RAFT-synthesized diblock and triblock copolymers: thermally-induced supramolecular assembly in aqueous media†
Charles L.
McCormick
*a,
Brent S.
Sumerlin
*b,
Brad S.
Lokitz
a and
Jonathan E.
Stempka
a
aDepartment of Polymer Science, University of Southern Mississippi, Hattiesburg, Mississippi, 39406, USA. E-mail: charles.mccormick@usm.edu; Fax: +1 601-266-5504; Tel: +1 601-266-4872
bDepartment of Chemistry, Southern Methodist University, Dallas, Texas 75275-0314, USA. E-mail: bsumerlin@smu.edu; Fax: +1 214-768-4089; Tel: +1 214-768-8802
First published on 4th July 2008
Abstract
This review highlights recent advances in the synthesis of functional, temperature-responsive, water-soluble block copolymers, including particular focus on the results obtained by employing reversible addition–fragmentation chain transfer (RAFT) polymerization. The applicability of the RAFT process for the polymerization of functional monomers under a diverse range of experimental conditions has facilitated the synthesis of water-soluble (co)polymers that were previously inaccessible. Unprecedented control afforded by RAFT in homogeneous aqueous media allows well-defined polymeric systems to be prepared without stringent purification techniques and under increasingly “green” conditions while maintaining the ability to tailor many of the macromolecular characteristics (molecular weight, chain topology, copolymer composition, functionality, etc.) that affect self-assembly in solution. Block copolymer formation and postpolymerization modification utilizing crosslinking and copper-catalyzed azide–alkyne “click” chemistry are described, with attention being paid to their ability to control copolymer structure for subsequent self-assembly in response to changes in temperature.
 Charles McCormick | Charles McCormick received his PhD in chemistry from the University of Florida. He is currently a Bennett Distinguished Research Professor at the University of Southern Mississippi with appointments in the Department of Polymer Science and the Department of Chemistry and Biochemistry. His research focuses on stimuli-responsive, water-soluble and amphiphilic copolymers with precisely designed architecture prepared by controlled/“living” free radical polymerization. |
 Brent Sumerlin | Brent Sumerlin received his BS from North Carolina State University in 1998 and his PhD in Polymer Science and Engineering from the University of Southern Mississippi in 2003 under the direction of Prof. Charles McCormick. He then served as a visiting assistant professor in the group of Prof. Krzysztof Matyjaszewski at Carnegie Mellon University. In 2005 he joined the Department of Chemistry at Southern Methodist University as an assistant professor where his research is dedicated to responsive block copolyers and polymer–protein conjugates. |
Introduction
Although the utility of stimuli-responsive, synthetic (co)polymers and biological macromolecules for speciality applications has been widely recognized for over fifty years,2 only recently have synthetic techniques become available that can yield (co)polymers with the requisite architectures, molecular weights, and narrow molecular weight distributions necessary for specific technological applications. Among the most significant are the controlled/living radical polymerization (CLRP) techniques3 which include stable free radical polymerization (SFRP),4 atom transfer radical polymerization (ATRP),5 and reversible addition–fragmentation chain transfer (RAFT) polymerization.6,7Since initial reports by the CSIRO group in 1998,8,9 the RAFT process has proven to be perhaps the most versatile of the CLRP methods, since virtually all types of vinyl monomers can be polymerized in bulk or with a variety of solvents under simple reaction conditions. Recognizing the potential of the technique for preparing homopolymers, block copolymers, and post-reaction-modified polymers, several groups have successfully prepared a range of water-soluble systems based on RAFT. Our group reported many of the first examples of polymerization of anionic, cationic, zwitterionic, and non-ionic monomers (Fig. 1) under conditions, often directly in water, that required no protecting groups.10 We have recently written a comprehensive review detailing our work and that of a rapidly growing number of other research groups concerning water-soluble (co)polymers prepared via RAFT.11
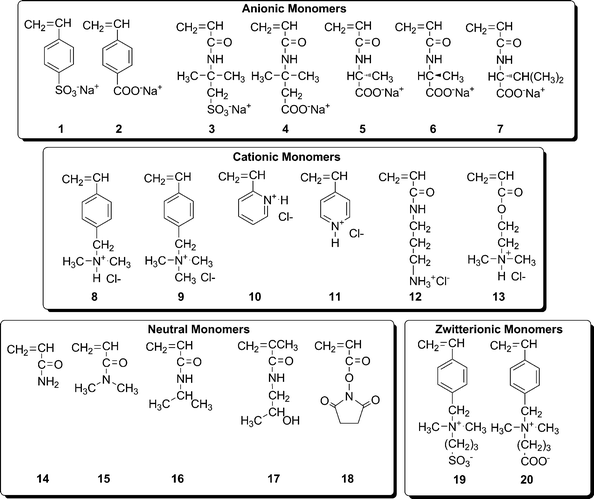 | ||
| Fig. 1 Selected water-soluble monomers polymerized via RAFT. | ||
In this review we focus on advances in the RAFT synthesis of functional, hydrolytically-stable, water-soluble block copolymers, including particular focus on the recent results obtained in our laboratories. Specifically, we discuss synthetic techniques for block copolymer formation as well as postpolymerization modification utilizing crosslinking and “click” chemistry. We also discuss the analytical methodology often utilized in ascertaining polymer structure and aqueous solution behavior. We demonstrate that the facile control over block copolymer structure allowed by RAFT is especially useful for self-assembly in aqueous media in response to external stimuli. We overview the self-assembly of selected block copolymers into micelles and vesicles in response to changes in temperature as well as the effects of crosslinking and/or postpolymerization modification on the inherent properties of the resulting materials. Based on the rapid pace of current RAFT technology development, it is likely that stimuli-responsive materials capable of being dissolved or dispersed in aqueous media or coated onto surfaces as thin films will find unprecedented application in biomedical, pharmaceutical, optical, and diagnostic areas.
Synthesis of block copolymersvia RAFT
RAFT polymerization (Scheme 1) is an especially facile method for preparing sequential blocks (e.g.AB, ABA, ABC) which may serve directly to alter solution viscosity or be “triggered” by an external stimulus such as pH, temperature, or ionic strength to form supramolecular assemblies. Among the various methods of CLRP, facile experimental setup and commercial availability of most reagents required for ATRP has resulted in its being employed in a majority of the literature reports concerning the synthesis of amphiphilic block copolymers. However, despite having been developed three years after the first reports of ATRP, RAFT has increasingly been adopted for the preparation of amphiphilic and responsive block copolymers that include functional groups not easily accommodated by ATRP (e.g., –COOH, –SO3H, etc.). Additionally, the exceptional ability of RAFT to control the polymerization of most acrylamido monomers facilitates the preparation of a wide variety of well-defined polymers that demonstrate temperature-responsive solubility. Herein, our primary focus is the synthesis and characterization of such temperature-responsive acrylamido polymers prepared by RAFT polymerization.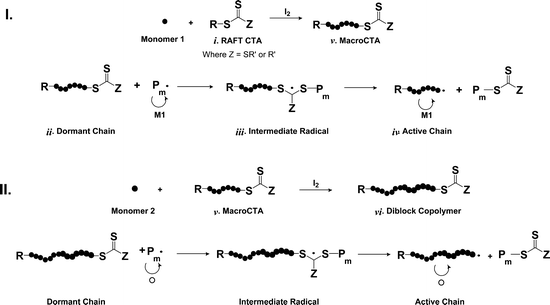 | ||
| Scheme 1 Mechanistic outline for RAFT homopolymerization (I) and block copolymerization (II). | ||
The ability to form block copolymer structures via RAFT with precise control of molecular weight, chain uniformity, and α,ω chain end functionality is a direct consequence of the use of a chain transfer agent, or CTA. The role of the CTA (i) is to suppress or limit the contribution of termination events that occur during free radical polymerization. This suppression is imposed through the establishment of an equilibrium between dormant (ii) and active chains (iv). The degenerative chain transfer process proceeds through an intermediate radical species (iii) which, once the main equilibrium has been established, possesses polymeric segments. Fragmentation of the intermediate radical in either direction facilitates the uniform extension of polymer chains. Although some aspects of the proposed mechanism (for example the fate and lifetime of the intermediate radicals)12 have not been resolved, proper selection of monomer, CTA, initiator, and reaction conditions can yield impressive results in terms of molecular weight control. Since the RAFT process is “living”, the reaction can be halted at predetermined times and the polymer isolated. The resultant CTA-functionalized polymer can then serve as a macroCTA (v) for block copolymer formation utilizing a second monomer in a manner analogous to that of homopolymerization. Further extension of the diblock macroCTA (vi) with a third monomer, yielding a controlled triblock, can also be accomplished.
