Continuous flow hollow fiber liquid-phase microextraction and monitoring of NSAIDpharmaceuticals in a sewage treatment plant effluent
Niklas
Larsson
*,
Estelle
Petersson
,
Marika
Rylander
and
Jan Åke
Jönsson
Division of Analytical Chemistry, Lund University, P.O. Box 124, 221 00, Lund, Sweden. E-mail: niklas.larsson@organic.lu.se; Fax: +46 46 222 82 09
First published on 7th September 2009
Abstract
A method for simultaneous extraction and quantification of four non-steroidal anti-inflammatory drugs (NSAIDs) based on continuous flow hollow fiber liquid-phase microextraction (CFHF-LPME) was developed. The effect of sample flow rate, acceptor flow rate, type of acceptor flow (continuous, semi-continuous or forward–backward), type of supported liquid membrane and sample volume was studied. The extraction of the final method was linear over an environmentally relevant concentration range and yielded high enrichment factors (720–940 times) in reagent water and (270–800 times) in sewage water for all analytes within 45 min. Repeatability was best (RSD 6–15%) during the first 30 min of extraction. The optimised method was used to monitor the occurrence and fate of the four NSAIDs in a Swedish sewage treatment plant (STP) effluent, which is discharged into a system of ponds before release into a river, during the period May–September 2008. All four analytes were detected at concentrations up to 0.92 µg L−1ketoprofen, 0.08 µg L−1naproxen, 0.43 µg L−1diclofenac and 0.25 µg L−1ibuprofen. A concentration drop during the summer was observed. For diclofenac and ketoprofen significant removal in the primary recipient pond system was observed. The presence of the studied pharmaceuticals in STP effluent together with concern about their environmental effects makes monitoring of their occurrence and knowledge of their environmental fate important. The proposed method provides a basis for automation of extraction towards on-site extraction using CFHF-LPME.
Introduction
Pharmaceuticals have become of increasing environmental concern during the last few years since several studies revealed that a number of different pharmaceutical compounds are frequently detected in surface waters.1,2 One major group of pharmaceuticals extensively used in many countries is non-steroid anti-inflammatory drugs (NSAIDs),2 including substances such as diclofenac, ibuprofen, ketoprofen and naproxen, which are widely used for treatment of pain and fever and therefore constitute the active ingredient in many common painkillers. After consumption by humans, pharmaceutical substances are excreted either as the parent compound or as metabolites and become part of the sewage water.1,3,4 Commonly used NSAIDs are acidic with a pKa in the range 4–55 and are water soluble at neutral pH, and this is why in sewage water adsorption to sludge is low and microbial degradation is the only relevant removal mechanism.6 The removal of several pharmaceuticals in sewage treatment plants (STPs) is often incomplete,3,7 and is why STPs are considered to be the main source of release of many pharmaceuticals into the environment.4,7NSAIDs belong to the pharmaceuticals most frequently detected in aquatic environments8 and concentrations of single NSAIDs found in STP recipient surface waters are usually in the range 0.01–1 µg L−1.2,4,7,9 Even though the concentrations thus are often low there is a continuous input of these substances into the environment.10There are indications that chronic effects from NSAIDs might occur at relevant environmental concentrations. For example, diclofenac has been shown to cause cellular damage in several organs such as liver, kidney and gills in rainbow trout, Oncorhynchus mykiss, at concentrations as low as 1 µg L−1.11 Heat shock protein 70 (hsp 70), a common agent in living cells for protection against toxic agents, is induced by ibuprofen in the liver of rainbow trout at an aqueous concentration of 1 µg L−1.12 It has been shown that toxicity of the NSAIDsdiclofenac, ibuprofen, naproxen and acetylsalicylic acid is additive since the mode of action (inhibition of the cyclooxygenase enzymes; COX-1 and COX-2) is the same for all substances.13 Thus, even if NSAIDs occur below their no observed effect concentration (NOEC), they can still together give rise to toxic effects. Studies also indicate that certain degradation products from diclofenac and naproxen seem to exhibit a higher toxic potency towards aquatic organisms than the parent compound themselves.10,14Diclofenac has been shown to bioaccumulate,11 and release of this compound into aqueous compartments poses an ecotoxicological risk for fish-eating birds. Diclofenac has been found to cause a widespread death of vultures.15
Based on their widespread consumption, occurrence in surface water and potentially hazardous environmental effects, monitoring of NSAIDs and knowing their environmental fate is an area of environmental concern. Since environmental water samples often are complex and NSAIDs often occurs at trace levels, extraction and pre-concentration methods are needed prior to final analysis.
