on silver staining Au NPs retained on filters has been demonstrated.77 Adding silver enhancement solution to Au NPs, Ag ions are deposited on their surfaces to form an Au–Ag core shell, leading to changes in the color of the spot from red to black. As a result, the contrast improves. This approach allows nanogram amounts of specific genomic DNA to be detected without conducting polymerase chain reactions .
Color changes due to growth of Apt-Au NPs after selective binding to target protein have been applied to the detection of protein.16 After the interaction between Apt-Au NPs (12.0 nm in diameter) and thrombin is equilibrated, Au NPs are subjected to growth in the presence of HAuCl4 and nicotinamide adenine dinucleotide on the glass surface. Upon increasing the concentration of thrombin in the parent solution, more Au NP aggregates form. As a result, the absorbance of the Au NPs after growth increases upon increasing thrombin concentration. A colorimetric assay using antibody functional DNA-Au NPs for signal reporter and amplification was demonstrated.17 On the first layer, the target protein is captured by the bifunctional Au NPs through antibody–antigen interaction on the plate surface. After capture, other DNA′-Au NPs (DNA′ and DNA are complementary) are used to increase the amount of Au NPs on the plate surface through DNA hybridization (second layer of DNA functionalized Au NPs), leading to amplified signals.
1.2 Non-cross-linking assays
The aggregation phenomenon of DNA-Au NP through non-cross linking induced by hybridization of target DNA at high salt concentrations has been demonstrated.8 Bare Au NPs (without capping with the probe DNA) immediately aggregate at 0.1 M NaCl. In contrast, the DNA-Au NP does not exhibit any visible change within the experimental range up to 2.5 M NaCl. In the presence of a target DNA with complementary sequence to the probe DNA, a clear colorimetric change from red into purples is immediately observed (<3 min) at a NaCl concentration higher than 0.5 M, representing the particle aggregation. At high salt concentration, Au NPs bioconjugated with double stranded DNA is less stable than those with single stranded DNA.8 This transition may reduce two parts of the repulsive interaction between NPs: (1) electrostatic repulsion possibly decreases by the screening effect because the tight conformation raises the binding constant with counterions, and (2) steric repulsion may reduce because the stiffening of the DNA lowers the entropic effect. This simple approach allows the detection of single-base mismatched DNA, with an LOD of 500 nM. The non-cross-linking aggregation process is much more rapid than that of the cross-linking systems, which takes several tens of minutes to hours at room temperature. In this non-cross-linking system, the aggregation is driven by the London–van der Waals attractive force between the NPs when the repulsive interaction is greatly reduced by formation of duplexes on their surfaces. On the other hand, in the cross-linking systems, the kinetics of the aggregation are dominated by random collisions between the NPs with relatively slow Brownian motion. In terms of specificity, cross-linking systems are advantageous over non-cross-linking systems, mainly due to their greater stability at high-ionic strength and ease of controlling optimum conformation on the surface of Au NPs.Apt-Au NPs are sensitive for the detection of small solutes such as adenosine triphosphate (ATP) and cocaine.21,22,27–29 An aptamer with a sequence (5′-T6ACCTGGGGGAGTATTGCGGAGGAAGGT-3′) that has a specific affinity for ATP was used to bioconjugate Au NPs for detection of ATP. In the absence of the analytes, the color of the Apt-Au NP solution changes from wine-red to purple as a result of salt-induced aggregation (Fig. 2). Binding of the analytes to the Apt-Au NPs induces folding of the aptamers on the Au NP surfaces into four-stranded tetraplex structures (G-quartet) and/or an increase in charge density. As a result, the Apt-Au NP solution is wine-red in color in the presence of the analytes under high salt conditions (150.0 mM NaCl; pH 7.4). The study suggests that the density of DNA modification on Au NPs is important in determining the sensitivity for ATP; 35 DNA molecules per Au NP is optimum. The sensitivity of this approach for ATP was further improved (100-fold) in the presence of polyethylene glycol and TOTO-3 (intercalator for DNA), mainly due to the greater binding between the aptamer and ATP. Polyethylene glycol promotes the structural rigidity of the G-quartet and TOTO-3 stabilizes the stem structure of the aptamer. The linear range and LOD of this sensing system for ATP are 20.0–100.0 nM and 10.0 nM, respectively. A similar study shows the detection of ATP using Au NPs bioconjugated with a DNA that has a complementary sequence to the adenosine-binding aptamer.27DNA-Au NPs hybridized with the aptamers are significantly more stable than DNA-Au NPs in the presence of MgCl2 at concentrations as high as 35 mM. Upon addition of the aptamer target (adenosine, 1 mM), the solution of DNA-Au NPs immediately changes from red to purple. The addition of adenosine leads to dehybridization of the aptamer from the DNA-Au NPs and decreases the resistant to induce a color change. In contrast, inosine, guanosine, and cytosine (all at 1 mM) do not show any color change.