A critical consideration in preparation of responsive polymers discussed in this review is control of segmental molecular weight (and molecular weight distribution) during homopolymerization (Scheme 1, I), diblock copolymerization (Scheme 1, II), or higher extension, for example to form triblocks. Under appropriate RAFT conditions, theoretical molecular weight, Mn,Th, can be estimated with eqn (1). Thus, segmental length can be targeted for each block by simply controlling the [monomer]0:[CTA]0 ratio and the conversion since the respective molecular weights of the monomer, MWmon, and CTA, MWCTA, are known.
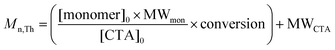 | (1) |
Stimuli-responsive block copolymersvia RAFT
Stimuli-responsive block copolymers typically contain both permanently hydrophilic blocks and “smart” blocks which are tunably hydrophilic/hydrophobic.13,14 The stimuli-responsive block copolymers undergo conformational changes in response to changes in external stimuli such as pH, electrolyte concentration, and/or temperature. The changes can cause the “smart” blocks to become hydrophobic and induce the self-assembly of the amphiphilic block copolymer into supramolecular structures, such as micelles and vesicles (Scheme 2).10,11,15–19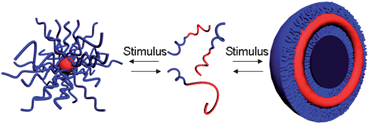 | ||
| Scheme 2 Idealized reversible aggregation in response to an external stimulus. | ||
Temperature-responsive block copolymersvia RAFT
The most common temperature-responsive polymers are prepared from N-alkyl acrylamide monomers. Of these, poly(N-isopropylacrylamide) (PNIPAM) from 16 (Fig. 1) has received the most attention due to its lower critical solution temperature (LCST) of ∼32 °C in water.20 With the temperature of the physiological fluids within the human body being 37 °C, NIPAM copolymers, including those with crosslinked modification, have been targeted for drug delivery applications.21–23To date there have been numerous reports detailing the successful RAFT polymerization of NIPAM in organic solvents.14 For example, Ganachaud et al.24 reported the AIBN-initiated solution polymerization of NIPAM employing both benzyl dithiobenzoate (in benzene) and cumyl dithiobenzoate (in 1,4-dioxane) at 60 °C. Subsequently, Schilli et al.25 disclosed the benzyl and cumyl dithiocarbamate-mediated polymerization of NIPAM, also in 1,4-dioxane at 60 °C. These experimental conditions led to polymers with polydispersity indices (PDIs) around 1.3. Winnik and coworkers have demonstrated the polymerization of NIPAM using a variety of trithiocarbonates and have recently investigated areas such as end group association,26 mesoglobule formation,27 and chain end modification.28 More recently, Ray and coworkers29 demonstrated the ability to control the tacticity in RAFT polymerizations of NIPAMvia the addition of a suitable Lewis acid such as Sc(OTf)3 or Y(OTf)3. RAFT-prepared PNIPAM has also been employed as a thermoresponsive stabilizing layer for gold nanoparticles/clusters30–32 following a synthetic procedure we reported earlier for macroCTA grafting onto noble metals.33,34
RAFT has also been utilized to synthesize NIPAM-based block copolymers. Yusa and Morishima35 studied the thermoresponsive aggregation of the block copolymer poly[2-(acrylamido)-2-methylpropanesulfonate (NaAMPS) (3)-b-NIPAM, Virtanen et al.36 studied the sequestration of fluorescent probes by PEO-b-NIPAM micelles, Liu and Perrier37 prepared block copolymers of DMA (15) and NIPAM in 1,4-dioxane, and Arotcarena and coworkers38,39 prepared doubly thermoresponsive block copolymers of 3-ammoniopropane sulfonate (SPP) and NIPAM. More recently, Voit et al.40 demonstrated the extension of NIPAM macroCTAs with a number of glycomonomers, resulting in sugar-containing responsive block copolymers. The comonomer content, glycomonomer spacer length, and chain architecture were shown to have a dramatic impact on the cloud points of the copolymers. Oupicky and coworkers41 utilized RAFT to synthesize heterobifunctional block copolymers of PEG-b-NIPAM with an internal lysine residue at the focal point and a terminal thiol group which was used to conjugate biotin. The copolymers demonstrated temperature-induced association and formed complexes with avadin.
The following sections focus on recent advances in the RAFT polymerization of temperature-responsive polymers reported by our respective research groups (McCormick at USM and Sumerlin at SMU) and several others within the field. Specifically, key elements of the aqueous RAFT polymerization of NIPAM and the synthesis of thermally responsive block copolymers directly in water,42,43 the facile preparation of temperature-responsive shell cross-linked (SCL) micelles,44,45 and the preparation of thermally responsive hyperbranches through the combination of RAFT and click chemistry46 will be presented.
Thermally responsive block copolymers of containing N-isopropylacrylamide (16)
While the RAFT polymerization of a diverse variety of monomers directly in water had previously been reported, the LCST of PNIPAM had generally been considered to preclude the possibility of living polymerization in homogeneous aqueous media because of the potential for precipitation at the high temperatures generally employed for RAFT. Thus, an important step toward the synthesis of thermally responsive, water-soluble block copolymers was accomplished when Convertine et al. conducted the room temperature RAFT polymerization of NIPAM directly in water.47,48 Employing an azo-initiator with an appropriately short half-life and the CTAs shown in Fig. 2 allowed controlled/living polymerization of NIPAM, as evidenced by pseudo-first-order kinetics, linear increase in molecular weight with conversion, and narrow molecular weight distributions (Fig. 3). | ||
| Fig. 2 Selected RAFT chain transfer agents (CTAs) and azo-initiators. | ||
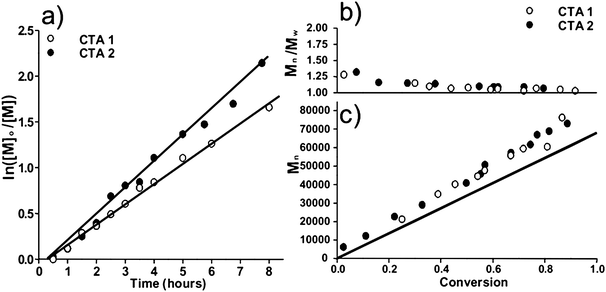 | ||
Fig. 3 a) Pseudo-first-order kinetic plot and b) Mn and c) Mw/Mnversus conversion for the aqueous homopolymerization of NIPAM (16) mediated by CTA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1 or CTA2 at 25 °C using I3 as a radical source. Monomer conversion and molecular weight distribution were determined viaNMR spectroscopy and size exclusion chromatography (SEC), respectively. Adapted with permission from ref. 42. Copyright 2006 American Chemical Society. 1 or CTA2 at 25 °C using I3 as a radical source. Monomer conversion and molecular weight distribution were determined viaNMR spectroscopy and size exclusion chromatography (SEC), respectively. Adapted with permission from ref. 42. Copyright 2006 American Chemical Society. | ||
The success of the controlled room temperature polymerization of NIPAM has allowed for the aqueous synthesis of a series of thermally responsive AB diblock and ABA triblock copolymers.42 These polymers contained hydrophilic DMA A blocks of fixed molecular weight and temperature-responsive NIPAM B blocks of varied chain length. These thermally responsive, water-soluble block copolymers were capable of reversibly forming micelles in response to changes in solution temperature, and the micelle size and transition temperature were dependent on both the NIPAM block length and the polymer architecture (diblock vs. triblock). Mono- and difunctional macroCTAs of DMA prepared from CTA1 and CTA2 (Fig. 2), respectively, were used for the chain extension of NIPAM in water at 25 °C to yield a range of AB diblock and ABA triblock copolymers (Scheme 3). The self-assembly of the thermally responsive, water-soluble block copolymers was followed by dynamic light scattering (DLS). Above the LCST, PNIPAM chains become dehydrated due to an entropy gain resulting from the release of water molecules upon association of the isopropyl groups.20,49 A reversible transition from molecularly dissolved unimers to aggregated micelles occurred above the critical micelle temperature (CMT).
 | ||
| Scheme 3 Synthetic route for preparation of diblock copolymers of DMA and NIPAMvia aqueous room temperature RAFT. | ||
Insight into association behavior was obtained by examining DLS data of the responsive block copolymers as a function of temperature and by static light scattering by estimating the number of unimers constituting the respective micelles.42 As shown in Fig. 4, increased NIPAM segment length led to lower CMTs and aggregates with larger hydrodynamic diameters. Interesting time-dependent reorganization (compaction) of structure was evident for intermediate block lengths. With sufficient time for reorganization, cycling between the unimeric (25 °C) and assembled (45 °C) states was realized for the P(DMA100-b-NIPAM460) block copolymers (Fig. 5).