Solid phase extraction (SPE) is today the prevailing extraction technique for extraction of NSAIDs from water matrices.8 Selectivity may be increased using molecularly imprinted SPE,16 but selectivity towards the desired analytes is often rather low since the possibility to tune the extraction by chemical additives is limited.17 For polar analytes, a breakthrough might also occur.17 González-Barreiro report enrichment factors in the range 10–100 for extraction of diclofenac, ketoprofen and naproxen from wastewater.18SPE also requires non-negligible amounts of solvent17 and is not applicable for continuous sampling in flowing systems.19
Solid-phase microextraction (SPME) has emerged as a popular alternative to SPE. Here the analytes are extracted onto a solid polymeric fiber placed on the needle of a syringe, followed by thermal desorption and GC analysis.20SPME has several advantages over SPE such as being faster, requiring smaller sample volume, can be automated easier, yields high enrichment factors and does not consume organic solvent.8 A major drawback with SPME is however problems with HPLC compatibility.21
SPE and SPME can thus suffer from certain drawbacks regarding extraction of polar organic compounds at trace levels in aqueous samples, and is why supported liquid membrane (SLM) extraction may be an attractive alternative. An SLM is a flat sheet membrane or a porous hollow fiber impregnated with an organic solvent, which is immobilised in the pores by capillary forces.17 Using hollow fibers (HF), analytical membrane extraction is often called liquid-phase microextraction (LPME).20LPME is based on the same chemical principle as LLE, but consumes only microliters of solvent. Ionisable analytes in neutral form can be extracted through the membrane and the extraction can be selectively tuned, depending mainly on pH in sample and extract. The addition of carrier to the sample or the membrane may increase extraction, and for some analytes the use of a carrier is mandatory.22 Charged species and macromolecules are excluded, while smaller neutral molecules are not trapped in the acceptor solution.17,20 Considerably cleaner extracts have been obtained with SLM than with SPE, leading to more reliable identification with the former method.23
Membrane extraction has previously been applied to NSAIDs in environmental samples. With batch-type LPME of five NSAIDs, enrichment factors of 56–154 were reported.24 For a toxic ibuprofen degradation product, enrichment factors of 425 and 295 were obtained in river and sewage water, respectively.25 Lee and coworkers extracted ibuprofen26 as well as ketoprofen and naproxen9 in wastewater, with two-step LPME. Even though high enrichment factors were obtained with two-step LPME (∼19009–1500026), it is not easily automated. Müller et al. developed a semi-automated HF membrane extraction method for pharmaceutical compounds, enriching ibuprofen from distilled water 187 times and from sat. NaCl solution 415 times.27
Though straightforward, spot sampling is time-consuming and requires care in sample preservation.28,29 In a survey of six STPs, the more time-representative approach of composite sampling was used to survey the occurrence of pharmaceuticals, including ibuprofen.4 However, both spot and composite sampling can miss episodic events, which can only be detected by either biological early warning systems or by on-line continuous measurements.29 Since NSAIDs are frequently sampled at STPs, it may be useful with a method for continuous sampling of these analytes. Continuous membrane extraction has the property of being applicable in systems with continuously flowing sample and extract solutions,17,19 with response times to changed sample concentrations within one hour.30 Depending on the sampling time of a continuous system, either concentration peaks or time-weighted average (TWA) concentrations may be detected. On-site TWA sampling based on SLM extraction has been applied by Knutsson et al. to six phenoxy acid herbicides from an agricultural area.28 Automated on-site TWA sampling of triazine herbicides has also been performed in our group (unpublished data). Using continuous flow hollow fiber LPME with an aqueous acceptor, enrichment factors up to 500 times have been demonstrated for haloacetic acids in reagent water.31
In the current work we report the method development of continuous flow hollow fiber LPME (CFHF-LPME) for simultaneous extraction of the four NSAIDsketoprofen, naproxen, diclofenac and ibuprofen from treated sewage water. The developed method was applied to monitor the occurrence of these substances in STP effluent. Källby STP in the city of Lund, Sweden, was used as the study object. The environmental fate of NSAIDs in the primary recipient pond system of the STP was studied.
Theory
Sample and acceptor (extract) solutions are separated by the organic solvent immobilised in the fiber pores. A three-phase system (aq–org–aq) for continuous flow HF-LPME is thereby realised with sample flowing on the outside of the fibers (shell side) and the acceptor solution inside the fibers (lumen side).The concentration gradient of an analyte in neutral, i.e. extractable, form is the driving force for the extraction from the sample (subscript S or I), over the membrane and into the acceptor (subscript A). The rate of mass transfer is proportional to the concentration difference or concentration gradient, ΔC, between the two aqueous compartments, expressed as
| ΔC = αSCS − αACAKA/KS | (1) |
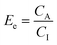 | (2) |
The amount (n) of analyte extracted is the extraction efficiency (E), expressed as
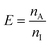 | (3) |
In this work, the sample is continuously recirculated. (This is due to practical reasons in developmental work, and for on-site extraction recirculation of the sample is not needed.) The recirculated sample will be continuously stripped of analyte and the effect is demonstrated by expanding the E parameter:
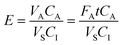 | (4) |
Methods
Chemicals
Ibuprofen, ketoprofen and naproxen (all 98% pure) were purchased from Sigma Aldrich Chemie GmbH (Steinheim, Germany). Diclofenac sodium salt was purchased from Sigma Aldrich Inc (St Louis, USA). HPLC grade methanol, formic acid (>98% pure) and ammonium carbonate (containing 30–33% NH3) were obtained from Sigma Aldrich Chemie GmbH. Sulfuric acid and di-n-hexyl ether (DHE; >97%) were purchased from Fluka Chemie GmbH (Buchs, Switzerland). Silicone oil (no. 146153) was purchased from Sigma-Aldrich. Ultra pure water from a MilliQ waterpurification system (Millipore, Bedford, MA, USA) was used. Silicon glue (Glassilikon®) and epoxy glue (Strong epoxy rapid®) from Akzo Nobel Deco Int AB, Casco (Stockholm, Sweden) were used.Individual analyte stock solutions were prepared in methanol and mixed working stock solutions containing 10 mg L−1 of the four analytes were diluted in water. 1 L samples in reagent water were prepared by dilution from the mixed stock solution with addition of ∼24 mL 1 M sulfuric acid, achieving a pH of 1.5–1.9. In method development, reagent water samples with each analyte at a concentration of 10 µg L−1 (unless otherwise noted) were extracted. The acceptor buffer consisted of 0.1 M ammonium carbonate solution with pH 9.5. Calibration solutions were prepared from mixed stock solution by dilution with acceptor buffer.
Membrane contactors
Polypropylene Celgard® hollow fiber membranes from Membrana (Wuppertal, Germany) were used. Properties of these membranes include a 30 µm wall thickness, 240 µm id and 0.1 µm pore size. To construct the membrane contactors30,31 (Fig. 1), six fibers were pulled through (i) a Y-tapered T-connector (120°, no. (70)527, Kartell Labware Division, Noviglio (MI), Italy), (ii) a 1 m Teflon® tube followed by (iii) a second Y-connector. To fix the construction and to create two separate channels for the donor and acceptor solutions, the ends with the hollow fibers in the Y-connectors were sealed around the fibers with glue. As glue, either glass silicone (no. 2985 Akzo Nobel Deco International AB, Casco, Stockholm, Sweden) or epoxy glue (no. 2806, Casco Strong Epoxy Rapid®) was used. Previously, contamination of extracts was experienced when using epoxy glue, which is why glass silicone which gave no significant interferences was used.30 However, glass silicone is more difficult to apply so the contactor is leak-tight, while contactors constructed with epoxy glue are more robust. With the epoxy glue employed in this study no contamination problem was observed, in spite of the fact that the contactors were used after only minimal time for curing. We therefore recommend the epoxy glue used in this study.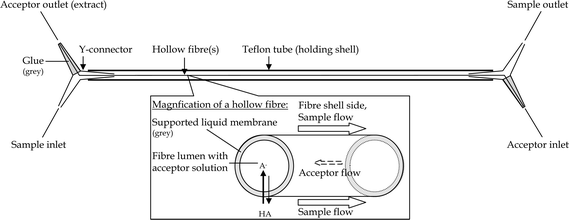 | ||
| Fig. 1 Schematics of the HF membrane module.30 | ||
After construction, the fibers were impregnated with DHE or silicone oil. Before the impregnation, the contactor sample compartment was filled with water. A syringe containing the organic solvent was attached to one of the Y-connectors (acceptor side). For the viscous silicone oil, the syringe was fixed with a constant overpressure overnight. Excess organic phase in the fiber lumen was removed using a gentle flow of pressurised air. Membrane contactors impregnated with DHE were stored with water in the sample channel to prevent evaporation of the membrane liquid. The stability of the SLM was monitored by measuring the pH of the outgoing acceptor solution using a Twin pH Compact pH meter B-212 from Horiba (Kyoto, Japan).