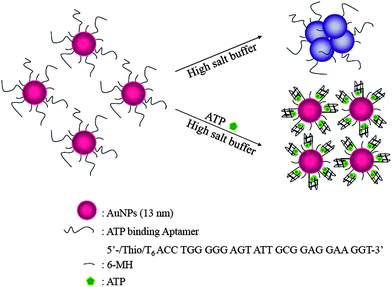 | ||
| Fig. 2 Schematic representation of the sensing mechanism of Apt-Au NPs for the colorimetric determination of ATP. Reprinted with permission from ref. 28. | ||
Unlike bioconjugation of DNA to Au NPs through Au–S bonding, ss-DNA can bind to citrate-capped Au NPs through DNA base-gold electrostatic interactions.9,10 As a result, DNA-Au NPs are stable in high-salt solution (up to 50 mM NaCl). In contrast, ds-DNA that is formed by the hybridization of ss-DNA and its complementary DNA target, shows little binding affinity to citrate-capped Au NPs, because the DNA bases are not free to bind to Au NPs. As a result, ds-DNA-Au NPs are not as stable as ss-DNA-Au NPs in high-salt solution. By carefully controlling salt concentration and selection of DNA sequences, this approach allows distinction of SNPs, with an LOD of 4.3 nM.10
A simple and cost-effective assay —using thymine (T)33 and Au NPs—has been demonstrated for the sensitive and selective detection of Hg2+ ions.30 The sensing mechanism takes advantage of the formation of DNA–Hg2+ complexes through T–Hg2+–T coordination to control the negative charge density of DNA strands—thereby varying their structures—adsorbed onto Au NPs (Fig. 3). The stability of the DNA-Au NPs (Tn/Au NPs; non-covalent bonding) increases in the order T80/Au NPs > T33/Au NPs > T7/Au NPs. Of the three DNA solutions, the degree of Hg2+-induced aggregation of Au NPs in the presence of T7 is small, mainly because of the difficulty encountered by T7 in forming folded structures. The solution of Au NPs is more sensitive toward Hg2+ in the presence of T33 than in the presence of T80, mainly because of the greater capability of T80 to stabilize the Au NPs. This approach provides an LOD of 250 nM for Hg2+ by the naked eye and the responds selectively toward Hg2+ ions—by a factor of 50-fold or more relative to the other metal ions. This approach avoids the need for conducting tedious modification and purification processes and use of expensive thiol-functionalized DNA molecules.
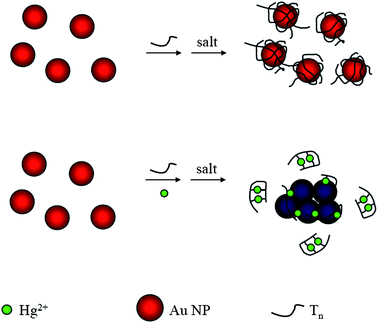 | ||
| Fig. 3 Schematic representation of Hg2+ nanosensors. Reprinted with permission from ref. 30. | ||
Similarly, a label-free Hg2+ sensor was developed, in which thymine-rich DNA molecules affect Au NP stability differently in the presence and absence of the analyte.31 Several similar strategies have also been demonstrated for sensing various analytes, including K+, Pb2+, ATP, and thrombin using Apt-Au NPs and DNAzyme-Au NPs.15,29,32,33
1.3 De-cross-linking assays
De-cross-linking (or called disassembly of Au NPs aggregation) means that the aggregation of Au NPs is reversible and the color of the solution could also turn from blue/purple to red. Recently, some de-cross-linking assays have been developed.7,20,22,25,26 For example, a reverse color change of DNA-Au NPs was demonstrated for SNP study under controlled temperature.7 Two Au NPs are sandwiched between the target DNA and DNA-Au NPs, which can be linked by the DNA ligase. The assay process involves three steps: a hybridization reaction that allows two Au NP-tagged probes to hybridize with the target DNA strand, a ligase reaction that generates the ligation between perfectly matched probes while no ligation occurs between mismatched ones, and a thermal treatment at a temperature (75 °C) that discriminates the ligation of probes. When the reaction mixture is heated to denature the formed duplex, the purple color of the perfect-match solution can not revert to red, while the mismatch gives a red color as the assembled Au NPs disparted. The identification of a single-base mutation in codon 12 of a K-ras oncogene shows the potential of this approach for colorectal cancer diagnosis.Two different thiol-modified oligonucleotide strands, DNA-1 and DNA-2, were used to prepare DNA-1-Au NPs and DNA-2-Au NPs, respectively. DNA-1 and DNA-2 are complementary to each other. Therefore, DNA-1-Au NPs and DNA-2-Au NPs can hybridize to form a cross-linked network of the Au NPs, leading to a color change from red to purple. As the endonuclease degrades the DNA-duplex interconnects, particles are released, regenerating a red color due to the dispersed Au NPs. The DNA-Au NPs aggregates were used to evaluate the enzymatic activity of deoxyribonuclease (DNase) I.20 As a result, the nucleic acid hydrolysis catalyzed by DNase I can be measured by measuring the absorbance at 520 nm. The reaction rate increases upon increasing enzyme concentration, which can be followed in real time.
Apt-Au NPs have been developed for the detection of adenosine and cocaine based on reversed sensing mechanisms (from aggregation to suspension).22 The sensor is based on aggregation of DNA-Au NPs (3′- and 5′- thiol-modified DNA) and a linker aptamer molecule that can be divided into two segments including hybridized and recognized sites. In the presence of adenosine, the aptamer changes its structure to bind adenosine, leading to disruption of aggregates and thus a change in the solution color from purple to red at room temperature. Under the optimized conditions, the approach provides an LOD of 20 µM for the adenosine by the naked eye. Similar approaches have been demonstrated for detection of Pb2+ ions within 10 min.25,26 The design of the lead biosensor is depicted in Fig. 4. The system consists of 5′-thiol-modified 12-mer DNA attached to 13-nm-diameter Au NPs, a DNAzyme (17E), and its substrate (SubAu). The sequence of the SubAu is designed so that it can hybridize specifically to a DNA-Au on each end, while maintaining the 17E recognition portion. These hybridizations cause aggregation of Au NPs and result in a blue color. However, in the presence of Pb2+ ions, the 17E catalyzes hydrolytic cleavage of SubAu and prevents the formation of NP aggregates. A red color appears as a result. The LOD of this sensor system is down at the sub-micromolor level for Pb2+ ions.