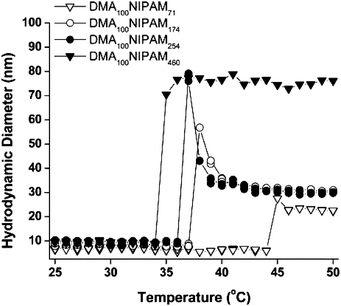 | ||
| Fig. 4 Hydrodynamic diameter (Dh) as a function of temperature for a series of diblock copolymers, as measured by dynamic light scattering. Adapted with permission from ref. 42. Copyright 2006 American Chemical Society. | ||
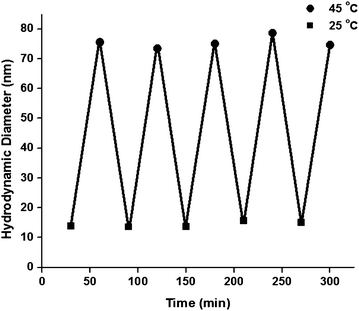 | ||
| Fig. 5 Temperature-induced reversible association of P(DMA100NIPAM460) block copolymers as measured by DLS upon cycling between 25 and 45 °C at 30 min intervals. Adapted with permission from ref. 42. Copyright 2006 American Chemical Society. | ||
In contrast to ABA triblock copolymers, BAB triblock copolymers can form flower micelles at low concentrations and physical gels at moderate to high concentrations (Fig. 6). Armes et al. have reported the synthesis of several temperature- and pH-responsive triblock copolymers utilizing ATRP.50–54 Several of the copolymer gels have potential applications as controlled release substrates53 and cell growth scaffolds.52 Recently, we reported the aqueous RAFT polymerization of BAB triblock copolymers, where the B blocks consist of PNIPAM and the A block consists of PDMA.55 At moderate to high concentrations, the triblock copolymers P(NIPAM455-DMA210-b-NIPAM455) and P(NIPAM455-DMA277-b-NIPAM455) could form physical gels under physiological conditions (140 mM NaCl/20 mM phosphate buffer, pH 7.4, 37 °C). In addition, the mechanical properties were similar to collagen, a commonly used cell growth platform.55
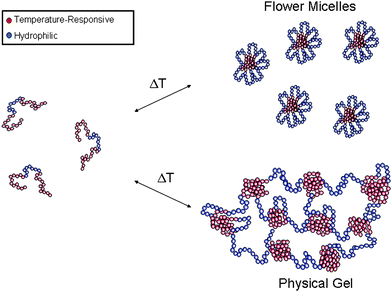 | ||
| Fig. 6 The formation of flower micelles and physical gels from BAB triblock copolymers. | ||
Müller and Barner-Kowollik et al. reported another example of responsive systems based on PNIPAM being prepared directly in homogeneous aqueous media. Block copolymers of NIPAM and acrylic acid (AA) were prepared by aqueous RAFT initiated with γ-irradiation.56 The temperature-responsive nature of the PNIPAM block, coupled with the pH-responsive characteristics of the poly(acrylic acid) (PAA), give rise to block copolymers with potential doubly responsive or “schizophrenic” behavior.57,58 RAFT has also been utilized for the synthesis of block copolymers with a PNIPAM segment and a second segment demonstrating the opposite temperature response.59 Because the second block was composed of zwitterionic repeat units, it demonstrated upper critical solution temperature (UCST) aqueous solution behavior that facilitated the preparation of micelles with PNIPAM coronas at low temperatures.
ABC triblock copolymers incorporating the active monomer N-acryloxysuccinimide (18) and the formation of shell cross-linked (SCL) micelles
When solutions of polymer micelles are diluted below the critical micelle concentration (CMC), dissociation to unimers occurs. Therefore, under conditions of high dilution (e.g., in vivo), polymeric micelle stability is compromised, and the potential for controlled-delivery applications is reduced. To circumvent this limitation, several groups have explored stabilization of the micelle corona through chemical or physical cross-linking. These stabilized micelles, commonly referred to as shell cross-linked (SCL) micelles, were first reported by Wooley and coworkers60 in 1996 and are the subject of a recent review by Armes et al.61 SCL micelles have potential applications in drug delivery, emulsification, sequestration of metabolites, and entrapment of environmental pollutants.62–68 Recently, we44 synthesized ABC triblock copolymers of poly(ethylene oxide) (PEO)-b-P((DMA-stat-N-acryloxysuccinimide (NAS))-b-NIPAM) (Scheme 4) and demonstrated the facile formation of SCL micelles. One hydrophilic segment of the ABC triblock copolymer was comprised of PEO, selected on the basis of demonstrated biocompatibility and dual solubility in both aqueous and organic media. Incorporation of PEO segments into block copolymers prepared by RAFT is generally accomplished by first preparing a macroCTA from ω-hydroxy PEO.69,70Polymerization in the presence of such a RAFT agent allows in situblock copolymer formation. A procedure similar to that reported by Perrier and coworkers was employed to prepare the PEO macroCTA necessary for synthesis of PEO-b-P((DMA-stat-NAS)-b-NIPAM) (Scheme 4).71 NAS units in the resulting block copolymer demonstrated minimal susceptibility to hydrolysis and served as internal crosslinking sites for reaction with difunctional primary amines.72 The thermoresponsive block of the copolymer was prepared by RAFT polymerization of NIPAM.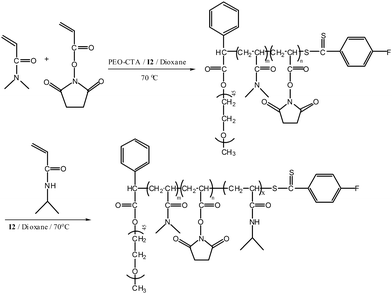 | ||
| Scheme 4 Pathway for the synthesis of the PEO-b-(DMA-s-NAS)-b-NIPAM triblock copolymers. | ||
The aqueous self-assembly behavior of these NIPAM-based triblock copolymers was investigated73 utilizing DLS (Fig. 7). The CMT for the block copolymers decreased with increasing NIPAM block length, consistent with previous reports.43,48,74 In contrast to the previously discussed P(DMA-b-NIPAM) copolymers, hydrodynamic dimensions were also dependent on the NIPAM/NAS ratio within the middle block, as typically observed for statistical copolymers with hydrophilic and hydrophobic segments.75
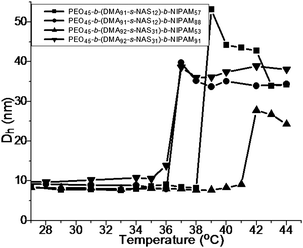 | ||
| Fig. 7 Hydrodynamic diameter vs. temperature for the PEO-b-P((DMA-stat-NAS)-b-NIPAM) triblock copolymers in aqueous solution. Adapted with permission from ref. 73. Copyright 2006 American Chemical Society. | ||
Incorporation of NAS provided a facile and efficient strategy for formation of SCL micelles. This was accomplished by addition of a diamine to the aqueous micelle solution (Scheme 5). The cross-linking of the hydrophilic shell with ethylenediamine was rapid, resulting in over 95% completion within 2 h. Notably, Wooley and coworkers76 adopted this cross-linking procedure to prepare SCL micelles based on the amphiphilic block copolymer poly((methyl acrylate)-b-(NAS-co-(N-acryloylmorpholine))). In our studies, aggregate structure of the SCL micelles was conserved after reduction of the solution temperature; instead of dissolution to unimers, the NIPAM cores swelled after being rendered hydrophilic below the CMT. Atomic force microscopy (AFM) images of the SCL micelles prepared from an example of this triblock showed the spherical shape and relative uniformity of the micelles (Fig. 8).