Extraction method and equipment
The sample is on the fiber shell side and the acceptor is in the lumen of the fibers. The contactor is oriented so that sample flows upwards, in order to ensure that no air bubbles disturb the contact between sample and membrane in the contactor.Sample flow at 100 mL min−1 (unless otherwise noted) was delivered with a valveless rotary piston dispensing pump model REGLO-CPF Digital equipped with a RH0 pump head (ISMATEC SA, Glattbrugg, Switzerland). In on-site continuous sampling, there is no limitation in sample volume, while in developmental work, a finite sample volume must be used for practical reasons. The 1 L sample was recirculated from the sample bottle, through the contactor and back to the bottle. The sample was stirred using a magnetic stirrer (MR 1000, Heidolph Instruments GmbH, Schwabach, Germany). To avoid heating of the sample from the magnetic stirrer, the sample bottle was placed 2–3 cm above the stirrer using a wooden plate as support.
The acceptor was continuously pumped counter currently to the sample flow and delivered with a peristaltic pump, Minipuls 3, from Gilson S.A. (Villiers le Bel, France), with Acidflex (id 0.889 mm) peristaltic tubing from Elkay Laboratory Products (Shrewsbury, UK). In a few experiments, the acceptor solution was pumped alternately forward and backward. This was done using the Minipuls 3 interface, which was connected to a computer via an electronic connector built in our laboratory. When the sample flow was turned on, the acceptor typically needed ∼10 min to stabilise, and then FA was set to the desired value. During extractions, FA was continually monitored by weighing each vial before and after extraction. At the acceptor outlet, extracts of the acceptor solution were collected with regular time intervals. Usually, extracts were collected into one vial during 15 min.
In order to ensure no carry-over between extractions, the sample side was washed before and after each extraction with ∼300 mL buffer followed by ∼1 L water. For the same reason, and also in order to keep the acceptor channel conditioned for subsequent extraction, the acceptor buffer was continuously delivered at a low flow rate between extractions. No carry-over was confirmed by analysing solutions from both the sample and acceptor side of the membrane prior to an extraction.
Sampling site
Källby STP is located in the southern part of Lund and receives sewage water from the city as well as surrounding villages. During 2008, the total number of persons connected to the plant was estimated as 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.34 The STP daily treats about 30000 m3 of sewage water, depending on water consumption and precipitation. The sewage treatment is made up of four major steps: screen raking and sand catch, primary settling, biological degradation with activated sludge and finally chemical treatment. The sewage water retention time in the plant is about 24 h. Before release into the river recipient, Höje å, the water also passes six serially connected ponds, in which some passive treatment such as further denitrification and sedimentation takes place. About half of the effluent is released into the first of these ponds and the other half into the middle of the second pond. The total area of the pond system is ∼88000 m2.7 Effluent retention is 24 h in pond 2 and an additional 43 h in ponds 3–6. The ponds attract many bird species and before a draining of the ponds in 2008, the ponds were home to many fishes, especially common carp (Cyprinus carpio). Due to the size of the pond system and the associated ecological life, the pond system may be seen as a primary environmental recipient of surface water. The rather small recipient river, which has a water flow ranging from 0.1 to 15 m3 s−1 depending on season, is described in more detail elsewhere.7,35
000.34 The STP daily treats about 30000 m3 of sewage water, depending on water consumption and precipitation. The sewage treatment is made up of four major steps: screen raking and sand catch, primary settling, biological degradation with activated sludge and finally chemical treatment. The sewage water retention time in the plant is about 24 h. Before release into the river recipient, Höje å, the water also passes six serially connected ponds, in which some passive treatment such as further denitrification and sedimentation takes place. About half of the effluent is released into the first of these ponds and the other half into the middle of the second pond. The total area of the pond system is ∼88000 m2.7 Effluent retention is 24 h in pond 2 and an additional 43 h in ponds 3–6. The ponds attract many bird species and before a draining of the ponds in 2008, the ponds were home to many fishes, especially common carp (Cyprinus carpio). Due to the size of the pond system and the associated ecological life, the pond system may be seen as a primary environmental recipient of surface water. The rather small recipient river, which has a water flow ranging from 0.1 to 15 m3 s−1 depending on season, is described in more detail elsewhere.7,35
Bendz et al. studied the removal of certain substances in Källby STP. Removal efficiencies of NSAIDs were 22% for diclofenac, 96% for ibuprofen, 65% for ketoprofen and 93% for naproxen.7 All four substances were also detected in the effluents of ponds 2 and 6 in concentrations between 0.1 and 0.7 µg L−1.