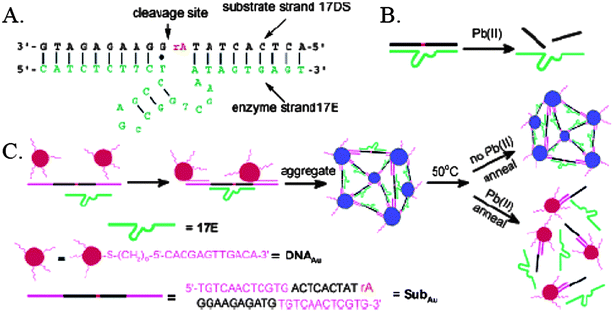 | ||
| Fig. 4 (A) Secondary structure of the “8–17” DNAzyme system that consists of an enzyme strand (17E) and a substrate strand (17DS). The cleavage site is indicated by a black arrow. Except for a ribonucleoside adenosine at the cleavage site (rA), all other nucleosides are deoxyribonucleosides. (B) Cleavage of 17DS by 17E in the presence of Pb2+. (C) Schematics of DNAzyme-directed assembly of Au NPs and their application as biosensors for metal ions such as Pb2+. In this system, the 17DS has been extended on both the 3′ and 5′ ends for 12 bases, which are complementary to the 12-mer DNA attached to the 13-nm gold nanoparticles (DNA-Au NPs). Reprinted with permission from ref. 25. | ||
2. Fluorescence detection
Fluorescence detection is more sensitive than absorption detection when using traditional dyes. Although Au NPs having sizes larger than 3 nm do not fluoresce, detection of analytes using Au NPs based on fluorescence quenching has been realized.11,18,19,34,35,74 These successful studies take advantage of Au NPs that act as super quenchers for fluorophores through both energy-transfer and electron-transfer processes. Au NPs have a Stern–Volmer quenching constant (KSV) that is greater with several (>5) orders of magnitude than that of typical small molecule dyequencher pairs.78–80 This super quenching property of Au NPs allows them to be employed as effective proximal quenchers in optical detection.Molecular beacons (MBs) conjugated with Au NPs have become interesting in DNA detection.81,82 MBs are DNA sequences composed of one target-recognition region of about 15–30 bases flanked by two short complementary stem sequences, leading to formation of a stem–loop structure in the absence of a target.83 To allow the monitoring of conformation changes in MB upon interactions with targeted DNA, a fluorophore and a quencher are covalently conjugated at the termini of each MB strand. They act as fluorescence resonance energy transfer-based switches that are normally in the closed or “fluorescence off” state, but switch to the open or “fluorescence on” state in the presence of target (complementary) DNA strands. However, a drawback of MBs is the low quenching efficiency of the molecular quencher . When the molecular quencher is replaced with Au NPs, the quenching is much more efficient, resulting in a more sensitive probe.81DNA-Au NPs (1.4 nm diameter) and fluorophores including fluorescein , rhodamine 6G, texas red, and Cy5 were used for the detection of DNA. In the absence of the target DNA, the fluorescence is highly quenched by the NPs through a distance dependent process. The fluorescence of this hybrid molecule increases by a factor of as many as several thousand as it binds to a complementary ssDNA. The probes have higher single base mismatch selectivity (25-fold) when compared to conventional MBs (4-fold). Unlike use of MBs, dye (fluorescein and tetramethylrhodamine) labeled thiol-oligonucleotides (complementary sequence) bioconjugated Au NPs were used for detection of DNA.82 The fluorophores nonspecifically bind to the gold surface in the absence of target DNA, resulting in no fluorescence. When DNA-Au NPs recognize perfectly matched DNA, hybridization occurs, leading to a longer distance between Au NPs and fluorophores. As a result, the solution fluoresces more strongly, allowing a detection of DNA down to 40 nM.
DNA-Au NPs as fluorescence quenchers have been demonstrated for the detection of proteins.18,19 An Apt-Au NP-based molecular light switching sensor was prepared for the analysis of PDGFs and their receptors in homogeneous solutions. The PDGF bindingaptamer has a unique structure with a triple-helix conformation that allows N,N-dimethyl-2,7-diazapyrenium dication (DMDAP) and PDGF bindings (Fig. 5).18 The fluorescence of DMDAP is almost completely quenched by Apt-Au NPs when it intercalates with the aptamers. Owing to high magnitudes of increases (up to 40-fold) in the turn-on fluorescence signals of DMDAP/Apt-Au NP upon PDGF binding, the approach is highly sensitive for the detection of PDGFs.