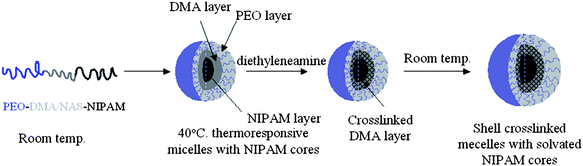 | ||
| Scheme 5 Self-assembly into micelles and shell cross-linked micelles of PEO-b-P((DMA-stat-NAS)-b-NIPAM) triblock copolymers. Adapted with permission from ref. 73. Copyright 2006 American Chemical Society. | ||
 | ||
| Fig. 8 Tapping-mode AFM images of PEO-b-P((DMA-s-NAS)-b-NIPAM) micelles after cross-linking with ethylenediamine. (A) Height image; (B) Phase image. Samples were prepared by drop deposition (5 μL, 0.01% concentration) onto freshly cleaved mica and allowed to dry in air. Adapted with permission from ref. 73. Copyright 2006 American Chemical Society. | ||
Reversible SCL micelles and model compound release
A major concern for drug delivery with SCL micelles is the possibility of in vivo accumulation of non-degradable materials in the kidneys and other organs. One approach to overcome this problem is to provide chemical or physical cross-linking entities functionally labile in the physiological environment. To demonstrate the feasibility of such a process, micelle-forming triblocks of PEO45-b-P((DMA98-stat-NAS30)-b-NIPAM87) were prepared by a procedure similar to that described in the previous section.77 After heating a solution of the block copolymer to 45 °C (above the CMT, 37 °C), the resulting micelles were cross-linked with cystamine, a disulfide-containing diamine, in an equimolar ratio with the NAS units in the statistical block (Scheme 6).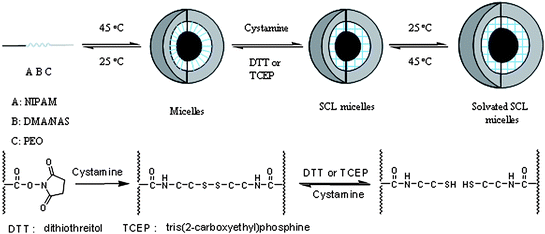 | ||
| Scheme 6 Formation of reversible shell cross-linked micelles from PEO-b-P((DMA-stat-NAS)-b-NIPAM) triblock copolymers by reaction with cystamine. Adapted with permission from ref. 77. Copyright 2006 American Chemical Society. | ||
Disulfide bonds can be readily cleaved using a thiol-exchange reaction with dithiol compounds such as dithiothreitol (DTT)78,79 or tris(2-carboxyethyl)phosphine hydrochloride (TCEP).80 When DTT was used to cleave the disulfide bonds at 45 °C, complete reaction was achieved within 10 h. TCEP, a more efficient reducing agent, led to complete cleavage within 30 min. In both cases unimer formation was confirmed by DLS and by SEC. After removal of excess reducing agent from the solution of cleaved micelles, addition of cystamine caused reconstitution of the SCL structure via the resulting thiol/disulfide exchange reaction. Fig. 9 shows the respective DLS size distributions for PEO45-b-(DMA98-s-NAS30)-b-NIPAM87 unimers at 25 °C (peak 1), micelles at 45 °C (peak 2), swollen SCL micelles at 25 °C (peak 3), unimers at 25 °C after cleavage with DTT (peak 4), micelles at 45 °C after cleavage with DTT (peak 5), and SCL micelles after a second cross-linking reaction with cystamine at 25 °C (peak 6).
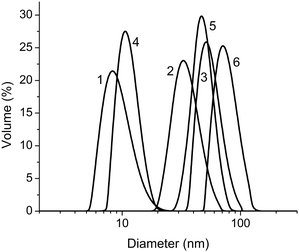 | ||
| Fig. 9 Size distribution of 0.5 % aqueous solution of PEO45-b-P((DMA98-s-NAS30)-b-NIPAM87) at (1) 25 °C; (2) 45 °C; (3) SCL micelles at 25 °C; (4) SCL micelles at 25 °C after cleavage with DTT; (5) SCL micelles at 45 °C after cleavage with DTT; (6) SCL micelles at 25 °C after cross-linking the cleaved micelles with cystamine. Adapted with permission from ref. 77. Copyright 2006 American Chemical Society. | ||
The drug delivery potential of the reversible SCL micelles was assessed by release of a model drug, dipydridamole (DIP). DIP was loaded into the hydrophobic micelle core at 45 °C. Lowering the solution temperature to 25 °C caused the micelles to dissociate into unimers and resulted in burst release of the drug, as monitored by UV absorption of DIP at 415 nm (Fig. 10). Cross-linking the micelle core with cystamine led to significant retardation of release, both at 25 and 45 °C. These results suggest that drug-release rate can be tuned by the degree of cross-linking. While leading to irreversible cleavage, H2O2 can also be used to degrade the micelle cross-links.81 Because H2O2 is produced in mammalian immune systems,82SCL micelle cleavage in situ could potentially facilitate return to unimers and subsequent elimination from the body.
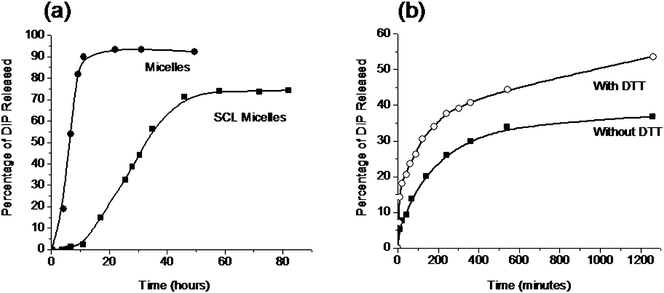 | ||
| Fig. 10 Cumulative DIP release to PBS buffer from (a) shell cross-linked (SCL) and un-cross-linked micelles at 25 °C and (b) SCL micelles at 37 °C in the presence of DTT (○) and without DTT (■). Adapted with permission from ref. 77. Copyright 2006 American Chemical Society. | ||
SCL micelles from block copolymers containing an unprotected amino acid based monomer
McCormick83–86 and others87–90 have reported the polymerization of N-acryloyl derivatives of amino acids to obtain a variety of water-soluble polymers. These monomers can be synthesized in a facile manner from readily available amino acids, and their amphiphilic nature allows for polymerization, purification, and characterization directly in aqueous solution. In addition, the chiral structures produced from polymerization of the respective D and L enantiomeric monomers have potential optical and pharmaceutical applications.The RAFT polymerization of AAL (6) based on L-alanine was accomplished directly in water using initiator I3 and CTA1 and CTA2 (Fig. 2) and proceeded in a controlled manner as evidenced by low polydispersities and linear increases in molecular weight with conversion.43 Having established conditions for controlled/living homopolymerization, thermally responsive triblock and pentablock copolymers with NIPAM and DMA were prepared (Scheme 7). The initial hydrophilic, neutral block was synthesized from DMA with a monofunctional (Pathway A) or difunctional (Pathway B) CTA. Sequential block copolymerization with AAL followed by NIPAM yielded well-defined block copolymers that were subsequently characterized in solution.
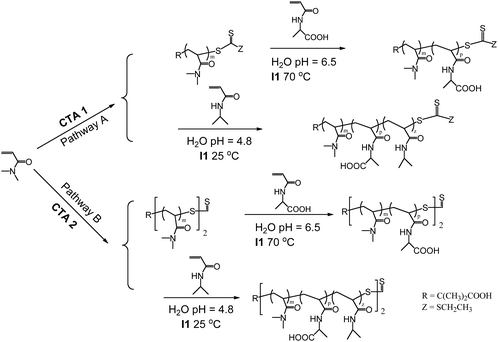 | ||
| Scheme 7 Synthetic routes for preparation of ABC (Pathway A) and ABCBA (Pathway B) block copolymersvia aqueous RAFT. | ||
For both the tri- and pentablock copolymers, raising the solution temperature above the CMT led to a transition from molecularly dissolved unimers to micelles. Fig. 11 shows the temperature-induced changes in the hydrodynamic volume for block copolymers with varying composition. As the PNIPAM block length increased, CMT decreased and average aggregate size increased, as described earlier.48
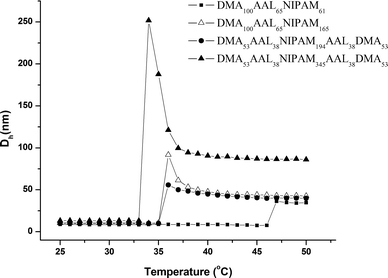 | ||
| Fig. 11 Apparent hydrodynamic diameters (Dh) for the block copolymers measured by dynamic light scattering (1.0 g L−1) as a function of temperature. Adapted with permission from ref. 43. Copyright 2006 American Chemical Society. | ||
Block copolymers with AAL units also allowed the formation of SCL micelles. The presence of the anionic carboxylate groups within the AAL blocks of the thermally-assembled micelles allowed ionic crosslinking91 (Scheme 8) through the addition of an equimolar (by repeat unit) amount of poly((ar-vinylbenzyl)trimethylammonium chloride) PVBTAC (9). Electrostatic interaction between the coronal AAL polyanion blocks and the polycation unimers led to interpolyelectrolyte cross-linked micelles that remained intact at reduced temperatures. The slight decrease in micelle size observed when the solution was cooled to room temperature was attributed to reduced electrostatic repulsion of the carboxyl groups, facilitating more efficient packing of the AAL segments.