Sampling
A collection of STP effluent samples was carried out approximately every week during May–September 2008. Samples were collected at the ends of ponds 2 and 6. GPS locations were N 55° 41. 862′, E 13° 09. 441′ for pond 2 and N 55° 42. 068′, E 13° 08. 879′ for pond 6; the locations can also be seen on a map presented by Bendz et al.7 Samples were taken with a Ruttner collector and transferred to 1 L glass bottles. All bottles were rinsed with sample two times before being filled and were transported in a cooled box. In the laboratory, samples were preserved with ∼24 mL 1 M sulfuric acid. One sample (from the same pond) was used to fill up the sample being preserved to zero headspace. Samples were then stored in the dark at 4 °C until analysis. From the beginning of September (and for the last three sampling occasions), samples were only gathered from pond 2, since the STP drained ponds 3–6 to remove sediments as a part of their maintenance work. Linearity in the environmental matrix was investigated using a 5 L sample collected from pond 2 (on the 8th of October), which was divided into 1 L aliquots for standard addition extraction.Extraction of environmental samples
Extraction of the samples was performed in a random order with respect to sample date. Prior to the extractions all samples were allowed to equilibrate to room temperature in the dark. STP samples were extracted using the same conditions as for spiked samples, using the determined Ee-peak time.Solid phase extraction
The SPE method used was based on Gentili et al.8 and references therein. As for membrane samples, the samples to be extracted by SPE were acidified to pH ∼2. SPE samples were filtered to avoid clogging of cartridges. Extractions were carried out using 3 mL Oasis HLB cartridges from Waters Corporation, (Milford MA, USA) containing 60 mg of sorbent. Conditioning was performed with 3 mL 100% methanol at a flow rate of ∼3 mL min−1, sample loading at a flow rate of ∼15 mL min−1, washing with 3 mL 5 : 95% (v/v) methanol : water at a flow rate of ∼1 mL min−1 and analyte elution with 5 mL 100% methanol at a flow rate of ∼1 mL min−1. Prior to HPLC analysis, the extracts were evaporated to a final volume of around 300 µL. The exact volume was determined by weighing the extract.Chromatography
Chromatographic analysis was performed on a 1100 series HPLC instrument from Agilent Technologies (Waldbronn, Germany) controlled via the Chemstation software and equipped with on-line vacuum degasser, diode array detector (DAD) and fluorescence detector (FLD). The used column was a Zorbax XDB-C8 (4.6 × 150 mm and particle size 5 µm) from Agilent Technologies, thermostatted at 20 °C and protected by a Security Guard KJ0-4282 precolumn from Phenomenex (Torrance, USA). The autosampler was thermostatted at 4 °C. Usually an injection volume of 25 µL was used. The mobile phase consisted of methanol and 0.1% formic acid in water. Gradient elution was performed at a flow of 1 mL min−1, starting with 60% methanol and increasing to 90% in 15 min, followed by isocratic elution with 90% methanol for 3 min for a column wash and finishing with 5 min for equilibration to initial conditions prior to subsequent analysis. Separation of the four analytes was achieved in 11 min. Using the diode array detector (DAD), ketoprofen was detected at 255 nm, naproxen at 230 nm and diclofenac and ibuprofen at 220 nm. For all analytes, the signal bandwidth was 12 nm and the reference signal was 600 nm with a 20 nm bandwidth. The response time was 1 s and slit width 4 nm. FLD was used to qualitatively identitify naproxen at low concentrations, but was not used for quantitative purposes. Calibration was performed by analysing standard solutions containing the four analytes from 750 to 12000 µg L−1 (for samples in reagent water) or from 75 to 500 µg L−1 (for STP samples) with r > 0.9899.
Results and discussion
Composition of sample and acceptor phases
For extraction of organic acids such as NSAIDs the sample pH is adjusted below the pKa of the analytes, which thereby will become uncharged and thus can be extracted into the SLM. The acceptor pH is kept well above pKa meaning that the analytes will become deprotonated and since only neutral species can diffuse through the membrane, they will become trapped and thus enriched in the acceptor phase. Therefore, pHS was set to 1.5–2.0 and pHA to 9.5.Sample flow
The effect of sample flow rate, FS, was investigated by performing an extraction with a flow rate of 30 mL min−1 instead of 100 mL min−1, which was used for most of the extractions. With this decrease of the sample flow rate, the enrichment was more than halved. The decrease in Ee may be due to the fact that less analyte passed the contactor per unit time. It could also be due to a more laminar flow at the lower flow rate (Reynolds number Re being 155 instead of 518), leading to a thicker diffusion layer on the shell side of the fibers and thereby decreased extraction into the membrane. FS could not be increased beyond 100 mL min−1, since this was the maximum of the pump employed.Acceptor flow
The acceptor flow, FA, is an important parameter in continuously flowing membrane extraction. Decreasing FA increases the residence time of the acceptor in the fiber lumen, which is beneficial for obtaining high enrichment. By using several fibers, e.g. six instead of one, the flow per fiber is decreased. A net FA of 2.5 µL min−1 was shown to yield higher enrichment and smoother extraction curves than 5 and 10 µL min−1. However, the pump used for the acceptor was operated close to its minimum speed, and because of this FA could not be reproducibly decreased below 2.5 µL min−1.Since acceptor solution continuously leaves the contactor, there will be a limit to how high enrichment factors can be obtained. Using a stagnant acceptor, which is harvested after extraction (a semi-continuous approach), the extract should become more concentrated. Enrichment factors up 1600 times were indeed obtained when the acceptor was kept stagnant during 90 min extraction followed by extract harvest with a flow rate of 25 µL min−1. However, this setup had practical problems. In multiple experiments, it proved hard to keep the acceptor perfectly stagnant. This may be due to vibration of the membrane contactor, caused by the pistonless pump used for the sample solution. Therefore, we suspected extraction using stagnant acceptor would be hard to reproduce (with the pumps used).
Pumping the acceptor alternately forward and backward may decrease the diffusion layer inside the fibers and thus lead to higher mass transfer from the membrane to the acceptor bulk. Controlling FA with a computer makes this possible. When the acceptor solution was pumped alternately forward and backward with a net forward flow of 2.5 µL min−1, the obtained Ee were higher for ketoprofen, naproxen and ibuprofen (15–30%), but lower for diclofenac (20%), compared to using regular forward flow. Within-experiment enrichment was less precise for alternately forward–backward flow compared to forward flow only.
For future work, other pumps should be used for the acceptor in order to achieve lower FA. We expect better precision in FA would be obtained using a syringe pump, because of more accurate dispensing in itself, but also because the peristaltic tubing got worn rather quickly. A more precise FA could also aid achievement of more precise results for semi-continuous and alternately forward–backward acceptor flow methods.