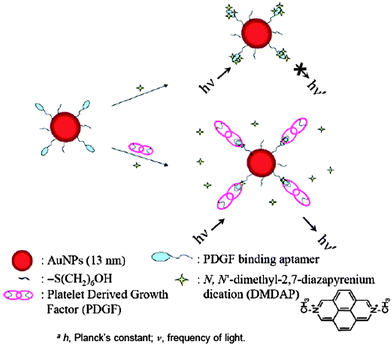 | ||
| Fig. 5 Schematic representations of PDGF nanosensors that operate based on modulation of the fluorescence resonance energy transfer between DMDAP and Apt-Au NPs. Reprinted with permission from ref. 18. | ||
Alternatively, two differently sized Au NPs, acting separately as donor and acceptor, were used for detection of PDGF in homogeneous photoluminescence quenching assays .19 Introduction of PDGF AA to a solution of 11-mercaptoundecanoic acid-protected, 2.0 nm photoluminescent Au nanodots (LAuND) led to the preparation of PDGF AA-LAuND as the donor. Thiol derivative PDGF bindingaptamers and 13 nm spherical Au NPs were used to synthesize the Apt-QAuNP acceptor. The photoluminescence of PDGF AA-LAuND at 520 nm decreases when photoluminescence quenching occurs between Apt-QAuNP and PDGF AA-LAuND. The PDGF AA-LAuND/Apt-QAuNP-based molecular light switching system has been applied to analyze PDGFs and PDGFR-β in separate homogeneous solutions. In the presence of PDGFs, the interaction between Apt-QAuNP and PDGF AA-LAuND decreases as a result of competitive reactions between the PDGFs and Apt-QAuNP (Fig. 6). Similarly, the interaction between Apt-QAuNP and PDGF AA-LAuND reduces as a result of competitive reactions between PDGFR-β and PDGF AA-LAuND. The LODs for PDGF AA and PDGFR-β are 80 pM and 0.25 nM, respectively, resulting from a low background photoluminescence signal. When using the Apt-QAuNP as selectors for (a) the enrichment of PDGF AA and (b) the removal of matrixes possessing intense background fluorescence from cell media and urine samples, the LOD for PDGF AA is 10 pM.
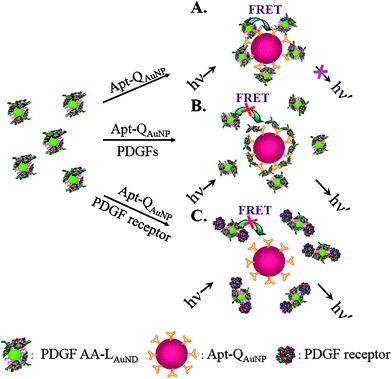 | ||
| Fig. 6 Schematic representations of PDGF and PDGF receptor nanosensors that operate based on the modulation of the photoluminescence quenching between PDGF AA-LAuND and Apt-QAuNP. Reprinted with permission from ref. 19. | ||
A new highly selective and sensitive technique has been demonstrated for the detection of Hg2+ using DNA-Au NPs and OliGreen.34 This system is the first that allows the detection of Hg2+ based on the release of DNA molecules, induced by conformational changes on Au NP surfaces, and its sensitivity is highly dependent upon surface DNA density. When Hg2+ ions interact with the thymine units of the DNA molecules bound to the Au NPs through Au–S bonds (more than 60 DNA per Au NPs), the conformations of these DNA derivatives change from linear to hairpin structures, causing the release of some of the DNA molecules from the surface of the Au NPs into the bulk solution to react with OliGreen. The fluorescence of OliGreen–DNA complexes increases upon increasing concentration of Hg2+, providing an LOD of 25 nM for Hg2+. A linear correlation exists between the fluorescence intensity and the concentration of Hg2+ over the range 0.05–2.5 µM (R2 = 0.98). A mercury sensor using thymine bioconjugated Au NPs was demonstrated for the detection of Hg2+ down to 40 nM in aqueous solution.35 The dye-tagged ssDNA containing thymine sequences was chosen as Hg2+ acceptor. At high ionic strength, the formation of a T–Hg2+–T complex reduces the capability to stabilize bare Au NPs against salt-induced aggregation, remaining a blue in the color of the solution, but the fluorescence signal increases upon increasing Hg2+ concentration.
3. Scattering detection
Unlike fluorescence detection, fluorophores are not needed in light scattering approaches. In addition, photobleaching is less problematic. Scattering detection approaches are simply based on analyte-induced aggregation of Au NPs, leading to increased scattering light. We point out that scattering intensity and wavelength is dependent on the nature, size and shape of Au NPs. Although scattering approaches are quite simple and sensitive, their quantitative results are less reproducible.Light scattered by Au NPs is strong and stable. This property allows the detection of 10−16 M Au NPs with an average diameter of 118 nm.84 Many scattering-based techniques such as the scanometric method, surface plasmon resonance (SPR), laser diffraction, surface enhanced Raman spectroscopy (SERS), colorimetric scattering on a glass waveguide using Au NPs have been demonstrated for DNA detection.5,85–87 These approaches can push the LODs of target DNA to picomolar, and even femtomolar values. However, each of these assays needs to detect Au NPs on a solid phase and inevitably needs a sophisticated process and an expensive instrument. A simple and sensitive light-scattering technique using DNA-Au NPs has been developed for detecting DNA in homogeneous solution.88 One feature of homogeneous scattering assays is their capability for monitoring the synthesis of specific nucleic acids in real time. A homogeneous scattering assay using DNA-Au NPs was developed for detecting DNA, a 30-base fragment of human p53 gene (exon 8).89 The assay relies on the observation of greatly enhanced light scattering that originated from the aggregation of DNA-Au NPs directed by the target DNA. Probe 1 and 2, both made of 15 bases, are complementary to part of the target DNA. The quantitative assessment of the proposed light-scattering assay for DNA hybridization is based on monitoring the light-scattering intensity at 315 nm that is dependent on the target-DNA concentration. The corresponding calibration plot of light-scattering intensity versus the target-DNA concentration is linear over the range of 0.7–119 pM with an LOD of 0.1 pM. A scattering approach using DNA-Au NPs was developed for the identification of two different DNA targets in a solution.90 The immobilized DNA probe, the target, and the DNA-Au NPs were designed to cohybridize in a three-component sandwich assay . The 50 and 100 nm-diameter DNA-Au NPs were used to identify two different target sequences. Green and orange light are observed when 50 and 100 nm Au NPs are attached to the surface, respectively. Based on the features, oligonucleotide arrays can be fabricated to simultaneously detect many DNA molecules in one solution.