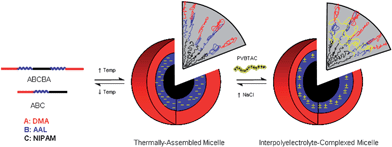 | ||
| Scheme 8 Temperature-responsive micellization of block copolymers and reversible interpolyelectrolyte-complexed micelle formation.43 | ||
The self-assembled morphology of the block copolymers has been confirmed by transmission electron microscopy (TEM) which shows interpolyelectrolyte cross-linked micelles with diameters between 30 and 40 nm, in reasonable agreement with that of 34 nm determined by DLS.
The stability/reversibility of the ionically SCL micelles was investigated by introducing simple electrolytes (Fig. 12). Micelles remained intact in water at NaCl concentrations as high as 0.3 M; the dissociation into unimers was observed at a NaCl concentration of 0.4 M. Thus, dissolution of the aggregates demonstrated the reversibility of the interpolyelectrolyte crosslinks. Interestingly, above 0.8 M NaCl, aggregates were reformed, as the NIPAM block was “salted out.” The facility of formation and the reversibility of these interpolyelectrolyte-complexed micelles suggest the possible utility of such structures in pharmaceutical applications.
![Apparent hydrodynamic diameters (Dh) as a function of sodium chloride concentration ([NaCl]) for ionically cross-linked DMA100-b-AAL65-b-NIPAM165triblock copolymer micelles.43](/image/article/2008/SM/b719577j/b719577j-f12.gif) | ||
| Fig. 12 Apparent hydrodynamic diameters (Dh) as a function of sodium chloride concentration ([NaCl]) for ionically cross-linked DMA100-b-AAL65-b-NIPAM165triblock copolymer micelles.43 | ||
Vesicles from thermally responsive block copolymers
Vesicles composed of lipid molecules play important roles in several biological functions, including the storage and transportation of small molecules.92,93Vesicles prepared from block copolymers or “polymersomes” have been extensively studied for biological applications due to possible increased integrity arising from intermolecular chain entanglements.94–97 Methods of vesicle formation from typical amphiphilic block copolymers involve the use of organic cosolvents such as THF,96DMF,94 or dioxane.98 Such polymersome solutions generally require extensive purification processes which can be time-consuming and problematic. A more versatile route is the stimuli-responsive self-assembly of asymmetric block copolymers in water.99–102Polymersomes were conveniently prepared by the self-assembly of poly(N-(3-aminopropyl) methacrylamide hydrochloride (12))-b-NIPAM) block copolymers directly in water (Scheme 9).45 These diblock copolymers readily dissolved in aqueous solution at room temperature. Increasing the solution temperature above the LCST of the NIPAM block led to uniform aggregates with diameters of approximately 280 nm. The large, uniform sizes and TEM images are indicative of polymersomes (Fig. 13). Increasing the hydrophilic AMPA block length while keeping the NIPAM block length constant resulted in an increase in the phase transition temperature.45 A similar effect was observed by Xia et al. for NIPAM homopolymers with differing end group hydrophilicites.103
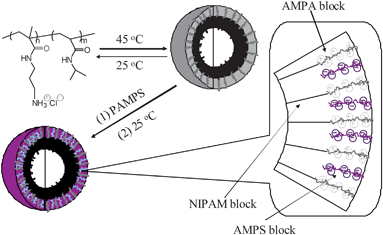 | ||
| Scheme 9 Formation of vesicles from PAPMA-PNIPAM diblock copolymers and their subsequent ionic cross-linking. PAPMA: poly(N-(3-aminopropyl) methacrylamide hydrochloride). Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission from ref. 45. | ||
 | ||
| Fig. 13 Transmission electron microscopy images of (A) vesicles prepared from PAMPA88-PNIPAM50via rapid increase of solution temperature from 25 °C to 45 °C (magnification 25k). (B) Single vesicle (magnification 50k). Copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission from ref. 45. | ||
A sufficiently slow heating rate (0.1 °C min−1) during the morphological transition from unimers to vesicles was needed in order to obtain aggregates with uniform size. Once above the transition temperature, polymersome size remained constant, suggesting self-assembly was kinetically controlled. Solution concentration also affects size, as relatively low block copolymer concentrations (<0.5 mg ml−1) led to more uniform size distributions. Since AMPA is pH-responsive, the stability of the polymersomes between pH 0 and 11 was investigated. Over this range, the structures remained intact, while varying in size with solution pH. The changes in size (310 nm at pH 3.0 and 220 at pH 10.8) were consistent with the expected degree of protonation of the AMPA units.
Interpolyelectrolyte complexation can also be used to “fix” or cross-link the shells of the polymersomes. Addition of an anionic polyelectrolyte, poly(sodium 2-acrylamido-2-methylpropanesulfonate) (AMPS, 3) to a solution of PAPMA-PNIPAM diblock copolymer resulted in stabilized “crosslinked” structures. Upon introduction of the oppositely charged AMPS homopolymer, vesicle size decreased from 270 to 140 nm due to charge neutralization upon complexation. After cross-linking, the solution temperature was reduced to 25 °C without loss in vesicle integrity. The resulting ionically cross-linked vesicles were stable over a wide pH range and moderate electrolyte concentration. Raising the electrolyte concentration above 0.8 M NaCl caused vesicle dissociation, thereby demonstrating cross-link reversibility.
Another method for the “fixing” of the shell of polymersomes has recently been reported.104Gold nanoparticle-decorated polymersomes can be synthesized by mixing a solution of poly[2-dimethylamino)ethyl methacrylate (13)-b-(N-isopropylacrylamide) with a solution of NaAuCl4 above the LCST (Scheme 10). The formation of gold nanoparticles in the PDMAEMA domains functions as a cross-linking agent due to the anchoring of multiple chains to the surface of the nanoparticles. In Fig. 14, the transition from unimers at 25 °C (a) with hydrodynamic diameter (Dh) below 8 nm to polymersomes (b) with average Dh of 140 nm. After formation of the gold nanoparticles, the polymersome size and size distribution at 50 °C increased slightly (b to c), which is attributed to increased protonation of the PDMAEMA segments during equilibration and gold complex reduction. When the gold nanoparticle-decorated polymersomes are cooled to 25 °C, the polymersomes remain intact and the size again increases due to the increased hydrophilicity of the PNIPAM block.
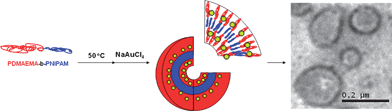 | ||
| Scheme 10 Formation of thermally responsive vesicles decorated with gold nanoparticles. Adapted with permission from ref. 104. Copyright 2007 American Chemical Society. | ||
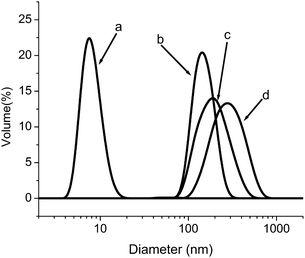 | ||
| Fig. 14 Dynamic light scattering size distribution of a 0.01 wt% PDMAEMA73-b-PNIPAM99diblock copolymer solution: a) 25 °C; b) 50 °C; c) 50 °C after in situreduction of NaAuCl4; d) after in situreduction of NaAuCl4 upon lowering temperature to 25 °C. Adapted with permission from ref. 104. Copyright 2007 American Chemical Society. | ||
RAFT and click chemistry
Clearly, the efficiency provided by CLRP allows control of molecular weight, molecular weight distribution, and chain end functionality and has served to significantly advance the field of controlled architecture polymers. The versatility of RAFT polymerization and its utility in aqueous media facilitates preparation of stimuli-responsive block copolymers that were only recently considered inaccessible. In addition to the functional group tolerance and efficiency inherent with this technique, postpolymerization modification remains a valuable tool by which additional functionality may be incorporated. In particular, transformation of polymer end groups is a useful method for preparation of surface-immobilized polymers,33,34 fluorescently-labeled chains,105–107 and bioconjugates.108,109 The inherent low concentration of end groups and the possibility of side reactions with other functional groups contained in the polymer require reactions with high efficiency and fidelity for successful and specific polymer modification.Recently developed synthetic techniques have demonstrated great promise for precise polymer functionalization. CuI-catalyzed azide–alkyne coupling (CuAAC) (Scheme 11) results in highly specific and efficient preparation of 1,4-disubstituted 1,2,3-triazole products.110,111 This particular coupling process can be conducted under moderate reaction conditions, in aqueous or organic media, and with few or no side reactions. The practicality and versatility of CuAAC has led to its inclusion in the class of efficient and specific organic reactions, commonly termed “click” chemistry, as coined by Sharpless et al.112
 | ||
| Scheme 11 General scheme for CuI-catalyzed azide–alkyne coupling. | ||
Several groups have reported the synthesis of highly functional (co)polymers by CLRP and subsequent azide–alkyne coupling reactions, although these reports predominately concern the modification of (co)polymers prepared by ATRP or SFRP.113–121 For instance, we reported the preparation of ω-(meth)acryloyl macromonomers viaATRP and CuAAC.122 While this approach proved an efficient means to prepare highly branched polymers from any monomer polymerizable by ATRP, we aimed to expand the method to other monomer classes polymerizable by RAFT.