Organic solvent
Di-n-hexyl ether yielded high enrichment factors (see Fig. 2), and was in this aspect satisfactory as an SLM solvent. However, even though DHE has low water solubility, it was eventually washed out of the membrane pores. SLM deterioration could be observed directly as very small droplets of organic solvent in the sample after extraction. Decreased precision of the delivered FA, increased FA or increases in outgoing pHA could also be used as an indicator of when the SLM deteriorated. When pHA dropped more than 0.5 pH units below the initial buffer pH, either a new contactor was made or the membrane was reimpregnated. Before reimpregnation, the contactor was washed with reagent water at both sample and acceptor sides to remove e.g. salt deposits from acceptor buffer or sample to avoid an inhomogeneous distribution of the membrane liquid. Typically, reimpregnation was required after approximately 15 extractions.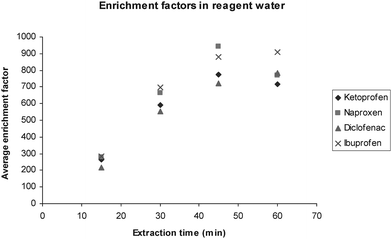 | ||
| Fig. 2 Average enrichment for the four NSAIDs extracted from reagent water in continuous CFHF-LPME. Enrichment peaks at 45–60 min (1 L sample). | ||
Fast diffusion of the analytes and thereby high enrichment is favored by a less viscous membrane liquid while the stability of the membrane is favored using a less polar liquid with higher viscosity.36 In CFHF-LPME extraction, the organic membrane is exposed to greater strain compared to batch-type LPME with individual fibers, due to the fast flowing sample (and in our case also vibrations from the sample pump). Silicone oil has been incorporated in membrane contactors and has the advantages of being more stable due to higher viscosity, lower water solubility and lower vapour pressure.37 However, using a silicone oil, much lower enrichment factors were obtained. The relative Ee in silicone oil compared to DHE ranged from 1% (diclofenac) to 14% (ibuprofen). However, reimpregnation was never necessary for contactors with silicone oil.
1-Octanol is commonly used as SLM solvent24,26 and has previously been found optimal to extract NSAIDs with LPME.9,27 However, Müller et al. called for a more stable membrane liquid than 1-octanol.27 Even though DHE is less water-soluble than 1-octanol, we arrive at a similar conclusion. Due to the high enrichment using DHE and the excellent stability of silicone oil, we conclude that a membrane liquid with the optimal relation between stability and enrichment properties still needs to be found.
Enrichment and extraction time
Enrichment factors (Ee) were determined according to eqn (2) and their maximum values were around 800 for all analytes for spiked samples in reagent water (Fig. 2). Enrichment was linear during 30–45 min, whereafter it started to decline, due to depletion of CS. As detailed above, this decrease is expected when a limited sample volume is reextracted with a continuously flowing acceptor. Extraction efficiency (eqn (4)) was about 10% during the first 45 min. Ee-peak time was 45 min for ketoprofen and naproxen and 60 min for diclofenac and ibuprofen. The later Ee-peak for diclofenac and ibuprofen is probably due to the fact that these analytes have somewhat higher log Kow values5 and therefore are transferred more slowly from the membrane into the acceptor.38The repeatability of the method was investigated by extracting four spiked samples consecutively using the same membrane contactor and without any reimpregnation. During the linear regime, i.e. the first 30 min, the repeatability was rather good but decreased as peak-Ee was reached (Table 1). The reason for the decrease in repeatability with time is believed to be (non-constant) vibrations caused by the used sample pump. The (variation in) vibration is believed to affect the diffusion of the analytes, stability of the SLM, acceptor flow rate and thereby the enrichment. Otherwise the used pump is advantageous since it has minimal surface which may sorb analytes (compared to pumps with peristaltic tubes) and it can deliver high sample flows continuously (compared to syringe pumps). A more stable setup can probably be achieved, e.g. by stabilising the contactor in Styrofoam or moulding it into plastic.
| t/min | Ketoprofen | Naproxen | Diclofenac | Ibuprofen |
|---|---|---|---|---|
| 15 | 263 (7%) | 280 (6%) | 218 (15%) | 282 (8%) |
| 30 | 592 (12%) | 664 (13%) | 555 (14%) | 699 (12%) |
| 45 | 772 (28%) | 940 (12%) | 720 (18%) | 881 (22%) |
| 60 | 717 (24%) | 769 (25%) | 786 (25%) | 910 (24%) |
Reproducibility was checked by extracting three spiked samples using a different contactor for each extraction and was similar to the repeatability. At 45 min, reproducibility (RSD) for enrichment was 19% for ketoprofen, 22% for naproxen, 19% for diclofenac and 8% for ibuprofen.
Linearity and matrix effects
Matrix effects and linearity for the method in STP effluent were studied by extracting 5 samples originating from the same batch of environmental sample. 1 L aliquots were spiked with 0, 0.1, 0.5, 1 and 10 µg L−1 of each NSAID, thus covering also STP inlet concentrations.4,7 The chromatogram from 0.5 µg L−1 standard addition is presented in Fig. 3. The obtained Ee from 45 min were plotted versus the added sample concentration, and the slope represented the enrichment factors in STP effluent. The method showed good linearity (r2 > 0.9984) in the studied range.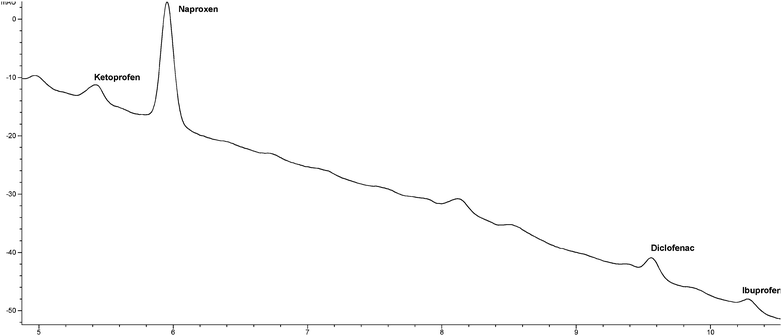 | ||
| Fig. 3 Chromatogram (DAD signal 230 nm) from analysis of a real sample spiked with 0.5 µg L−1 of each analyte. | ||
For analyte quantification and in order to increase accuracy for lower and more relevant concentrations in STP effluent, the highest concentration (10 µg L−1) was excluded when obtaining the Ee (the slope in the standard addition curve) to be used for determination of real samples in the monitoring campaign. r2-values and Ee for the range 0–1 µg L−1 are presented in Table 2, where enrichment from STP effluent is also compared with enrichment from reagent water. In the environmental matrix, enrichment ranged from 35 to 91% of the enrichment in reagent water. When the sample is acidified, the analytes are rendered uncharged and dissolved organic matter such as humic matter also has a lower total charge. Lipophilic interaction between analytes and humic matter could therefore be expected, but no correlation with analyte log Kow was observed. By contrast, ketoprofen, which has the lowest log Kow (however similar to the log Kow of naproxen), was the analyte affected most by the matrix.
| Analyte | Linearity (r2) in 0–1 µg L−1 range | Enrichment (Ee) in STP water | Relative matrix effect (Ee in STP water/Ee in reagent water, %) | MDL/µg L−1 | MQL/µg L−1 |
|---|---|---|---|---|---|
| Ketoprofen | 0.9952 | 270 | 35 | 0.05 | 0.18 |
| Naproxen | 0.9997 | 615 | 74 | 0.01 | 0.03 |
| Diclofenac | 0.9946 | 595 | 83 | 0.05 | 0.17 |
| Ibuprofen | 0.9979 | 805 | 91 | 0.03 | 0.11 |
The STP effluent varies in composition, in terms of e.g. dissolved/total organic carbon (TOC), organic micropollutants and inorganic species, due to differences in incoming sewage water and STP treatment processes. Thus, even though matrix effects are (to some degree) accounted for by using Ee from the standard addition for one of the environmental samples (Table 2), this procedure is not totally accurate in quantification of other samples from different occasions. For future work, method robustness toward TOC concentration should be studied, including 8.3 and 15 mg L−1, which are the average and maximum TOC in effluent water from this STP, respectively.34
The chloride concentration is expected to fluctuate in discharged water, because phosphate is precipitated with ferric chloride in the STP. The conductivity in STP samples was 0.5–0.7 mS cm−1. Due to the addition of sulfuric acid, for preservation and adjustment of pH < 2, conductivity increased to around 12 mS cm−1, which made initial variations in conductivity between samples negligible. Since the addition of sulfuric acid dominates over the original salt content in samples, the effect of salt was not studied further. No correlation between the initial conductivity in the environmental samples and the detected concentrations of NSAIDs was observed.
Comparison with SPE and method limit of detection
CFHF-LPME was compared with SPE by extracting spiked (5 µg L−1) and unspiked 0.5 L STP samples with both techniques. Lower enrichment factors were obtained for SPE extracts (Table 3). By evaporating the elution solvent and concentrating the SPE extracts, higher enrichment factors were however achieved for SPE. However, enrichment factors may be also increased in CFHF-LPME, using e.g. longer contactors or decreasing FA. Extracts from membrane extraction can also be readily injected without a concentration step prior to analysis. Cleaner extracts were obtained with the membrane extraction, which makes it easier to detect lower concentrations.| Ketoprofen | Naproxen | Diclofenac | Ibuprofen | |
|---|---|---|---|---|
| CFHF-LPME | 238 (90%) | 261 (97%) | 291 (100%) | 301 (100%) |
| SPE | 176 (95%) | 158 (99%) | 89 (100%) | 163 (100%) |
The method detection limit (MDL) for each analyte was determined by dividing the obtained LOD from the HPLC with the enrichment factors from the standard addition in the STP effluent matrix. Method quantification limits (MQL) were determined as 10/3 × MDL. MDLs and MQLs are presented in Table 2. Better MDLs and MQLs are expected by switching to LC-MS or (for naproxen and ibuprofen) fluorescence detection. Currently, MDL is probably improved more by switching to a more sensitive detection method, than trying to increase the enrichment.
Sample volume and perspective for on-site extraction
In on-site extraction of matrices such as STP or surface water, sample volume is not limiting. However, in this developmental work, a lower sample volume was used for practical reasons. The limited volume eventually caused an expected depletion of CS. With a larger sample volume, more analyte is introduced into the system and extraction increases. It was observed that doubling the sample volume gave enrichment up to 25% higher at Ee-peak time. Thus, higher enrichment and thus lower limit of detection can be obtained when sample volume is not limiting. Also, doubling the sample volume seemed to give a stable equilibrium (for at least 30 min) between acceptor and sample. This level of equilibrium may represent a situation where the extraction over the membrane equals the amount currently leaving the acceptor channel of the contactor, i.e. a dynamic enrichment equilibrium. If extraction is performed under conditions where the sample volume is not limiting, the enrichment factor needs some more investigation. Either the uptake rate constant in the initial linear extraction regime and/or the level of the dynamic enrichment equilibrium need to be determined.Monitoring in the STP effluent
For analysis of STP samples, quantification was based on the extracts collected after 45 min extraction and matrix effects were taken into account using Ee in Table 2. Concentrations in effluent samples are reported in Table 4. Maximum detected concentrations were 0.92 µg L−1ketoprofen, 0.08 µg L−1naproxen, 0.43 µg L−1diclofenac and 0.25 µg L−1ibuprofen. Taking into account STP discharge (Table 4), this gave daily load amounts into the receiving ponds up to 23 g ketoprofen, 2 g naproxen, 6 g ibuprofen and 12 g diclofenac. The higher concentrations of ketoprofen and diclofenac may be due to higher consumption, but also due to the rather low removal of these substances in the STP.7| Date | Ketoprofen | Naproxen | Diclofenac | Ibuprofen | Flow/m3 day−1 | ||||
|---|---|---|---|---|---|---|---|---|---|
| Pond 2 | Pond 6 | Pond 2 | Pond 6 | Pond 2 | Pond 6 | Pond 2 | Pond 6 | ||
| a nd = not detected, below MDL; * = data missing. | |||||||||
| 13-May | 0.39 | 0.26 | nd | nd | 0.43 | 0.19 | 0.03 | 0.15 | 26886 |
| 22-May | 0.33 | 0.24 | nd | 0.02 | 0.21 | 0.17 | 0.09 | 0.16 | 28194 |
| 29-May | 0.65 | 0.31 | 0.04 | 0.04 | 0.24 | 0.17 | 0.21 | 0.22 | 26727 |
| 7-Jun | nd | nd | 0.01 | nd | 0.19 | nd | 0.24 | nd | 22686 |
| 12-Jun | 0.23 | nd | nd | nd | 0.16 | nd | 0.16 | nd | 31103 |
| 19-Jun | nd | nd | nd | nd | nd | nd | nd | nd | 24061 |
| 26-Jun | 0.92 | nd | 0.08 | nd | 0.42 | 0.11 | 0.22 | nd | 25187 |
| 3-Jul | 0.17 | nd | nd | nd | nd | nd | 0.15 | nd | 21978 |
| 10-Jul | nd | 0.05 | nd | nd | nd | nd | nd | nd | 28321 |
| 17-Jul | 0.41 | nd | 0.04 | nd | 0.35 | nd | 0.13 | nd | 18707 |
| 24-Jul | 0.10 | nd | nd | nd | nd | nd | nd | nd | 17438 |
| 16-Aug | 0.23 | nd | 0.01 | nd | nd | nd | nd | nd | 18957 |
| 21-Aug | 0.43 | 0.22 | 0.02 | 0.03 | 0.13 | 0.15 | 0.17 | 0.10 | 21835 |
| 28-Aug | 0.38 | 0.16 | 0.02 | 0.01 | 0.16 | 0.10 | 0.13 | 0.11 | 26490 |
| 4-Sep | 0.22 | * | 0.02 | * | 0.28 | * | 0.25 | * | 25192 |
| 9-Sep | nd | * | 0.02 | * | nd | * | nd | * | 21793 |
| 8-Oct | 0.34 | * | 0.04 | * | 0.23 | * | nd | * | 22390 |
| Maximum | 0.92 | 0.31 | 0.08 | 0.04 | 0.43 | 0.19 | 0.25 | 0.22 | |
| Median | 0.23 | nd | 0.01 | nd | 0.19 | nd | 0.13 | nd | |
| Detection frequency (%) | 76 | 43 | 59 | 29 | 65 | 43 | 65 | 36 | |
Some of the reported values actually fall between MDL and MQL, especially for naproxen. Although this adds an uncertainty to statistical tests, all values were included and values < MDL were set to zero in ANOVA.
Monthly variations in NSAID concentrations were observed (p = 0.0037). Overall, analytes dropped during the summer and concentrations below MDL were most frequent during the summer. Due to less data in September and October, the difference for these months compared to the others was generally not signficant. Therefore, the only significant difference was between May and July. However, the average concentrations during the monitoring period generally follow a trend with decreasing concentrations in May and June, minimum in July and increasing concentrations thereafter.
Many inhabitants (i.e. students) leave the city in the beginning of June and come back around mid-August. The number of inhabitants and their consumption pattern change both the amount of pharmaceuticals and water reaching the plant, and because of this a decrease in population may however not yield a lower effluent concentration. No correlation between STP discharge and NSAID concentrations in pond 2 could be observed.
The seasonal variation agrees with observations by Sacher et al.,3 who observed that concentrations of diclofenac and ibuprofen in the river Rhine were significantly lower during the period April–September than the rest of the year. The authors attributed this to a more effective removal process at sewage treatment plants during the warmer months since no correlation to changes in the river flow could be seen. With increasing temperature, microbial activity and biodegradation increases. NSAID concentrations in the discharge pond (pond 2) were however not correlated to average temperature during the last 1–2 days prior to the sampling and the sampling day itself. The non-effect of temperature for pond 2 could possibly be related to the fact that energy is extracted from the STP effluent with heat pumps. The temperature effect for pond 6 was also tested with ANOVA, using the average temperature during the last 3, 4, 5 and 10 days prior to the sampling. 10 days is more than the hydrological retention time in the ponds, but was used since microbial growth during a longer period may be important for biodegradation. In all these cases, the dates of 21st and 28th August grouped together with the three dates 13th–29th of May, even though temperatures in August were similar to those in July. Thus, the seasonal variation in the investigated period may be due to the shifting population.
For the studied STP, it was previously concluded that no apparent degradation occurred in the primary recipient pond system, i.e. between pond 2 and 6.7 However, we observed a significant decline in concentrations between the ponds for diclofenac (p = 0.011) and ketoprofen (p = 0.001) using ANOVA. Also for ibuprofen and naproxen, a decline in concentrations between the ponds was observed, although the differences were not significant (p = 0.067 and 0.192, respectively). Thus, NSAIDs seem to be removed in the pond system, due to e.g. biodegradation and/or photodegradation.10,14 For future studies of the environmental fate of NSAIDs, it may be worth pointing out that ketoprofen had the highest relative matrix effect (35%, Table 4) and also had the most significant concentration decrease in the primary recipient pond system. Possibly there is a correlation between these observations.
Conclusions
A method for simultaneous extraction of four NSAIDs from treated sewage water utilising continuous flow hollow fiber liquid phase micro extraction (CFHF-LPME) followed by HPLC analysis has been developed. The extraction material is cheap, easy-to-use and consumes only minor amounts of organic solvent. The method was shown to be well applicable for the intended analytes in an STP effluent matrix with linear extraction in an environmentally relevant concentration range. Enrichment factors from 270 to 800 were achieved in 45 min for spiked treated sewage water samples. The proposed method was used to monitor the occurrence of the four NSAIDs in effluent from a Swedish STP and showed the continuous release of these xenobiotics into the environment. In the primary recipient, removal was shown for two of the NSAIDs and indicated for the two others.In future work, the membrane liquid ought to be optimised with concern to the special considerations in CFHF-LPME. CFHF-LPME is suited for automated extraction. With automated on-site extraction, concentration peaks can be detected, which may be useful in a more thorough study on the release of pharmaceuticals into the environment. For this purpose, a more sensitive detection than DAD is also desired, in order to decrease detection and quantification limits.
Acknowledgements
The PhD Programme of Environmental Chemistry, Microbiology and Toxicology (RECETO) is greatly acknowledged for financial support. The authors would also like to express their gratitude to Dr Philipp Mayer at NERI, Denmark. Jan Svensson and Michael Ljunggren at Källby STP/VA Syd are acknowledged for providing information about the STP. Ellen Ingre-Khans and Tatjana Trendafilova are acknowledged for assisting in a sub-project. The Swedish Meteorological and Hydrological Instititue (SMHI) is acknowledged for providing temperature data.References
- K. Kümmerer, Pharmaceuticals in the Environment, Springer-Verlag, Berlin, 2nd edn, 2004 Search PubMed.
- B. Halling-Sørensen, S. N. Nielsen, P. F. Lanzky, F. Ingerslev, H. C. H. Lützhøft and S. E. Jørgensen, Chemosphere, 1998, 36, 357–393 CrossRef CAS.
- F. Sacher, M. Ehmann, S. Gabriel, C. Graf and H. J. Brauch, J. Environ. Monit., 2008, 10, 664–670 RSC.
- R. Kanda, P. Griffin, H. A. James and J. Fothergill, J. Environ. Monit., 2003, 5, 823–830 RSC.
- CHEMnetBASE, http://www.chemnetbase.com/.
- M. Carballa, F. Omil, J. M. Lema, M. Llompart, C. Garcia-Jares, I. Rodriguez, M. Gomez and T. Ternes, Water Res., 2004, 38, 2918–2926 CrossRef CAS.
- D. Bendz, N. A. Paxéus, T. R. Ginn and F. J. Loge, J. Hazard. Mater., 2005, 122, 195–204 CrossRef CAS.
- A. Gentili, Anal. Bioanal. Chem., 2007, 387, 1185–1202 CrossRef CAS.
- J. Wu and H. K. Lee, J. Chromatogr., A, 2005, 1092, 182–190 CrossRef CAS.
- M. Isidori, M. Lavorgna, A. Nardelli, A. Parrella, L. Previtera and M. Rubino, Sci. Total Environ., 2005, 348, 93–101 CrossRef CAS.
- R. Triebskorn, H. Casper, A. Heyd, R. Eikemper, H.-R. Kohler and J. Schwaiger, Aquat. Toxicol., 2004, 68, 151–166 CrossRef CAS.
- A. Gravel and M. M. Vijayan, Aquat. Toxicol., 2007, 81, 73–78 CrossRef.
- M. Cleuvers, Ecotoxicol. Environ. Saf., 2004, 59, 315–319.
- M. Schmitt-Janssen, P. Bartels, N. Adler and R. Altenburger, Anal. Bioanal. Chem., 2007, 387, 1389–1396 CrossRef.
- J. L. Oaks, M. Gilbert, M. Z. Virani, R. T. Watson, C. U. Meteyer, B. A. Rideout, H. L. Shivaprasad, S. Ahmed, M. J. I. Chaudhry, M. Arshad, S. M. Ali and A. A. Khan, Nature, 2004, 427, 630–633 CrossRef CAS.
- S. Zorita, B. Boyd, S. Jönsson, E. Yilmaz, C. Svensson, L. Mathiasson and S. Bergström, Anal. Chim. Acta, 2008, 626, 147–154 CrossRef CAS.
- J. Å. Jönsson and L. Mathiasson, J. Sep. Sci., 2001, 24, 495–507 CrossRef CAS.
- C. González-Barreiro, M. Lores, M. C. Casais and R. Cela, J. Chromatogr., A, 2003, 993, 29–37 CrossRef CAS.
- K. Hylton and S. Mitra, J. Chromatogr., A, 2007, 1152, 199–214 CrossRef CAS.
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr., A, 2008, 1184, 132–142 CrossRef CAS.
- H. L. Lord, J. Chromatogr., A, 2007, 1152, 2–13 CrossRef CAS.
- S. Hultgren, N. Larsson, B. F. Nilsson and J. Å. Jönsson, Anal. Bioanal. Chem., 2009, 393, 929–937 CrossRef CAS.
- N. Megersa, T. Solomon and J. Å. Jönsson, J. Chromatogr., A, 1999, 830, 203–210 CrossRef CAS.
- J. B. Quintana, R. Rodil and T. Reemtsma, J. Chromatogr., A, 2004, 1061, 19–26 CrossRef CAS.
- S. Zorita, T. Barri and L. Mathiasson, J. Chromatogr., A, 2007, 1157, 30–37 CrossRef CAS.
- X. Wen, C. Tu and H. K. Lee, Anal. Chem., 2004, 76, 228–232 CrossRef CAS.
- S. Müller, M. Möder, S. Schrader and P. Popp, J. Chromatogr., A, 2003, 985, 99–106 CrossRef.
- M. Knutsson, G. Nilvé, L. Mathiasson and J. Å. Jönsson, J. Agric. Food Chem., 1992, 40, 2413–2417 CrossRef CAS.
- I. J. Allan, G. A. Mills, B. Vrana, J. Knutsson, A. Holmberg, N. Guigues, S. Laschi, A.-M. Fouillac and R. Greenwood, Trends Anal. Chem., 2006, 25, 704–715 CrossRef CAS.
- N. Larsson, K. Utterback, L. Toräng, J. Risberg, P. Gustafsson, P. Mayer and J. Å. Jönsson, Chemosphere, 2009, 76, 1213–1220 CrossRef CAS.
- X. Wang, D. Kou and S. Mitra, J. Chromatogr., A, 2005, 1089, 39–44 CrossRef CAS.
- J. Å. Jönsson, P. Lövkvist, G. Audunsson and G. Nilvé, Anal. Chim. Acta, 1993, 277, 9–24 CrossRef.
- L. Chimuka, N. Megersa, J. Norberg, L. Mathiasson and J. Å. Jönsson, Anal. Chem., 1998, 70, 3906–3911 CrossRef CAS.
- Environmental report Källby STP (in Swedish), http://www.lund.se/upload/29701/text%20Kallby202007.doc, accessed 2008-09-01 Search PubMed.
- R. Berndtsson, Water Resour. Res., 1990, 26, 1549–1558 CAS.
- G. Audunsson, Anal. Chem., 1986, 58, 2714–2723 CrossRef CAS.
- S. V. Ho, P. W. Sheridan and E. Krupetsky, J. Membr. Sci., 1996, 112, 13–27 CrossRef CAS.
- L. Chimuka, L. Mathiasson and J. Å. Jönsson, Anal. Chim. Acta, 2000, 416, 77–86 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2009 |