Several special scattering techniques using DNA-Au NPs have been developed for detecting DNA. Like fluorescence correlation spectroscopy, a scattering technique allowing one to obtain scattering-light intensities and a diffusion time of Au NPs was demonstrated.91 In the presence of target DNA, the intensity of the scattering light sharply increases and the diffusion time is prolonged. This result indicates that new structures with bigger hydrodynamic radii (hybridization products) form during the hybridization reaction. Based on the diffusion time and Stokes–Einstein equation, the average size of Au NPs was calculated to be 23 nm. The average sizes of aggregated Au NPs induced by single-base mismatch and perfect complementary target hybridization are about 32 and 61 nm, respectively. A linear relationship between target concentration and diffusion time, τ (s), exists at low target concentrations (<64 nM). A hyper-Rayleigh scattering technique using DNA-Au NPs was developed for the detection of DNA.92Hyper-Rayleigh scattering can be observed from fluctuations in symmetry, caused by rotational fluctuations, after the addition of target DNA into DNA-Au NPs (5′-GGCAACCTGAGGACCC-3′; 50 nM). The scattering intensity does not change when the target DNA with one base-pair mismatch with respect to the probe DNA is added. This is owing to the fact that in the presence of ss-DNA, the oligonucleotides adsorb onto the Au NPs through van der Waals electrostatic forces and provide negative charges that enhance the repulsion between the Au NPs. The electrostatic forces are due to dipolar interactions and depend on the configuration and orientation of the ss-DNA. As transient structural fluctuations permit short segments of the ss-DNA to uncoil, the bases face the Au NPs. Attractive electrostatic forces between the Au NPs and nucleotides cause the ss-DNA to adsorb onto the Au NPs. After hybridization, ss-DNA forms ds-DNA, resulting in the fact that the ds-DNA cannot uncoil sufficiently (unlike ss-DNA) to expose its bases toward the Au NPs. Repulsion between the charged phosphate backbone of ds-DNA and the citrate ions from the Au NPs dominates the electrostatic interaction and does not allow the ds-DNA to adsorb onto the Au NPs. As a result, Au NPs undergo aggregation in the presence of NaCl. The scattering signal changes even in the presence of a low concentration (10 nM) of complementary DNA.
The potential use of Apt-Au NPs for cancer cell detection has been recognized.36 Through specific binding of the aptamers toward PDGF, MDA-MB-231 and Hs578T cells (breast cancer cells) that over-express PDGF interact with Apt-Au NP to a greater extent than do H184B5F5/M10 cells (normal cells). Aggregation of the Apt-Au NPs in the cytoplasm of the cancer cells leads to the generation of an intense scattered light upon photo-illumination; this phenomenon allows the differentiation of cancer cells from normal cells using a dark field optical microscope. In addition, the presence of Apt-Au NPs suppress the proliferation of MDA-MB-231 cancer cell, but not H184B5F5/M10 cells.
4. Conclusions
The promise of DNA-Au NPs for the detection of various analytes based on absorption, fluorescence, and scattering detection is presented. DNA-Au NPs are stable, biocompatible, and easy to prepare. In addition, optical approaches using DNA-Au NPs are sensitive and selective as listed in Table 1. The excellent performance of the DNA-Au NPs-based sensor systems is due to highly selective interactions of the DNA with the target analytes and high amplification of signals. In addition, the multivalent effect due to the ultrahigh densities of DNA on the local surface of the Au NPs increases the binding constant more than two orders and improves the sensitivity.| Method | Probe | Target | Amplification | LOD | Time | Real sample | Ref. |
|---|---|---|---|---|---|---|---|
| a EXPAR: exponential nucleic acid amplification reaction. b Ab: antibody. c PSA: prostate-specific antigen. d ADDL: amyloid-β-derived diffusible ligand. e IgG: immunoglobulin G. f LOD without conducting polymerase chain reaction (PCR ). | |||||||
| Colorimetric cross-link | DNA-Au NPs | DNA | — | 10.0 nM | 15 min | — | 4 |
| DNA-Au NPs & DNA-MMPs | DNA | Bio-bar code | 500.0 zM | 3–4 h | — | 5 | |
| DNA-Au NPs | DNA | EXPARa | 100.0 fM | — | — | 6 | |
| Abb -Au NPs-DNA & Ab-MMPs | PSAc | Bio-bar code & PCR | 3.0 aM | — | — | 12 | |
| Ab-Au NPs-DNA & Ab-MMPs | ADDLd | Bio-bar code & PCR | <200 aM | — | CSF | 13 | |
| Apt-Au NPs | PDGF | — | 2.5 nM | 2 h | — | 14 | |