Previously, Hawker and Wooley et al. reported the RAFT block copolymerization of a protected acetylene-containing monomer.123 After deprotection, the resulting block polymers were employed to prepare shell-crosslinked micelles with cores susceptible to functionalization with low molecular weight azides. The same authors reported alkynyl-functionalized RAFT agents for preparation of surface-decorated micelles that were reacted with azido compounds.124
CuAAC has also been employed for end group functionalization of RAFT-derived polymers.46,125,126 We prepared two azido-functionalized CTAs, namely 2-dodecylsulfanylthiocarbonylsulfanyl-2-methylpropionic acid 3-azidopropyl ester (CTA3) and 4-cyano-4-methyl-4-(thiobenzoylsulfanyl)butyric acid 3-azidopropyl ester (CTA4) (Fig. 15). These novel compounds were used to prepare PDMA homo- and block copolymers that were subsequently functionalized with a variety of low molecular weight alkynes.46 Preparing end functional polymers or functional CTAs by modification via alternative reaction pathways might have less applicability due to limited efficiency and orthogonality. However, the fidelity associated with click chemistry allows facile preparation of a range of functional macromolecules.
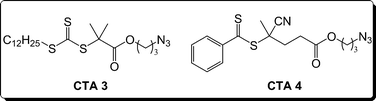 | ||
| Fig. 15 Structures of novel azido CTAs employed to prepare functional telechelic polymers by RAFT. | ||
Postpolymerization modification of temperature-responsive (co)polymers by CuAAC has allowed us to prepare a range of functional systems with biological relevance. For instance, azido-terminated P(DMA-b-NIPAM) was efficiently coupled with propargyl folate to yield a diblock copolymer capable of temperature-responsive self-assembly in aqueous media (Scheme 12).127 Because the folate residue was incorporated on the end group of the hydrophilic PDMA block, the resulting micelles were candidates for tumor-specific drug delivery.
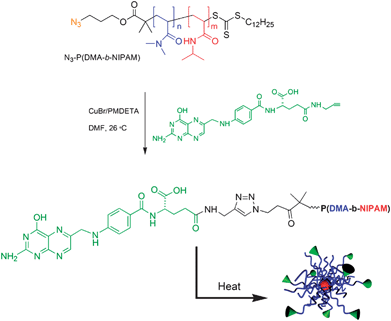 | ||
| Scheme 12 Folate-terminated P(DMA-b-NIPAM) block copolymers and polymeric micelles.127 | ||
Azido-terminated polymers prepared by RAFT can also be readily conjugated to biomacromolecules, as we recently demonstrated. PNIPAM-N3 was efficiently coupled with a model alkyne-functionalized protein by CuAAC.128 Bovine serum albumin was labeled with an alkyne by reaction of its lone free cysteine with propargyl maleimide. The resulting activated protein was coupled with azido-terminated PNIPAM to yield well-defined polymer–protein bioconjugates capable of temperature-responsive self-assembly (Scheme 13).
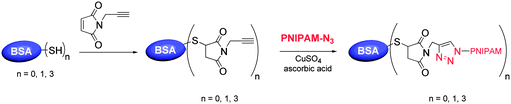 | ||
| Scheme 13 Polymer–protein bioconjugates prepared by CuAAC of alkyne-functionalized bovine serum albumin (BSA) and azido-terminated PNIPAM.128 | ||
CuAAC has also proven to be a feasible method for functionalization of low molecular weight CTAs prior to polymerization. Whereas other methods of CTA modification are limited because of potential side reactions with the thiocarbonyl or other susceptible moieties, the orthogonal nature of click chemistry facilitates specific CTA functionalization. Recently, a CTA prepared by this method was utilized to synthesize hyperbranched PNIPAM.46CTA3 was reacted with propargyl acrylate to yield an acryloyl trithiocarbonate (Scheme 14, CTA4) that contained both monomer and CTA functionality.
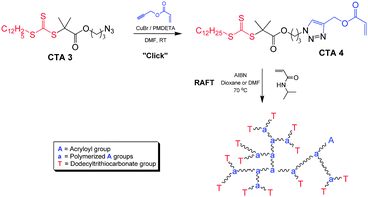 | ||
| Scheme 14 Synthesis of an acryloyl trithiocarbonate chain transfer agent (CTA4) and subsequent branched PNIPAM preparation via RAFT. Adapted with permission from ref. 46. Copyright 2007 CSIRO. | ||
Homopolymerization of AB* monomers containing both a polymerizable double bond (A) and an initiating moiety (B*) can lead to hyperbranched polymersvia a process often termed self-condensing vinylpolymerization (SCVP).129 Accordingly, the acryloyl trithiocarbonate was copolymerized with NIPAM to yield thermoresponsive hyperbranches. By virtue of the RAFT process, the individual branches were well-defined with length and number depending on the ratio of [NIPAM]:[CTA4], and each chain end contained the dodecyltrithiocarbonate moiety derived from the original CTA.
The aqueous thermoresponsive nature of these copolymers was investigated since highly branched polymers often demonstrate significantly different solubility behaviors from their linear counterparts. In addition to reduced chain entanglement, end group effects can be particularly important since branched polymers contain multiple termini.130 We observed transition temperatures significantly lower than the typical values. For example, PNIPAM (Mn = 11![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000 g mol−1, 4% branching) prepared with CTA4 showed a dramatic increase in hydrodynamic diameter, indicative of intermolecular aggregation at just 25 °C (Fig. 16). As the degree of branching increased, the LCST of the hyperbranched polymer decreased. For instance, PNIPAM with 10% branching was soluble in water only below 2 °C, an observation attributed to the enhanced hydrophobic contributions of the dodecyl chain termini. Indeed, end group cleavage by radical induced reduction yielded PNIPAM hyperbranches with hydrogen termini and the expected LCST of 32 °C.
000 g mol−1, 4% branching) prepared with CTA4 showed a dramatic increase in hydrodynamic diameter, indicative of intermolecular aggregation at just 25 °C (Fig. 16). As the degree of branching increased, the LCST of the hyperbranched polymer decreased. For instance, PNIPAM with 10% branching was soluble in water only below 2 °C, an observation attributed to the enhanced hydrophobic contributions of the dodecyl chain termini. Indeed, end group cleavage by radical induced reduction yielded PNIPAM hyperbranches with hydrogen termini and the expected LCST of 32 °C.
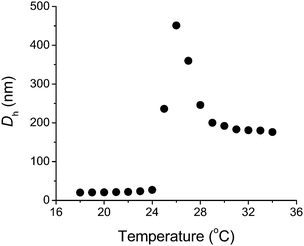 | ||
| Fig. 16 Hydrodynamic diameter (Dh) versus temperature for a 5% w/v aqueous solution of highly branched PNIPAM (11000 g mol−1, 4% branching). Adapted with permission from ref. 46. Copyright 2007 CSIRO. | ||
4. Conclusion/future work
The applicability of the RAFT process for the polymerization of functional monomers under a diverse range of experimental conditions has facilitated the synthesis of responsive, hydrolytically-stable, water-soluble (co)polymers that were previously inaccessible. Unprecedented control afforded by RAFT in homogeneous aqueous media allows well-defined polymeric systems to be prepared without stringent purification techniques and under increasingly “green” conditions while maintaining the ability to tailor many of the macromolecular characteristics (molecular weight, chain topology, copolymer composition, functionality, etc.) that affect self-assembly in solution.We have highlighted the work in our groups and others detailing block copolymer formation by RAFT, as well as postpolymerization modification utilizing crosslinking and other highly efficient and orthogonal synthetic methods. Particular attention was paid to temperature-responsive systems, but the facile control over block copolymer structure afforded by RAFT is useful for self-assembly in aqueous media in response to a variety of other external stimuli. This flexibility, combined with the capacity of RAFT to readily control block copolymer composition, which generally dictates self assembled aggregate morphology (micelles, vesicles, etc.), offers great potential to prepare controlled and targeted drug delivery vehicles, biocompatible hydrogels, responsive polymer–protein bioconjugates, and many other advanced materials with biological relevance.1 Indeed, RAFT polymerization and many of the postpolymerization modification approaches described herein have facilitated access to “smart” materials with unprecedented opportunities in biomedical, pharmaceutical, and diagnostic areas.
Acknowledgements
CLM gratefully acknowledges financial support provided by the Department of Energy (DE-FC26-01BC15317) and the MRSEC program of the National Science Foundation (DMR-0213883). BSS acknowledges the Donors of the American Chemical Society Petroleum Research Fund (45286-G7), Oak Ridge Associated Universities (Ralph E. Powe Junior Faculty Enhancement Award), and the Defense Advanced Research Projects Agency (HR0011-06-1-0032).References
- Part 129: A. W. York, S. E. Kirkland and C. L. McCormick, Adv. Drug Delivery Rev., 2008, 60, 1018 Search PubMed.
- A. B. Lowe, C. L. McCormick and N. Ayres, Encyclopedia of Polymer Science and Technology, John Wiley and Sons, Hoboken, NJ, 2007 Search PubMed.
- K. Matyjaszewski and T. Davis, Handbook of Radical Polymerization, John Wiley and Sons, Inc., Hoboken, IL, 2002 Search PubMed.
- C. J. Hawker, A. W. Bosman and E. Harth, Chem. Rev., 2001, 101, 3661 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2006, 59, 669 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559 CrossRef CAS.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, Y. K. Chong, G. Moad and S. H. Thang, Macromolecules, 1999, 32, 6977 CrossRef CAS.
- C. L. McCormick and A. B. Lowe, Acc. Chem. Res., 2004, 37, 312 CrossRef CAS.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283 CrossRef CAS.
- C. Barner-Kowollik, M. Buback, B. Charleux, M. L. Coote, M. Drache, T. Fukuda, A. Goto, B. Klumperman, A. B. Lowe, J. B. Mcleary, G. Moad, M. J. Monteiro, R. D. Sanderson, M. P. Tonge and P. Vana, J. Polym. Sci., Part A: Polym. Chem., 2006, 5809 CrossRef CAS.
- L. Chang and A. S. Hoffman, J. Controlled Release, 1990, 21.
- F. Kohori, K. Sakai, T. Aoyagi, M. Yokoyama, Y. Sakurai and T. Okano, J. Controlled Release, 1998, 87 CrossRef CAS.
- J. N. Cambre, D. Roy, S. R. Gondi and B. S. Sumerlin, J. Am. Chem. Soc., 2007, 129, 10348 CrossRef CAS.
- C. L. McCormick, S. E. Kirkland and A. W. York, J. Macromol. Sci., Part C: Polym. Rev., 2006, 46, 421 Search PubMed.
- C. Allen, D. Maysinger and A. Eisenberg, Colloids Surf., B, 1999, 16, 3 CrossRef CAS.
- S. Liu and S. Armes, Curr. Opin. Colloid Interface Sci., 2001, 6, 249 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2008, 2477 RSC.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 1441 CrossRef CAS.
- L. C. Dong and A. S. Hoffman, J. Controlled Release, 1990, 13, 21 CrossRef CAS.
- F. M. Winnik, A. J. Alexander and K. Hirowi, Can. J. Chem., 1995, 73, 2030 CrossRef CAS.
- G. Chen and A. S. Hoffman, Nature, 1995, 373, 49 CrossRef CAS.
- F. Ganachaud, M. J. Monteiro, R. G. Gilbert, M.-A. Dourges, S. H. Thang and E. Rizzardo, Macromolecules, 2000, 33, 6738 CrossRef CAS.
- C. Schilli, M. G. Lanzendörfer and A. H. E. Müller, Macromolecules, 2002, 35, 6819 CrossRef CAS.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341 CrossRef CAS.
- P. Kujawa, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 3048 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromol. Rapid Commun., 2006, 27, 1648 CrossRef CAS.
- B. Ray, Y. Isobe, K. Morioka, S. Habaue, Y. Okamoto, M. Kamigaito and M. Sawamoto, Macromolecules, 2003, 36, 543 CrossRef CAS.
- J. Raula, J. Shan, M. Nuopponen, A. Niskanen, H. Jiang, E. Kauppinen and H. Tenhu, Langmuir, 2003, 3499 CrossRef CAS.
- J. Shan, M. Nuopponen, H. Jiang, E. Kauppinen and H. Tenhu, Macromolecules, 2003, 36, 4526 CrossRef CAS.
- J. Shan, M. Nuopponen, H. Jiang, T. Viitala, E. Kauppinen, K. Kontturi and H. Tenhu, Macromolecules, 2005, 38, 2918 CrossRef CAS.
- A. B. Lowe, B. S. Sumerlin, M. S. Donovan and C. L. McCormick, J. Am. Chem. Soc., 2002, 124, 11562 CrossRef CAS.
- B. S. Sumerlin, A. B. Lowe, P. A. Stroud, P. Zhang, M. W. Urban and C. L. McCormick, Langmuir, 2003, 19, 5559 CrossRef CAS.
- S. L. Yusa, Y. Shimada, Y. Mitsukami, T. Yamamoto and Y. Morishima, Macromolecules, 2004, 35, 7507 CrossRef.
- J. Virtanen, S. Holappa, H. Lemmetyinen and H. Tenhu, Macromolecules, 2002, 35, 4763 CrossRef CAS.
- B. Liu and S. Perrier, J. Polym. Sci., Polym. Chem., 2005, 43, 3643 CrossRef CAS.
- M. Arotcarena, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787 CrossRef CAS.
- J. Virtanen, M. Arotcarena, B. Heise, S. Ishaya, A. Laschewsky and H. Tenhu, Langmuir, 2002, 18, 5360 CrossRef CAS.
- Z. Ozyurek, H. Komber, S. Gramm, D. Schmaljohann, A. H. E. Muller and B. Voit, Macromol. Chem. Phys., 2007, 208, 1035 CrossRef.
- Y.-Z. You and D. Oupicky, Biomacromolecules, 2007, 8, 98 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724 CrossRef CAS.
- B. S. Lokitz, A. J. Convertine, R. G. Ezell, A. Heidenreich, Y. Li and C. L. McCormick, Macromolecules, 2006, 39, 8594 CrossRef CAS.
- Y. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2006, 39, 81 CrossRef CAS.
- Y. Li, B. S. Lokitz and C. L. McCormick, Angew. Chem., Int. Ed., 2006, 45, 5792 CrossRef CAS.
- A. P. Vogt, S. R. Gondi and B. S. Sumerlin, Aust. J. Chem., 2007, 60, 396 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, A. B. Lowe, C. W. Scales, L. J. Myrick and C. L. McCormick, Macromol. Rapid Commun., 2005, 26, 791 CrossRef CAS.
- A. J. Convertine, B. S. Lokitz, Y. Vasilieva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 5, 1724 CrossRef.
- E. C. Cho, J. Lee and K. Cho, Macromolecules, 2003, 36, 9929 CrossRef CAS.
- V. Castelletto, I. W. Hamley, Y. Ma, X. Bories-Azeau, S. P. Armes and A. L. Lewis, Langmuir, 2004, 20, 4306 CrossRef CAS.
- C. Li, N. J. Buurma, I. Haq, C. Turner, S. P. Armes, V. Castelletto, I. W. Hamley and A. L. Lewis, Langmuir, 2005, 21, 11026 CrossRef CAS.
- C. Li, Y. Tang, S. P. Armes, C. J. Morris, S. F. Rose, A. W. Lloyd and A. L. Lewis, Biomacromolecules, 2005, 6, 994 CrossRef CAS.
- Y. Ma, Y. Tang, N. C. Billingham, S. P. Armes and A. L. Lewis, Biomacromolecules, 2003, 4, 864 CrossRef CAS.
- J. Madsen, S. P. Armes and A. L. Lewis, Macromolecules, 2006, 39, 7455 CrossRef CAS.
- S. E. Kirkland, R. M. Hensarling, S. D. McConaughy, Y. Guo, W. L. Jarrett and C. L. McCormick, Biomacromolecules, 2008, 9, 481 CrossRef CAS.
- P. E. Millard, L. Barner, M. H. Stenzel, T. P. Davis, C. Barner-Kowollik and A. H. E. Muller, Macromol. Rapid Commun., 2006, 27, 821 CrossRef CAS.
- S. Liu and S. P. Armes, Angew. Chem., Int. Ed., 2002, 41, 1413 CrossRef CAS.
- S. Liu and S. P. Armes, Langmuir, 2003, 19, 4432 CrossRef CAS.
- M. Arotcarena, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787 CrossRef CAS.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1996, 118, 7239 CrossRef.
- E. S. Read and S. P. Armes, Chem. Commun., 2007, 3021 RSC.
- K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 1996, 7239.
- K. B. Thurmond, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1997, 6656 CrossRef CAS.
- H. Huang, E. E. Remsen, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 1999, 3805 CrossRef CAS.
- Q. Ma, E. E. Remsen, T. Kowalewski and K. L. Wooley, J. Am. Chem. Soc., 2001, 4627 CrossRef CAS.
- T. Sanji, Y. Nakatsuka, S. Ohnishi and H. Sakurai, Macromolecules, 2000, 8524 CrossRef CAS.
- R. S. Underhill and G. Liu, Chem. Mater., 2000, 2082 CrossRef CAS.
- S. Fujii, Y. Cai, J. V. M. Weaver and S. P. Armes, J. Am. Chem. Soc., 2005, 7304 CrossRef CAS.
- Y. K. Chong, T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1999, 32, 2071 CrossRef CAS.
- C.-E. Hong, Y.-Z. You and C.-Y. Pan, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4873 CrossRef CAS.
- S. Perrier, P. Takolpuckdee, J. Westwood and D. M. Lewis, Macromolecules, 2004, 37, 2709 CrossRef CAS.
- P. Relogio, M. T. Charreyre, J. P. S. Farinha, J. M. G. Martinho and C. Pichot, Polymer, 2004, 45, 8639 CrossRef CAS.
- Y. Li, B. S. Lokitz and C. L. McCormick, Macromolecules, 2006, 39, 81 CrossRef CAS.
- Y. Xia, X. Yin, N. A. D. Burke and H. D. H. Stover, Macromolecules, 2005, 38, 5937 CrossRef CAS.
- J. Pietrasik, B. S. Sumerlin, R. Y. Lee and K. Matyjaszewski, Macromol. Chem. Phys., 2007, 208, 30 CrossRef CAS.
- Y. Li, I. Akiba, S. Harrison and K. L. Wooley, Adv. Funct. Mater., 2008, 18, 551 CrossRef CAS.
- Y. Li, B. S. Lokitz, S. Armes and C. L. McCormick, Macromolecules, 2006, 39, 2726 CrossRef CAS.
- N. V. Tsarevsky and L. Matyjaszewski, Macromolecules, 2002, 35, 9009 CrossRef CAS.
- H. A. Aliyar, P. D. Hamilton and N. Ravi, Biomacromolecules, 2005, 6, 204 CrossRef CAS.
- D. J. Cline, S. E. Redding, S. G. Brohawn, J. N. Psathas, J. P. Schneider and C. Thorpe, Biochemistry, 2004, 43, 15195 CrossRef CAS.
- Y. Li and S. P. Armes, Macromolecules, 2005, 38, 8155 CrossRef CAS.
- M. B. Grisham, Free Radical Biol. Med., 2004, 36, 1479 CrossRef CAS.
- R. G. Ezell, I. Gorman, B. S. Lokitz, N. Ayres and C. L. McCormick, J. Polym. Sci., Part A: Polym. Chem., 2006, 3125 CrossRef CAS.
- R. G. Ezell, I. Gorman, B. S. Lokitz, N. D. Treat, S. D. McConaughy and C. L. McCormick, J. Polym. Sci., Part A: Polym. Chem., 2006, 4479 CrossRef CAS.
- R. G. Ezell and C. L. McCormick, J. Appl. Polym. Sci., 2007, 104, 2812 CrossRef CAS.
- B. S. Lokitz, J. E. Stempka, A. W. York, Y. Li, H. K. Goel, G. R. Bishop and C. L. McCormick, Aust. J. Chem., 2006, 749 CrossRef CAS.
- I. D. Chung, P. Britt, D. Xie, E. Harth and J. Mays, Chem. Commun., 2005, 1046 RSC.
- H. Mori, H. Iwaya, A. Nagai and T. Endo, Chem. Commun., 2005, 4872 RSC.
- H. Mori, M. Matsuyama, K. Sutoh and T. Endo, Macromolecules, 2006, 4351 CrossRef CAS.
- H. Mori, K. Sutoh and T. Endo, Macromolecules, 2005 Search PubMed.
- J. V. M. Weaver, Y. Tang, S. Liu, P. D. Iddon, R. Grigg and S. P. Armes, Angew. Chem., Int. Ed., 2004, 43, 1389 CrossRef CAS.
- H. R. Petty, Molecular Biology of Membranes, Structure and Function, Plenum, New York, 1993 Search PubMed.
- Y. Morishima, Angew. Chem., Int. Ed., 2007, 1370 CrossRef CAS.
- B. M. Discher, Y. Won, D. S. Ege, J. C. M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 1143 CrossRef CAS.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2004, 8, 323.
- J. Wu and A. Eisenberg, J. Am. Chem. Soc., 2006, 128, 2880 CrossRef CAS.
- L. Zhang and A. Eisenberg, Science, 1995, 268, 1728 CrossRef CAS.
- P. L. Soo and A. Eisenberg, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 923 CrossRef CAS.
- F. Checot, S. Lecommandoux, Y. Gnanou and H. A. Klok, Angew. Chem., Int. Ed., 2002, 41, 1339 CrossRef CAS.
- J. Du and S. Armes, J. Am. Chem. Soc., 2005, 127, 12800 CrossRef CAS.
- J. Du, Y. Tang, A. L. Lewis and S. P. Armes, J. Am. Chem. Soc., 2005, 127, 17982 CrossRef CAS.
- J. Rodriguez-Hernandez and S. Lecommandoux, J. Am. Chem. Soc., 2005, 127, 2026 CrossRef CAS.
- Y. Xia, X. Yin, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2005, 38, 5937 CrossRef CAS.
- Y. Li, A. E. Smith, B. S. Lokitz and C. L. McCormick, Macromolecules, 2007, 40, 8524 CrossRef CAS.
- G. Mantovani, V. Ladmiral, L. Tao and D. M. Haddleton, Chem. Commun., 2005, 4702 RSC.
- C. W. Scales, A. J. Convertine and C. L. McCormick, Biomacromolecules, 2006, 7, 1389 CrossRef CAS.
- A. W. York, C. W. Scales, F. Huang and C. L. McCormick, Biomacromolecules, 2007, 8, 2337 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372 CrossRef CAS.
- D. Bontempo, R. C. Li, T. Ly, C. E. Brubaker and H. D. Maynard, Chem. Commun., 2005, 4702 RSC.
- V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS.
- C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2007, 28, 15 CrossRef CAS.
- R. A. Evans, Aust. J. Chem., 2007, 60, 384 CrossRef CAS.
- C. J. Hawker, V. V. Fokin, M. G. Finn and K. B. Sharpless, Aust. J. Chem., 2007, 60, 381 CrossRef CAS.
- M. J. Joralemon, R. K. O'Reilly, C. J. Hawker and K. L. Wooley, J. Am. Chem. Soc., 2005, 127, 16892 CrossRef CAS.
- J. F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018 CrossRef CAS.
- J. F. Lutz, H. G. Borner and K. Weichenhan, Macromol. Rapid Commun., 2005, 26, 514 CrossRef CAS.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57 RSC.
- R. K. O'Reilly, M. J. Joralemon, K. L. Wooley and C. J. Hawker, Chem. Mater., 2005, 17, 5976 CrossRef CAS.
- N. V. Tsarevsky, B. S. Sumerlin and K. Matyjaszewski, Macromolecules, 2005, 38, 3558 CrossRef CAS.
- A. P. Vogt and B. S. Sumerlin, Macromolecules, 2006, 39, 5286 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, Chem.–Eur. J., 2006, 12, 6776 CrossRef CAS.
- R. K. O'Reilly, M. J. Joralemon, C. J. Hawker and K. L. Wooley, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5203 CrossRef CAS.
- S. R. Gondi, A. P. Vogt and B. S. Sumerlin, Macromolecules, 2007, 40, 474 CrossRef CAS.
- D. Quemener, T. P. Davis, C. Barner-Kowollik and M. H. Stenzel, Chem. Commun., 2006, 5051 RSC.
- P. De, S. R. Gondi and B. S. Sumerlin, Biomacromolecules, 2008, 9, 1064 CrossRef CAS.
- M. Li, P. De, S. R. Gondi and B. S. Sumerlin, Macromol. Rapid Commun., 2008 DOI:10.1002/marc.200800073.
- J. M. J. Frechet, M. Henmi, I. Gitsov, S. Aoshima, M. R. Leduc and R. B. Grubbs, Science, 1995, 269, 1080 CrossRef CAS.
- R. Plummer, D. J. T. Hill and A. K. Whittaker, Macromolecules, 2006, 39, 8379 CrossRef CAS.
Footnote |
| † Water-soluble polymers. Part 130.1 |
| This journal is © The Royal Society of Chemistry 2008 |