| Apt-Au NPs | Thrombin | Enlargement | 20 nM | — | — | 16 | |
| Ab-DNA-Au NPs | IgGe | Multiple layers of Au NPs | 2.0 pM | 5 h | — | 17 | |
| Apt/DNA-Au NPs | Adenosine | — | 20 µM | — | human blood serum | 21 | |
| DNA-Au NPs | Hg2+ | — | 100.0 nM | — | — | 23 | |
| DNA-Au NPs | Hg2+ | — | 1.0 µM | — | — | 24 | |
| Colorimetric non-cross-link | DNA-Au NPs | DNA | — | 500.0 nM | <3 min | — | 8 |
| DNA/Au NPs | DNA | PCR | 4.3 nMf | <10 min | PCR product | 9,10 | |
| Apt/Au NPs | Thrombin | — | 0.83 nM | 30 min | — | 15 | |
| Apt/DNA-Au NPs | Adenosine | — | 10 µM | 1 min | — | 27 | |
| Apt-Au NPs | ATP | — | 10.0 nM | 40 min | Urine | 28 | |
| Apt/Au NPs | ATP | — | 0.6 µM | 40 min | — | 29 | |
| Apt/Au NPs | Hg2+ | — | 250 nM | 20 min | — | 30 | |
| Apt/Au NPs | Hg2+ | — | 10.0 nM | 20 min | — | 31 | |
| Au NPs & DNAzyme & substrate | Pb2+ | — | 3.0 nM | 10 min | — | 32 | |
| Apt/Au NPs | K+ | — | 1.67 mM | 10 min | — | 33 | |
| Colorimetric de-cross-link | DNA-Au NPs | DNA | — | 74 pM | 100 min | — | 7 |
| DNA-Au NPs | DNase I | — | 1.0 µM | 5 min | — | 20 | |
| Apt/DNA-Au NPs | Adenosine | — | 100.0 µM | 10 s | — | 22 | |
| Apt/DNA-Au NPs | Cocaine | — | 10.0 µM | 1 min | — | 22 | |
| DNA-Au NPs & DNAzyme & substrate | Pb2+ | — | 100.0 nM | 10 min | — | 25,26 | |
| Fluorescence turn on | Dye-DNA-Au NPs | DNA | — | 67 pM | <1 min | — | 11 |
| Apt-Au NPs & DMDAP | PDGF | — | 8.0 pM | 1 h | — | 18 | |
| Apt-Au NPs & PDGFR-Au NDs | PDGF | — | 80.0 pM | 1 h | Urine and cell media | 19 | |
| Apt-Au NPs & OliGreen | Hg2+ | — | 25.0 nM | 70 min | Pond water | 34 | |
| Au NPs & Dye-Apt | Hg2+ | — | 40.0 nM | 30 min | — | 35 | |
As well as DNA that recognizes target DNA through complementary hybridization, aptamers and DNAzymes extend the practicality of DNA-Au NPs to other targets, such as metal ions, small molecules, proteins, even the cells. It is expected that DNA-Au NPs will become more popular when more and more functional DNA molecules are found and synthesized. With significant progress in the preparation of different sizes and shapes of Au NPs,52–54,93–95 the use of anisotropic Au NPs bioconjugated with DNA becomes more interesting. Because of their greater absorption in the near infrared region when compared to those of spherical Au NPs, their use for bioassays are advantageous, including high sensitivity and low matrix interference. However, anisotropic Au NPs are usually prepared in the presence of detergents or polymers, which makes their bioconjugation difficult. In addition to more common non-fluorescent Au NPs, fluorescent Au NPs (<2 nm in diameter) have been proved sensitive for the detection of proteins and Hg2+ ions through fluorescence quenching.19,96,97 In order to further improve the sensitivity when using fluorescent Au NPs, improvements in their fluorescence quantum yields and photostability are extremely important.
We are grateful to the National Science Council of Taiwan for providing financial support to this study under contracts NSC 98-2627-M-002-013 and NSC 98-2627-M-002-014. Y.-W. L. thanks the National Taiwan University for PDF support (96R8044).
Notes and references
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- R. Wilson, Chem. Rev., 2008, 37, 2028–2045 CrossRef CAS.
- R. Elghanian, J. J. Storhoff, R. C. Mucic, R. L. Letsinger and C. A. Mirkin, Science, 1997, 277, 1078–1081 CrossRef CAS.
- J.-M. Nam, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932–5933 CrossRef CAS.
- E. Tan, J. Wong, D. Nguyen, Y. Zhang, B. Erwin, L. K. Van Ness, S. M. Baker, D. J. Galas and A. Niemz, Anal. Chem., 2005, 77, 7984–7992 CrossRef CAS.
- J. Li, X. Chu, Y. Liu, J.-H. Jiang, Z. He, Z. Zhang, G. Shen and R.-Q. Yu, Nucleic Acids Res., 2005, 33, e168 CrossRef.
- K. Sato, K. Hosokawa and M. Maeda, J. Am. Chem. Soc., 2003, 125, 8102–8103 CrossRef CAS.
- H. Li and L. J. Rothberg, J. Am. Chem. Soc., 2004, 126, 10958–10961 CrossRef CAS.
- H. Li and L. J. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14036–14039 CrossRef CAS.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS.
- J. M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884–1886 CrossRef CAS.
- C. D. Keating, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2263–2264 CrossRef CAS.
- C.-C. Huang, Y.-F. Huang, Z. Cao, W. Tan and H.-T. Chang, Anal. Chem., 2005, 77, 5735–5741 CrossRef CAS.
- H. Wei, B. Li, J. Li, E. Wang and S. Dong, Chem. Commun., 2007, 3735–3737 RSC.
- V. Pavlov, Y. Xiao, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2004, 126, 11768–11769 CrossRef CAS.
- P. Hazarika, B. Ceyhan and C. M. Niemeyer, Small, 2005, 1, 844–848 CrossRef CAS.
- C.-C. Huang, S.-H. Chiu, Y.-F. Huang and H.-T. Chang, Anal. Chem., 2007, 79, 4798–4804 CrossRef CAS.
- C.-C. Huang, C.-K. Chiang, Z.-H. Lin, K.-H. Lee and H.-T. Chang, Anal. Chem., 2008, 80, 1497–1504 CrossRef CAS.
- X. Xu, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46, 3468–3470 CrossRef CAS.
- J. Liu, D. Mazumdar and Y. Lu, Angew. Chem., 2006, 118, 8123–8127 CrossRef.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 90–94 CrossRef CAS.
- J.-S. Lee, M. S. Han and C. A. Mirkin, Angew. Chem., 2007, 119, 4171–4174 CrossRef.
- X. Xue, F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 3244–3245 CrossRef CAS.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2003, 125, 6642–6643 CrossRef CAS.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2004, 126, 12298–12305 CrossRef CAS.
- W. Zhao, W. Chiuman, M. A. Brook and Y. Li, ChemBioChem, 2007, 8, 727–731 CrossRef CAS.
- S.-J. Chen, Y.-F. Huang, C.-C. Huang, K.-H. Lee, Z.-H. Lin and H.-T. Chang, Biosens. Bioelectron., 2008, 23, 1749–1753 CrossRef CAS.
- J. Wang, L. Wang, X. Liu, Z. Liang, S. Song, W. Li, G. Li and C. Fan, Adv. Mater., 2007, 19, 3943–3946 CrossRef CAS.
- C.-W. Liu, Y.-T. Hsieh, C.-C. Huang, Z.-H. Lin and H.-T. Chang, Chem. Commun., 2008, 2242–2244 RSC.
- D. Li, A. Wieckowska and I. Willner, Angew. Chem., Int. Ed., 2008, 47, 3927–3931 CrossRef CAS.
- Z. Wang, J. H. Lee and Y. Lu, Adv. Mater., 2008, 20, 3263–3267 CrossRef CAS.
- L. Wang, X. Liu, X. Hu, S. Songa and C. Fan, Chem. Commun., 2006, 3780–3782 RSC.
- C.-W. Liu, C.-C. Huang and H.-T. Chang, Langmuir, 2008, 24, 8346–8350 CrossRef CAS.
- H. Wang, Y. Wang, J. Jin and R. Yang, Anal. Chem., 2008, 80, 9021–9028 CrossRef CAS.
- Y.-F. Huang, Y.-W. Lin, Z.-H. Lin and H.-T. Chang, J. Nanopart. Res., 2009, 11, 775–783 CrossRef CAS.
- J. K. Jaiswal, H. Mattoussi, J. M. Mauro and S. M. Simon, Nat. Biotechnol., 2003, 21, 47–51 CrossRef CAS.
- B. M. Lingerfelt, H. Mattoussi, E. R. Goldman, J. M. Mauro and G. P. Anderson, Anal. Chem., 2003, 75, 4043–4049 CrossRef CAS.
- E. R. Goldman, A. R. Clapp, G. P. Anderson, H. T. Uyeda, J. M. Mauro, I. L. Medintz and H. Mattoussi, Anal. Chem., 2004, 76, 684–688 CrossRef CAS.
- C.-W. Liu and H.-T. Chang, Open Anal. Chem. J., 2007, 1, 1–6 Search PubMed.
- K.-H. Lee, C.-K. Chiang, Z.-H. Lin and H.-T. Chang, Rapid Commun. Mass Spectrom., 2007, 21, 2023–2030 CrossRef CAS.
- L. Wang and W. Tan, Nano Lett., 2006, 6, 84–88 CrossRef CAS.
- H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893–3957 CrossRef CAS.
- D.-J. Kim, Y.-K. Lyu, H.-N. Choi, I. H. Min and W.-Y. Lee, Chem. Commun., 2005, 2966–2968 RSC.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- R. C. Mucic, J. J. Storhoff, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 12674–12675 CrossRef CAS.
- M. Famulok and A. Jenne, Curr. Opin. Chem. Biol., 1998, 2, 320–327 CrossRef CAS.
- M. Famulok and G. Mayer, Curr. Top. Microbiol. Immunol., 1999, 243, 123–136 Search PubMed.
- J. Hesselberth, M. P. Robertson, S. Jhaveri and A. D. Ellington, J. Biotechnol., 2000, 74, 15–25 CAS.
- C. Cretu and E. van der Lingen, Gold Bull., 1999, 32, 115–126 CAS.
- U. Kreibig, M. Vollmer, Optical Properties of Metal Cluster; Springer: Verlag, 1995 Search PubMed.
- C.-C. Huang, Z. Yang and H.-T. Chang, Langmuir, 2004, 20, 6089–6092 CrossRef CAS.
- Y.-F. Huang, Y.-W. Lin and H.-T. Chang, Nanotechnology, 2006, 17, 4885–4894 CrossRef CAS.
- Y.-F. Huang, K.-M. Huang and H.-T. Chang, J. Colloid Interface Sci., 2006, 301, 145–154 CrossRef CAS.
- S. Aryal, R. Bahadur, N. Bhattarai, C. K. Kim and H. Y. Kim, J. Colloid Interface Sci., 2006, 299, 191–197 CrossRef CAS.
- Y. Choi, N.-H. Ho and C.-H. Tung, Angew. Chem., 2007, 119, 721–723 CrossRef.
- A. T. Gates, S. O. Fakayode, M. Lowry, G. M. Ganea, A. Murugeshu, J. W. Robinson, R. M. Srongin and I. M. Warner, Langmuir, 2008, 24, 4107–4113 CrossRef CAS.
- S. Basu and T. Pal, J. Nanosci. Nanotechnol., 2007, 7, 1904–1910 CrossRef CAS.
- J. K. Lim and S.-W. Joo, Appl. Spectrosc., 2006, 60, 847–852 CrossRef CAS.
- K. Naka, H. Tanaka and Y. Chujo, Polym. J., 2007, 39, 1122–1127 CrossRef CAS.
- J.-Y. Shim and V. K. Gupta, J. Colloid Interface Sci., 2007, 316, 977–983 CrossRef CAS.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107, 4797–4862 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 605–609.
- N. Kohler and A. Lipton, Exp. Cell Res., 1974, 87, 297–301 CrossRef CAS.
- R. Ross, J. Glomset, B. Kariya and L. Harker, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1207–1210 CrossRef CAS.
- A. Hammacher, U. Hellman, A. Johnsson, A. Ostman, K. Gunnarsson, B. Westermark, A. Wasteson and C.-H. Heldin, J. Biol. Chem., 1988, 263, 16493–16498 CAS.
- S. P. Evanko, E. W. Raines, R. Ross, L. I. Gold and T. N. Wight, Am. J. Pathol., 1998, 152, 533–546 Search PubMed.
- H.-J. Yeh, I. Silos-Santiago, Y.-X. Wang, R. J. George, W. D. Snider and T. F. Deuel, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 1952–1956 CrossRef CAS.
- S. Katz, J. Am. Chem. Soc., 1952, 74, 2238–2245 CrossRef CAS.
- T. Yamane and N. Davidson, J. Am. Chem. Soc., 1961, 83, 2599–2607 CrossRef CAS.
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS.
- Y. Tanaka, S. Oda, H. Yamaguchi, Y. Kondo, C. Kojima and A. Ono, J. Am. Chem. Soc., 2007, 129, 244–245 CrossRef CAS.
- C.-C. Huang and H.-T. Chang, Chem. Commun., 2007, 1215–1217 RSC.
- C.-C. Huang and H.-T. Chang, Anal. Chem., 2006, 78, 8332–8338 CrossRef CAS.
- R. A. Reynolds III, C. A. Mirkin and R. L. Letsinger, Pure Appl. Chem., 2000, 72, 229–235 CrossRef CAS.
- R. Wilson, Y. Chen and J. Aveyard, Chem. Commun., 2004, 1156–1156 RSC.
- P. Upadhyay, M. Hanif and S. Bhaskar, Clin. Microbiol. Infect., 2006, 12, 1118–1122 Search PubMed.
- P. V. Kamat, S. Barazzouk and S. Hotchandani, Angew. Chem., Int. Ed., 2002, 41, 2764–2767 CrossRef CAS.
- E. Dulkeith, A. C. Morteani, T. Niedereichholz, T. A. Klar, J. Feldmann, S. A. Levi, F. C. J. M. van Veggel, D. N. Reinhoudt, M. Möller and D. I. Gittins, Phys. Rev. Lett., 2002, 89, 203002 CrossRef CAS.
- C. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297–6301 CrossRef CAS.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365–370 CrossRef CAS.
- D. J. Maxwell, J. R. Taylor and S. Nie, J. Am. Chem. Soc., 2002, 124, 9606–9612 CrossRef CAS.
- S. Tyagi and F. R. Kramer, Nat. Biotechnol., 1996, 14, 303–308 CrossRef CAS.
- J. Yguerabide and E. E. Yguerabide, Anal. Biochem., 1998, 262, 137–156 CrossRef CAS.
- T. A. Taton, C. A. Mirkin and R. L. Letsinger, Science, 2000, 289, 1757–1760 CrossRef CAS.
- Y. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS.
- V. Kattumuri, M. Chandrasekhar, S. Guha, K. Raghuraman, K. V. Katti, K. Ghosh and R. J. Patel, Appl. Phys. Lett., 2006, 88, 153114–153116 CrossRef.
- X. Xu, D. G. Georganopoulou, H. D. Hill and C. A. Mirkin, Anal. Chem., 2007, 79, 6650–6654 CrossRef CAS.
- B.-A. Du, Z.-P. Li and C.-H. Liu, Angew. Chem., 2006, 118, 8190–8193 CrossRef.
- T. A. Taton, G. Lu and C. A. Mirkin, J. Am. Chem. Soc., 2001, 123, 5164–5165 CrossRef CAS.
- K. Wang, X. Qiu, C. Dong and J. Ren, ChemBioChem, 2007, 8, 1126–1129 CrossRef CAS.
- P. C. Ray, Angew. Chem., Int. Ed., 2006, 45, 1151–1154 CrossRef CAS.
- Z. Yang and H.-T. Chang, Nanotechnology, 2006, 17, 2304–2310 CrossRef CAS.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS.
- W. He, C. Z. Huang, Y. F. Li, J. P. Xie, R. G. Yang, P. F. Zhou and J. Wang, Anal. Chem., 2008, 80, 8424–8430 CrossRef CAS.
- C.-C. Huang, Z. Yang, K.-H. Lee and H.-T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824–6828 CrossRef CAS.
- C.-C. Huang, C.-T. Chen, Y.-C. Shiang, Z.-H. Lin and H.-T. Chang, Anal. Chem., 2009, 81, 875–882 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |