 Open Access Article
Open Access ArticleNatural products as an inspiration in the diversity-oriented synthesis of bioactive compound libraries
Christopher
Cordier
ab,
Daniel
Morton
ab,
Sarah
Murrison
ab,
Adam
Nelson
*ab and
Catherine
O'Leary-Steele
ab
aSchool of Chemistry, University of Leeds, Leeds, LS2 9JT, UK
bAstbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
First published on 14th April 2008
Abstract
Covering: up to the end of August 2007
The purpose of diversity-oriented synthesis is to drive the discovery of small molecules with previously unknown biological functions. Natural products necessarily populate biologically relevant chemical space, since they bind both their biosynthetic enzymes and their target macromolecules. Natural product families are, therefore, libraries of pre-validated, functionally diverse structures in which individual compounds selectively modulate unrelated macromolecular targets. This review describes examples of diversity-oriented syntheses which have, to some extent, been inspired by the structures of natural products. Particular emphasis is placed on innovations that allow the synthesis of compound libraries that, like natural products, are skeletally diverse. Mimicking the broad structural features of natural products may allow the discovery of compounds that modulate the functions of macromolecules for which ligands are not known. The ability of innovations in diversity-oriented synthesis to deliver such compounds is critically assessed.
 Chris Cordier | Chris Cordier graduated in 2003 from the University of Leeds with a first class BSc (Hons) degree in Chemistry and Pharmacology. His PhD, under the supervision of Prof. Adam Nelson and Dr Stuart Warriner, concerned the synthesis of diverse compounds with natural product-like structural motifs. He has been awarded a Herchel Smith postdoctoral research fellowship in organic chemistry at the University of Cambridge. |
 Daniel Morton | Daniel Morton obtained his MChem from the University of East Anglia in 2001, where he remained for his PhD (supervised by Dr Rob Stockman and Prof. Rob Field) which concerned the synthesis of chiral aziridines. In 2005, he joined the Nelson group, where he has worked on the total synthesis of hemibrevetoxin B and the diversity-oriented synthesis of natural product-like molecules. |
 Sarah Murrison | Sarah Murrison obtained a first class MChem degree from the University of Aberdeen. Her final year involved an overseas placement at l’Université Blaise Pascal, Clermont-Ferrand. Sarah joined Prof. Nelson's group in 2006 as a PhD student. Her project involves the development of a scaffold-switching strategy for the diversity-oriented synthesis of polycyclic alkaloid-like compounds. |
 Adam Nelson | Adam Nelson was appointed at the University of Leeds in 1998, and is currently Professor of Chemical Biology and Deputy Director of the Astbury Centre. Prof. Nelson was awarded the RSC Meldola medal (in 2001), the Pfizer academic award (in 2002), an EPSRC advanced research fellowship (2004–2009) and the AstraZeneca award in organic chemistry (in 2005). |
 Catherine O’Leary-Steele | Catherine O'Leary-Steele graduated with a first class MChem degree from the University of Oxford in 2005, completing her Part II project under the supervision of Prof. Steve Davies. Catherine is currently a PhD student in Professor Nelson's group, working on the synthesis of skeletally diverse, natural product-like small molecules. |
1 Introduction
The interrogation of fundamental biological mechanisms is an immense challenge for the next decade and beyond. The functions of all proteins identified from the genome must be determined, the in vivo relevance of post-translation states and interaction targets recognised, and the effects on cellular physiology established. The field of chemical genetics is based on the premise that small molecules may be used to perturb the functions of specific biological macromolecules, thus revealing insights into their roles in complex biological mechanisms.1 Chemical genetics, and its impact on our understanding of fundamental biological mechanisms, has been comprehensively reviewed.1aThe use of small molecule probes in chemical genetic studies confers a number of advantages over conventional genetic manipulations which may be used to vary the structure, and hence the function, of encoded protein(s). First, the effects of small molecules are usually rapid, reversible and conditional. Second, by varying the concentration of the small molecule tool(s), the activity of a target macromolecule may be tuned in a refined way. Third, the chemical genetic approach can sometimes allow the many functions of individual proteins to be teased apart.2 Finally, by using more than one small molecule effector in combination, the interplay between proteins may be studied.3 The approach is particularly useful for investigating tightly coordinated, dynamic biological mechanisms, especially when classical genetics may render the organism inviable.4
1.1 Diversity-oriented synthesis
A key challenge in chemical genetics is the design and synthesis of libraries which span large tracts of biologically relevant chemical space.5 This challenge has spawned a new field in synthetic chemistry – diversity-oriented synthesis6 (DOS). The synthetic challenges of DOS are very different to target-oriented synthesis, where tailored methods may be developed to solve the synthetic problems raised. DOS methods must be sufficiently robust to allow diverse compounds to be prepared simultaneously, deliberately and combinatorially. In general, the assembly of diverse small molecule libraries must be accomplished in up to ca. five highly reliable synthetic steps, leaving little or no scope for protecting group chemistry.Chemical libraries prepared using diversity-oriented synthetic approaches can often be used to identify new lead molecules.1 In contrast, combinatorial chemistry is used to prepare much more focussed libraries which target narrower regions of chemical space. Indeed, such libraries are widely used in the pharmaceutical and agrochemical industries to optimise the structure, and hence the activity, of a particular ligand.
The diversity of chemical libraries may derive from the high substitutional†, stereochemical and skeletal diversity of its members. Combinatorial chemistry is focussed on the synthesis of chemical libraries with only high substitutional diversity. In general, combinatorial syntheses exploit a rather limited palette of chemical transformations to vary the substitution of library members: for example, amide and sulfonamide formation, the Mitsunobu reaction, and Pd-catalysed processes. The broad combinatorial chemistry approach can, however, be extended to the synthesis of libraries based on much more complex, natural product-like scaffolds.
Combinatorial variation of the configuration of library members is rather more challenging. However, a broad range of reliable asymmetric transformations has been developed over the past 25 years. In many cases, the high enantioselectivity of these transformations can dominate over substrate-based diastereoselectivity. Thus, by harnessing the power of modern asymmetric chemistry, highly stereochemically diverse chemical libraries may be prepared.
Preparing libraries with high skeletal diversity is an altogether more challenging proposition. A range of new synthetic innovations has been required to vary systematically the underlying scaffolds of ligands. In this review, we will place particular emphasis on the approaches which have been used to vary molecular scaffolds systematically. We have made no attempt to discuss every aspect of each synthetic strategy. In general, we have indicated in the starting materials the scope for substitutional diversity (through the use of generic substituents Rn). The ability of each approach to deliver alternative scaffolds is indicated (with a yield) for specific combinations of generic substituents. In most syntheses, however, each molecular scaffold was diversely substituted, and, thus, represented by more than one compound; the reader is directed to the primary literature for a full description of the synthetic chemistry undertaken.
1.2 Natural products as an inspiration in the design of compound libraries
Natural products necessarily reside in biologically relevant chemical space, since they bind both their biosynthetic enzymes and their target macromolecules. Natural product families are libraries of pre-validated, functionally diverse structures: these families are “privileged”7 since individual compounds selectively modulate unrelated targets. Indeed, some natural products, such as trichostatin,8 wortmannin9 and brefeldin A,10 have been directly exploited as valuable tools in chemical genetic studies. It is, therefore, no surprise that natural products have provided an inspiration for ligand library design.In this review, examples of diversity-oriented chemistry are described which have, to some extent, been inspired by the structures of natural products. The purpose of DOS, however, is to enable the discovery of small molecules with previously unknown biological functions. Natural products do not adhere to principles that have been formulated to guide the structures of drug molecules.11 Indeed, the only generally accepted principle uniting the structures of protein-binding small molecules is that conformational restriction is advisable. Natural products do, of course, adhere to this principle through the presence of a range of structural features: dense substitution, unsaturated functionality, and ring systems.
Although the inspiration which may be provided by natural products is useful, it is essential that it does not inform DOS chemistry to the extent that only compounds with the same biological functions as known natural products models emerge. We have deliberately omitted, however, studies in which the inherent biological activity of a natural product was optimised through the synthesis of derivatives.12 In some cases, the scaffold (or scaffolds) represented within a DOS library closely resemble those found in natural products. Inspiration from natural product scaffolds is valuable in DOS, as ligands based on specific natural product scaffolds have been shown to bind selectively to different proteins with similar folds.5b In other examples of DOS chemistry, the relationship to natural products is much looser, with perhaps just some of the broad structural features of a natural product family being retained. Given that natural products necessarily have the broad structural features needed to recognise macromolecules, this level of inspiration is appropriate: indeed, it can allow the identification of ligands with functions that natural products have not yet been found to possess.
2 Strategies for diversity-oriented synthesis
2.1 Synthesis of compound libraries with high substitutional diversity
The combinatorial chemistry approach may be extended to the diversification of complex natural product scaffolds. The scaffold, 1, of the alkaloid galanthamine was prepared in five steps from a solid-supported tyrosine derivative.13 A sequence of reactions was then used to vary four of the substituents (R1 to R4) in the final compounds (Scheme 1). Functionalisation of the phenol using a Mitsunobu reaction (→ 2) was followed by conjugate addition of a thiol to the α,β-unsaturated ketone (→ 3), and functionalisation of the secondary amine (→ 4). Finally, the ketones 4 were converted into imines or oxime ethers 5. Following release from solid support, the successful formation of 2527 of 2946 possible compounds was confirmed by mass spectrometry. A cell-based phenotypic assay was used to identify secramine 6 as a potent inhibitor of protein trafficking from the Golgi apparatus to the plasma membrane. The discovery of secramine underlines the validity of exploiting natural product scaffolds in the discovery of small molecules with novel functions: secramine’s structure was inspired by galanthamine, but it has a novel biological function.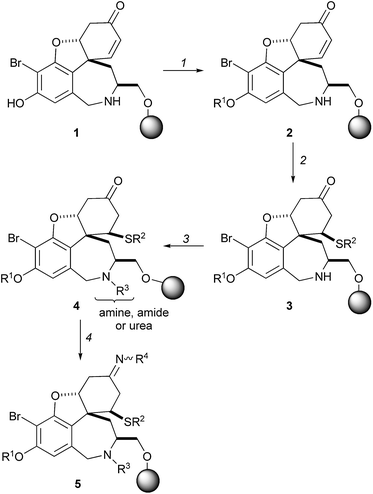 | ||
| Scheme 1 Shair's synthesis of galanthamine-like molecules with high substitutional diversity. Reagents and conditions: (1) R1OH, PPh3, DIAD, THF, 0 °C (×2); (2) R2SH, 2,6-lutidine, nBuLi, THF, 0→40 °C; (3) (i) R3CHO, AcOH, MeOH–THF; (ii) NaBH3CN, MeOH or R3COCl, 2,6-lutidine, CH2Cl2 or R3NCO, CH2Cl2; (4) R4NH2, AcOH, MeOH–CH2Cl2. | ||
A similar approach was used to prepare a library of conformationally restricted 1,3-dioxanes 10 with high substitutional diversity (Scheme 2).14 A split-pool approach was used to diversify three substituents in the final compounds 10. The supported epoxides 7 were opened with either a thiol or an amine nucleophile to yield 1,3-diols which were converted into Fmoc-substituted 1,3-dioxanes. Some of the nucleophiles used in the first step were hydroxy-substituted, leading to the formation of mixed acetals in the second step: these acyclic acetals were, therefore, hydrolysed by treatment of the resins with PPTS in THF–MeOH. Following removal of the Fmoc group (→ 9), the resulting free amines were converted into amides, ureas, thioureas or sulfonamides (→ 10), and the final compounds cleaved from the beads. Using material cleaved from a single bead, and with knowledge of the substituent added into the final step, it was possible to use mass spectrometry to identify possible combinations of substituents added in the first two diversification steps.
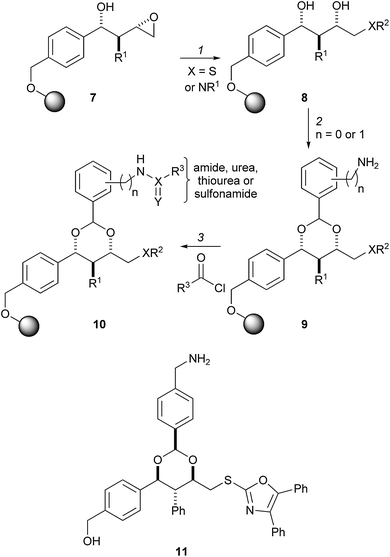 | ||
| Scheme 2 Schreiber's split-pool synthesis of 1,3-dioxanes with high substitutional diversity. Reagents and conditions: (1) R2SH or R1R2NH; (2) (i) ArCH(OMe)2, Me3SiCl, HCl, dioxane; (ii) PPTS, THF–MeOH; (iii) piperidine; (iv) Me3SiCl; (3) amide, urea, thiourea or sulfonamide formation. | ||
Using this general synthetic approach, a 3780-member library of small molecules was used to create a small molecule microarray.15 The microarray was then challenged with a fluorescently labelled derivative of Ure2 p, the central repressor of genes involved in nitrogen metabolism. The approach enabled the identification of the first small molecule ligand for Ure2 p: uretupamine, 11. Uretupamine was found to modulate only a subset of the functions of Ure2 p, and thus its effects were more specific than those resulting from deletion of the URE2 gene.
An asymmetric hetero-Diels–Alder reaction was used to prepare a library of dihydropyrancarboxamides which resemble sialic acid mimetics (structures 14 and their enantiomers) (Scheme 3).15 The library of 4320 structures was encoded using a binary encoding protocol which involved the attachment of tags to individual macrobeads. The asymmetric step (12 → 13) served a number of purposes: it generated the molecular scaffolds; it introduced some stereochemical diversity; and it incorporated the variable substituent R1. Deprotection of the ester, amide formation, and release from solid support gave the final compounds.
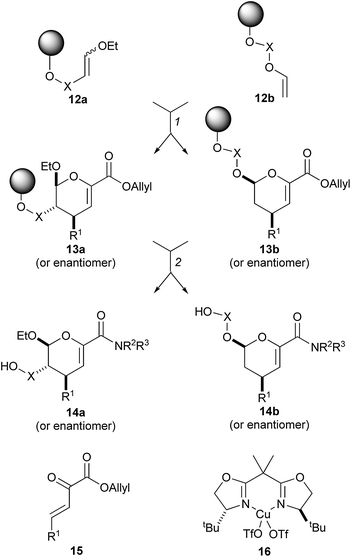 | ||
| Scheme 3 Schreiber's synthesis of dihydropyrans with high substitutional and stereochemical diversity. Reagents and conditions: (1) 15 or 20 mol% 16 (or enantiomer), THF, rt; (2) (i) Pd(PPh3)4, thiosalicylic acid, THF, rt; (ii) R2R3NH, PyBOP, iPr2NEt, CH2Cl2–DMF, rt; (iii) HF·pyridine, THF, rt, then Me3SiOMe. | ||
A small molecule microarray was created by immobolisation of three small molecule libraries (including the libraries of the 1,3-dioxanes 10 and the dihydropyrancarboxamides 14).16 The microarray was probed with a Hap3 p-GST fusion protein, and hits identified using a fluorescently-labelled anti-GST antibody. The approach enabled the discovery of haptamide A 14b, a ligand which targeted a subunit of the transcription factor, Hap3p. Haptamide B, an analogue of haptamide A, was subsequently discovered using a more traditional ligand optimisation approach.
In the three diversity-oriented syntheses described in Schemes 1–3, a library of substitutionally diverse small molecules was prepared. The inspiration provided by natural products, however, varied hugely. Each of the compounds 5 had the same skeleton as the alkaloid galanthamine, whereas the relationship between natural products and the other compounds was much looser. Schreiber has also described the preparation of a substitutionally diverse library of biaryl-containing medium-ring compounds (which have some similar structural features to biaryl-containing natural products such as vanomycin and pterocaryanin C).17
In the three examples described in detail (Schemes 1–3), a high-throughput screen enabled the identification of a small molecule with a novel function. A forward chemical genetic (phenotypic) screen was used to identify secramine (6) whereas reverse chemical genetic screens were used to identify small molecule partners (11 and 14b) for the proteins Ure2 p and Hap3p.‡ Substitutionally diverse libraries are, therefore, valuable tools which may be used to discover novel small molecules for interrogating biological mechanisms.
2.2 Use of ‘folding pathways’ to introduce skeletal diversity
The ‘folding pathway’ approach uses a common set of reaction conditions to transform a range of substrates into products with alternative molecular skeletons (Scheme 4). The substrates are encoded to ‘fold’ into the alternative scaffolds through strategically appended groups, known as ‘σ-elements’. This strategy leads skeletal diversification under substrate control.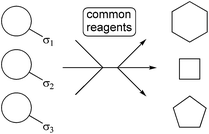 | ||
| Scheme 4 The folding pathway approach to generating skeletal diversity; the skeletal information is pre-encoded into the substrate through σ-elements which programme folding to yield distinct molecular skeletons under common reaction conditions. | ||
Schreiber has developed a folding pathway which exploits the Achmatowicz reaction (Scheme 5).18 The fate of oxidation of the furan substrates 17 depends on the functionalisation of groups elsewhere in the molecule (the σ-elements). Hence, oxidation of the furan 17c, which does not bear any free hydroxy groups, simply generated the ene-dione 18c. With suitably positioned nucleophilic groups in the starting material however, a cis-enedione intermediate could be intercepted. Hence, upon oxidation, the furyl alcohol 17b folded, and eliminated water, to yield the alkylidene-pyran-3-one 18b. In contrast, with two free hydroxyl groups, 17a folded to yield the bicyclic ketal 18a. The combination of solid-phase chemistry and common reaction conditions made the strategy amenable to split-pool synthesis, which significantly increased the efficiency of library generation.
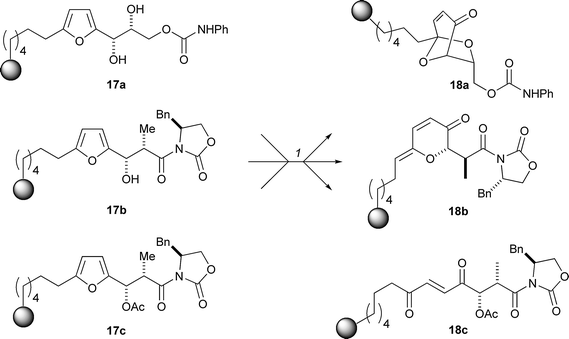 | ||
| Scheme 5 Schreiber's approach to pre-encoding skeletal information using the Achmatowicz reaction. Reagents and conditions: (1) N-bromosuccinimide, NaHCO3, NaOAc, THF–H2O (4 : 1), rt, 1 h; PPTS, CH2Cl2, 40–45 °C, 20 h. | ||
Schreiber used substrate-based control to create alternative indole alkaloid-like skeletons (Scheme 6).19 An α-diazo-β-keto-carbonyl group and an indole ring were strategically placed at different positions on the scaffolds of the starting materials (19). The densely functionalised starting materials were assembled either using an Ugi four-component coupling reaction or by alkylation of a common scaffold; subsequent conversion to a diazo compound yielded the cyclisation precursors. The substrates, 19, were treated with a catalytic amount of rhodium(II) octanoate dimer in benzene (80 °C). Presumably, generation of a carbonyl ylid was followed by intramolecular 1,3-dipolar cycloaddition with the indole ring to give the alternative polycyclic skeletons, 20.
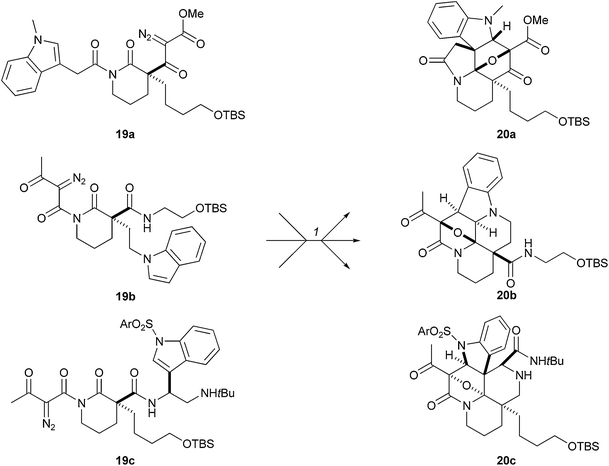 | ||
| Scheme 6 Schreiber's approach to pre-encoding skeletal information in rhodium-catalysed tandem cyclisation–cycloaddition cascade. Reagents and conditions: (1) Rh2(O2CC7H15)4, benzene, 50 °C; 20a: 74%; 20b: 73%; 20c: 57%. | ||
Panek and Porco used the cyclisation chemistry of radical intermediates to generate skeletal diversity (Scheme 7).20 The skeletons of the products 22 were pre-encoded in the substrates 21 by the location of the radical-initiating sites and the unsaturated groups. The tetrahydropyridine substrates 21 were prepared by stereoselective alkylation with a range of alkyl, alkenyl and alkynyl halides. The folding processes were triggered by treatment of the tetrahydropyridines 21 with tributyltin hydride and a substoichiometric amount of AIBN at 80 °C. Abstraction of a strategically-positioned bromine atom and cyclisation, yielded a range of distinct polycyclic alkaloid-like frameworks. Particularly high levels of skeletal complexity could be created when tandem radical cyclisation processes were possible (e.g. to yield 22a).
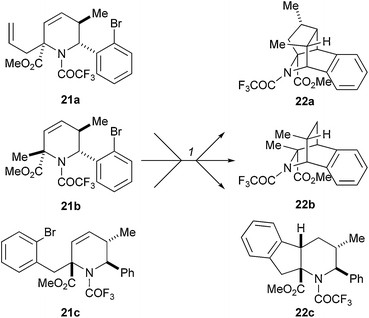 | ||
| Scheme 7 Panek and Porco's folding pathway using functionalised tetrahydropyridines. Reagents and conditions: (1) AIBN, Bu3SnH, 80 °C, 4 h, benzene, 22a: 65%; 22b: 89%; 22c: 81%. | ||
Schreiber has also developed an oligomer-based ‘folding’ strategy (Scheme 8).21 In this approach, metathesis substrates were assembled iteratively from monomer starting materials: Fukuyama–Mitsunobu reactions were used to prepare the substrates 23. The substrates 23 were folded by treatment with Grubbs' first-generation catalyst to yield 1,3-dienes 24. The 1,3-diene products 24 were substrates for a second diversification step: Diels–Alder reaction with a triazoline-3,5-dione dienophile gave the polycyclic products 25.
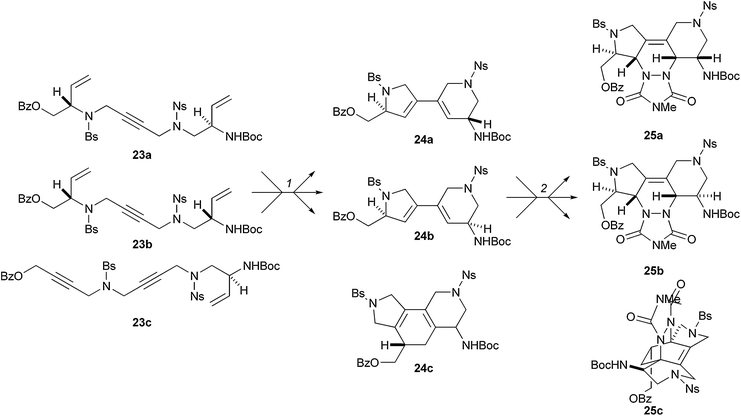 | ||
| Scheme 8 Oligomer-based approach to skeletal diversity employing metathesis cascades and Diels–Alder reactions as folding processes. Reagents and conditions: (1) Grubbs' first-generation catalyst (5 mol%), benzene, ethylene, reflux, 12–24 h; 24a: d.r. >20 : 1, 57%; 24b: d.r. >15 : 1, 74%; 24c: d.r. >20 : 1, 56%; (2) 4-methyl-1,2,4-triazoline-3,5-dione, CH2Cl2, 0 °C to rt.; 25a: 93%; 25b: 90%; 25c: 46%. Ns = 2-nitrobenzenesulfonyl, Bs = 4-bromobenzenesulfonyl. | ||
2.3 Assembly of alternative scaffolds using multi-component reactions
Innovative multi-component reactions have been developed to allow variation of the product scaffolds.§ In essence, alternative cyclisation precursors are assembled and ‘folded’ under common reaction conditions. The approach combines the power of multicomponent reactions with that of folding pathways.Dixon has used a one-pot gold(I)-catalysed cascade to prepare skeletally-diverse alkaloid-like small molecules (Scheme 9).22 Initially, Au(I) catalysed the cyclisation of an alkynyl carboxylic acid 26 to give a cyclic enol ether 27. Attack of an amine nucleophile 28 on the cyclic enol ether subsequently generated a ketoamide 29. In other words, the combination of alkynyl carboxylic acid 26 and amine 28 dictated the structure of the cyclisation precursor 29. Under the same reaction conditions, the cyclisation precursor was converted into an N-acyl iminium ion 30, which was then trapped by a tethered nucleophile (→ 31).
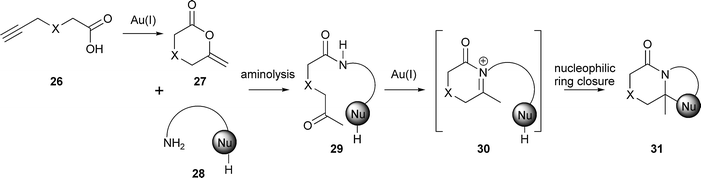 | ||
| Scheme 9 Outline of Dixon's approach for the synthesis of skeletally diverse, bicyclic heterocycles. X = O or CH2. | ||
The scope of Dixon's methodology is summarized in Scheme 10. Using combinations of the alkynyl carboxylic acids 26a–b, and the pyrrole- and indole-tethered amines 28a–b, a remarkable range of alternative alkaloid-like scaffolds were prepared.
![Dixon's Au(i)-catalysed cascade. Reagents and conditions: (1) AuPPh3Cl/AgOTf (1%), 26b, 28a, toluene, rt, 3 h, then reflux, 2 d, d.r. 2 : 1, 81% [R1 = H, R2 = CH2Ph, X = CH2]; (2) AuPPh3Cl/AgOTf (1%), 26a, 28a, toluene, rt, 3 h then reflux, 1 d, 91%; (3) AuPPh3Cl/AgOTf (1%), 26b, 28a, toluene, 75 °C, then reflux 1 d, d.r. 3 : 1, 64% [R1 = H, R2 = Me, X = O]; (4) AuPPh3Cl/AgOTf (1%), 26b, 28b, xylene, 75 °C, 3 h, then 125 °C, 1 d, d.r. 1 : 5 : 1, 97%, [R1 = n-pentyl, R2 = H, X = O]; (5) AuPPh3Cl/AgOTf (1%), 26b, 28b, toluene, 75 °C, 3 h, then reflux, 2 d, d.r. 1 : 1 : 5, 82%, [R1 = H, R2 = CH2Ph, X = CH2]; (6) AuPPh3Cl/AgOTf (1%), 26a, 28b, toluene, 75 C, 3 h, reflux, 1 d, 87%.](/image/article/2008/NP/b706296f/b706296f-s10.gif) | ||
| Scheme 10 Dixon's Au(I)-catalysed cascade. Reagents and conditions: (1) AuPPh3Cl/AgOTf (1%), 26b, 28a, toluene, rt, 3 h, then reflux, 2 d, d.r. 2 : 1, 81% [R1 = H, R2 = CH2Ph, X = CH2]; (2) AuPPh3Cl/AgOTf (1%), 26a, 28a, toluene, rt, 3 h then reflux, 1 d, 91%; (3) AuPPh3Cl/AgOTf (1%), 26b, 28a, toluene, 75 °C, then reflux 1 d, d.r. 3 : 1, 64% [R1 = H, R2 = Me, X = O]; (4) AuPPh3Cl/AgOTf (1%), 26b, 28b, xylene, 75 °C, 3 h, then 125 °C, 1 d, d.r. 1 : 5 : 1, 97%, [R1 = n-pentyl, R2 = H, X = O]; (5) AuPPh3Cl/AgOTf (1%), 26b, 28b, toluene, 75 °C, 3 h, then reflux, 2 d, d.r. 1 : 1 : 5, 82%, [R1 = H, R2 = CH2Ph, X = CH2]; (6) AuPPh3Cl/AgOTf (1%), 26a, 28b, toluene, 75 C, 3 h, reflux, 1 d, 87%. | ||
A Lewis acid-catalysed, multicomponent reaction involving an aniline 32, a cyclic enol ether 33 and a glyoxaldehyde 34, has been developed (Scheme 11).23 The cyclic enol ether 33 attacked an iminium ion derived from the aniline 32 and the aldehyde 34. The outcome of the reaction depended on the fate of the resulting oxocarbenium ion. Intramolecular capture can occur through participation of a pendant carboxylic acid (→ γ-lactone 35a), aryl ring (→ tetrahydroquinoline 35c), or alcohol (→ bicyclic acetal 35d). Alternatively, capture by the solvent (ethanol) yielded a THP acetal (35b). The scaffolds 35b–c were further skeletally diversified by oxidation to the quinolines 36 and 37.
![Latvilla's multicomponent reaction. Reagents and conditions: (1) Sc(OTf)3 (20%), 32a, 33, 34b, 4 Å MS, MeCN, Ar, 2 d, rt, d.r. 70 : 30, 83%, [R1,R2,R3 = H]; (2) Sc(OTf)3 (20%), 32b, 33, 34a, EtOH, d.r. 2 : 3 : 1, 82%, [R1,R2,R3=H]; (3) TFA (O2), MeCN–H2O, 71%; (4) Sc(OTf)3 (20%), 32b, 33, 34a, single stereoisomer, [R1,R2 =OAc; R3 =(R)-CH2OAc]; (5) CAN, MeCN, 58%; (6) Sc(OTf)3 (20%), 32a, 33, 34a, d.r. 2 : 5 : 1, 46%, [R1,R2 = H, R3= (R)-CH2OH].](/image/article/2008/NP/b706296f/b706296f-s11.gif) | ||
| Scheme 11 Latvilla's multicomponent reaction. Reagents and conditions: (1) Sc(OTf)3 (20%), 32a, 33, 34b, 4 Å MS, MeCN, Ar, 2 d, rt, d.r. 70 : 30, 83%, [R1,R2,R3 = H]; (2) Sc(OTf)3 (20%), 32b, 33, 34a, EtOH, d.r. 2 : 3 : 1, 82%, [R1,R2,R3=H]; (3) TFA (O2), MeCN–H2O, 71%; (4) Sc(OTf)3 (20%), 32b, 33, 34a, single stereoisomer, [R1,R2 =OAc; R3 =(R)-CH2OAc]; (5) CAN, MeCN, 58%; (6) Sc(OTf)3 (20%), 32a, 33, 34a, d.r. 2 : 5 : 1, 46%, [R1,R2 = H, R3= (R)-CH2OH]. | ||
Taylor and Raw have devised a multicomponent reaction which can yield skeletally diverse products (Scheme 12).24 The mechanism of formation of some of the products is shown in Scheme 13. Condensation of a carbonyl compound 39 with an amine 40 yields an enamine which can participate in an inverse electron-demand Diels–Alder with a triazine 38 (→ 42). Expulsion of molecular nitrogen, via a retro-Diels–Alder reaction, yields a 2-aza-diene 43. The outcome of the reaction depends on the fate of the 2-aza-diene 43. In the absence of a pendant alkene, the final product is usually a 2-aza-diene (e.g.41b, Scheme 12), although aromatisation to yield a highly substituted pyridine is also possible.24b,c If cyclobutanone is used as the carbonyl compound, electrocyclic ring-opening can occur to yield an eight-membered heterocycle (e.g.41c). However, with a suitably placed pendant alkene, an intramolecular Diels–Alder reaction is possible to generate yet more complex skeletons (e.g.41a, 41d or 41e). The skeletons 41 are diverse, and broadly resemble those of alkaloids.
![Taylor's cascade for the synthesis of polycyclic compounds. Reagents and conditions: (1) 4 Å MS, CHCl3, Δ; 41a: 96% [R1 = Py, R2,R3 = –(CH2)3–, R4,R5 = allyl]; 41b: 83% [R1 = Py, R2,R3 = –(CH2)3–, R4,R5 = –(CH2)4–]; 41c: 56% [R1= CO2Et, R2,R3 = –(CH2)2–, R4,R5= –(CH2)4–]; 41d: 67% [R1 = Py, R2 = trans-3-octane, R3 = H, R4, R5 = –(CH2)4–]; 41e: 39% [R1 = CO2Et, R2,R3 = –(CH2)3–, R4 = Bn, R5 = but-3-enyl]. Py = 2-pyridyl.](/image/article/2008/NP/b706296f/b706296f-s12.gif) | ||
| Scheme 12 Taylor's cascade for the synthesis of polycyclic compounds. Reagents and conditions: (1) 4 Å MS, CHCl3, Δ; 41a: 96% [R1 = Py, R2,R3 = –(CH2)3–, R4,R5 = allyl]; 41b: 83% [R1 = Py, R2,R3 = –(CH2)3–, R4,R5 = –(CH2)4–]; 41c: 56% [R1= CO2Et, R2,R3 = –(CH2)2–, R4,R5= –(CH2)4–]; 41d: 67% [R1 = Py, R2 = trans-3-octane, R3 = H, R4, R5 = –(CH2)4–]; 41e: 39% [R1 = CO2Et, R2,R3 = –(CH2)3–, R4 = Bn, R5 = but-3-enyl]. Py = 2-pyridyl. | ||
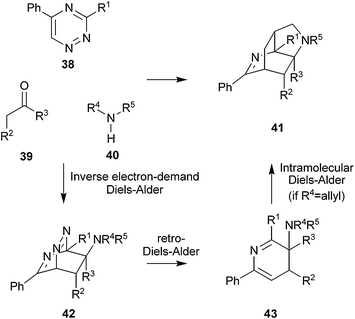 | ||
| Scheme 13 Mechanism of Taylor's reaction cascade. | ||
2.4 Exploitation of combinations of complexity-generating reactions
Using complexity-generating reactions in combination can lead to rapid increases in molecular complexity. Indeed, earlier in this review, a four-component Ugi reaction was used to prepare starting materials for Rh-catalysed dipolar cycloaddition reactions (Scheme 6).19 Furthermore, the use of metathesis cascade folding processes in combination with Diels–Alder reactions has also been described (Scheme 8).20 Both of these approaches were discussed in the context of folding pathways, but the use of two complexity-generating reactions in combination allowed some highly complex, polycyclic alkaloid-like molecules to be prepared. Here, we describe further examples of diversity-oriented syntheses in which complexity-generating reactions were used in combination.The Diels–Alder reactions of the solid-supported trienes 44 yielded skeletally diverse products (Scheme 14).25 The issue of regioselectivity is raised in the Diels–Alder reactions of the cross-conjugated trienes 44. With tri- and tetra-substituted dienophiles, one of the two available dienes reacted to yield a diene product (e.g.45a), which could then undergo a subsequent Diels–Alder reaction with a second dienophile (→ 46). Remarkably, however, with a disubstituted dienophile, such as N-ethyl maleimide, the other available diene reacted initially, and after a second Diels–Alder reaction, an alternative skeleton (e.g.45b) was obtained. The approach was used to prepare 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 polycyclic compounds with ten distinct molecular skeletons. Some of the compounds were immobilised on a small molecule microarray, which was then challenged with Cy5-conjugated calmodulin. The approach allowed novel sub-micromolar ligands for calmodulin to be discovered.
400 polycyclic compounds with ten distinct molecular skeletons. Some of the compounds were immobilised on a small molecule microarray, which was then challenged with Cy5-conjugated calmodulin. The approach allowed novel sub-micromolar ligands for calmodulin to be discovered.
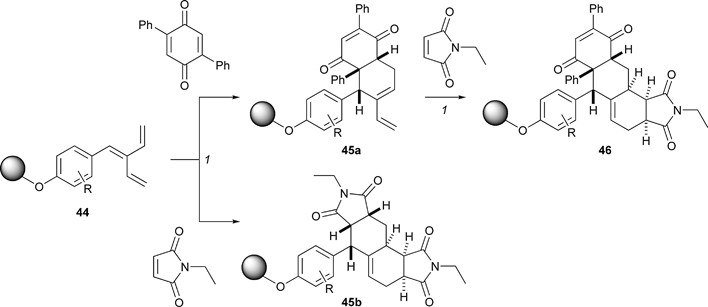 | ||
| Scheme 14 Summary of Schreiber's consecutive use of Diels–Alder reactions to construct 29400 discrete polycyclic compounds.Reagents and conditions: (1) toluene. | ||
Schreiber used the concept of ‘skeletal transformation’ to yield 4275 products with three underlying skeletons (Scheme 15).26 Each step of the synthetic route was adapted and optimised for use on macrobeads. To start with, the steroid-based epoxide 47 was opened, and then functionalised, to yield 171 distinct compounds with the scaffold 48. Methodology developed by Winterfeld was then used to sequentially transform the scaffold 48 into two alternative scaffolds, 49 and 50. First, a Diels–Alder reaction was used to introduce the variable substituent, R3, and to switch the scaffold (48 → 49); 2052 compounds with the scaffold 49 were prepared. Then, a retro-Diels–Alder reaction was used to fragment each of the compounds 49 to yield 2052 para-cyclophanes 50.
![An overview of the skeletal transformations applied by Schreiber in the preparation of >4000 skeletally diverse small molecules. Reagents and conditions: (1) amine, LiClO4 (or thiol); (2) alkylation, acylation or oxazolidone formation [for X = NR2]; (3) R3COC≡CH, Et2AlCl; (4) heat.](/image/article/2008/NP/b706296f/b706296f-s15.gif) | ||
| Scheme 15 An overview of the skeletal transformations applied by Schreiber in the preparation of >4000 skeletally diverse small molecules. Reagents and conditions: (1) amine, LiClO4 (or thiol); (2) alkylation, acylation or oxazolidone formation [for X = NR2]; (3) R3COC≡CH, Et2AlCl; (4) heat. | ||
Schreiber used the Ferrier reaction in combination with the Pauson–Khand reaction to prepare a library of polycyclic molecules (Scheme 16).27 Initially, a Ferrier reaction was used to condense a glycal (e.g.51) with a propargylic alcohol (e.g.52); the resulting enyne 53 was immobilised on a solid support and then further functionalised (→ 54). Intramolecular Pauson–Khand reactions were used to prepare fused cyclopentenones 55 to which nucleophiles could be added in a conjugate sense (not illustrated). In a pilot study, 2500 tricyclic compounds were prepared.
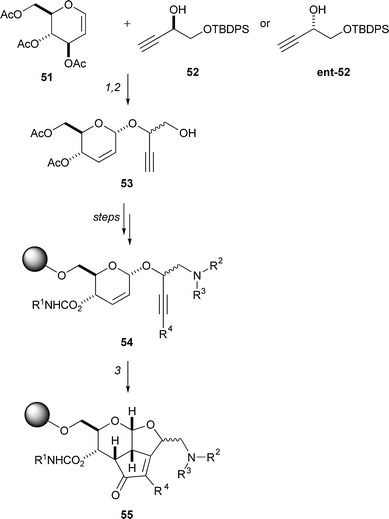 | ||
| Scheme 16 Summary of Schreiber's pairwise use of Ferrier and Pauson–Khand reactions for the synthesis of polycyclic skeletons. Reagents and conditions: (1) cat. BF3·OEt2, CH2Cl2, −78 to −20 °C, 92%; (2) TBAF, AcOH, THF; purification, 72%; (3) Co2(CO)8, CH2Cl2; NMO, CH2Cl2–CH3CN. | ||
Schreiber used three consecutive complexity-generating transformations in combination to prepare compounds of general structure 63 (Scheme 17):28 an Ugi reaction, a Diels–Alder reaction and a ring-closing metathesis. Thus, the Ugi reaction of the solid-supported amine 56, an isocyanide (e.g.59), furfural 57 and a fumaric acid derivative (e.g.58) yielded an initial adduct 60. However, the intramolecular Diels–Alder reaction of the furan and a dienophile – the fumaric acid derivative – was spontaneous, and the adduct 61 was obtained directly. Bis-allylation (→ 62), and ring-closing metathesis generated the final scaffold 63.
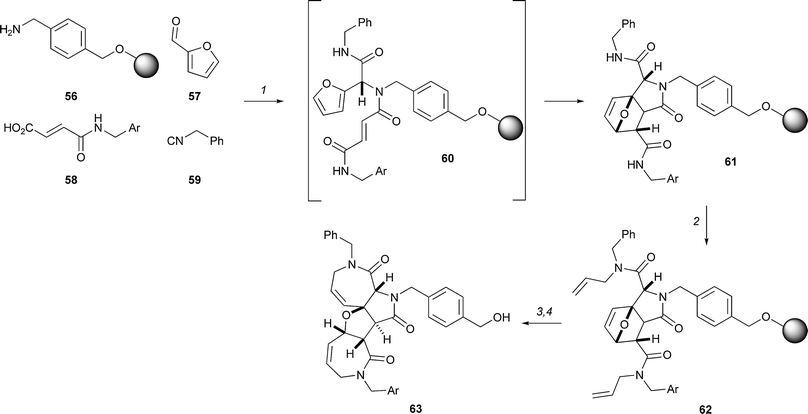 | ||
| Scheme 17 Schreiber's exploitation of the Ugi and Diels–Alder reactions and metathesis chemistry in combination. Reagents and conditions: (1) MeOH–THF, (2 : 1), 60 h; (2) KHMDS, allyl bromide; (3) Grubbs' 2nd-gen. cat., CH2Cl2, 40 °C, 60 h; (4) HF·pyridine, CH2Cl2, 2 h. Ar = m-bromophenyl. | ||
In a later adaptation of this approach, it was shown that the relative configuration of a metathesis precursor controlled the skeleton of the metathesis product (Scheme 18).29 Hence, the triene 64a underwent ring-closing metathesis to give the bridged macrocycle 65a in 87% yield. However, a diastereomeric triene 64b underwent a cascade of metathesis reactions to give the fused product 65b in 65% yield. This powerful example of a match–mismatch effect was exploited in the synthesis of a library of compounds based on the two very distinct product skeletons.
![Stereochemical control of skeletal diversity. Reagents and conditions: (1) 10 mol% Grubbs' 2nd-gen. cat., CH2Cl2, [65a: 87%; 65b: 65%].](/image/article/2008/NP/b706296f/b706296f-s18.gif) | ||
| Scheme 18 Stereochemical control of skeletal diversity. Reagents and conditions: (1) 10 mol% Grubbs' 2nd-gen. cat., CH2Cl2, [65a: 87%; 65b: 65%]. | ||
Fathi and Yang used a diversity-oriented synthetic approach to prepare a library of benzofurans, benzothiophenes and indoles (Scheme 19).30 A four-component Ugi reaction was used to prepare amides (e.g.70) which were substrates for intramolecular Diels–Alder reactions. After aromatisation, benzofurans, benzothiophenes or indoles were obtained. The approach was used to prepare 28 compounds with six distinct polycyclic frameworks.
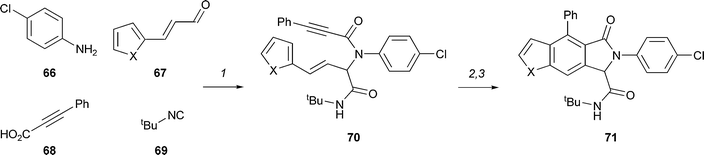 | ||
| Scheme 19 Summary of Yang's approach to the diversity-oriented synthesis of benzofurans and indoles. Reagents and conditions: (1) MeOH, 25 °C, 12 h; (2) xylene, 140 °C; (3) CH2Cl2–benzene (1 : 25), DDQ, (X = O, 87% over 3 steps; X = NMe, 61% over 3 steps). | ||
Zhu had previously used a conceptually similar approach to prepare oxa-bridged polyheterocycles 76 and pyrrolopyridines 77 (Scheme 20).31 Hence, after formation of an iminium ion (from 72 and 73), and nucleophilic addition of an isocyanide 74, the usual Ugi pathway was intercepted: the resulting nitrilium ion was attacked intramolecularly by the amide to yield oxazoles 75. The oxazoles 75 spontaneously underwent intramolecular hetero-Diels–Alder reactions to yield bridged products 76. Acid-catalysed ring-opening induced pyrrolopyridine formation (→ 77). In essence, the aromatisation step is a powerful skeletal transformation which converts one library of compounds (76) into another (77).
![Summary of Zhu's approach to the diversity-oriented synthesis of polyheterocycles and pyrrolopyridines. Reagents and conditions: (1) MeOH, rt, [R1 = Bn, R2 = CO2Et, R3 = C6H13, R4 = Bn, 92%]; (2) MeOH, TFA, −78 °C, [R1 = Bn, R2 = CO2Et, R3 = C6H13, R4 = Bn, 85%].](/image/article/2008/NP/b706296f/b706296f-s20.gif) | ||
| Scheme 20 Summary of Zhu's approach to the diversity-oriented synthesis of polyheterocycles and pyrrolopyridines. Reagents and conditions: (1) MeOH, rt, [R1 = Bn, R2 = CO2Et, R3 = C6H13, R4 = Bn, 92%]; (2) MeOH, TFA, −78 °C, [R1 = Bn, R2 = CO2Et, R3 = C6H13, R4 = Bn, 85%]. | ||
The examples described in this section are a testament to the power of exploiting combinations of complexity-generating reactions. The approach is particularly powerful when a skeletal transformation is used to convert one library of compounds into another library based on an entirely different scaffold (see Schemes 15 and 20).
2.5 Use of branching pathways to yield skeletal diversity
The ‘branching pathway’ strategy involves the conversion of common precursors into a range of distinct molecular scaffolds (Scheme 21). Ideally, it should be possible to design a range of flexible precursors which participate in complementary skeletal transformations. The development of a branching pathway may require considerable optimisation of the individual skeletal transforming steps. It is important, however, to ensure that the ethos of diversity-oriented synthesis is retained: i.e. that a library is prepared in a deliberate and simultaneous fashion. In some cases, the implementation of an intricate branching pathway, which exploits a very limited range of precursors, may cause the parallel nature of diversity-oriented synthesis to be lost.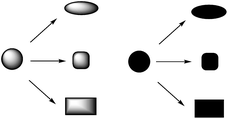 | ||
| Scheme 21 The ‘branching pathway' approach to skeletal diversity. Common precursors (circles) are converted, under different conditions, into distinct molecular skeletons. | ||
DNA-templated synthesis is emerging as a valuable tool in the implementation of branching pathways.32 By using a DNA-tagged substrate, a reaction sequence mediated by (DNA-tagged) reactants may be encoded. Remarkably, it is possible for a number of different reaction sequences to be undertaken in the same reaction vessel. Active compounds may be directly selected from the mixture of products, and identified by PCR-amplification of the DNA tag.
The branching pathway strategy has, however, largely been demonstrated using more conventional synthetic approaches. Schreiber has exploited the chemistry of boronic esters to prepare a polyketide-like library (Scheme 22).33 Trans-esterification of the boronic ester 78 with an allylic alcohol or a propargylic alcohol (→ 81), and in situ ring-closing diene or enyne metathesis, yielded the corresponding cyclic allylic boronic esters. After hydrolysis, these compounds were transformed into open-chain and cyclic hydroxylated derivatives. For example, oxidation of 82a (R1 = CH2OTIPS; R2 = Me) gave the corresponding diol 83a directly. However, 82a (R1 = CH2OTIPS; R2 = Me) could also be dihydroxylated and protected before the oxidation step to yield the protected tetraol derivative 83b. The diols 83 have polyketide-like structural features. In contrast, the dienes 82b were substrates for Diels–Alder reactions; subsequent oxidation then yielded products such as 84. Thus, a branching pathway strategy, which exploited a rather limited range of transformations, enabled skeletally diverse products to be prepared.
![Schreiber's branching pathway using boronic esters. Reagents and conditions: (1) 78 (3 eq.), Grubbs' 2nd-gen. cat. (20 mol%), CH2Cl2, reflux, then H2O; yield range: 78–92%; (2) H2O2, NaOH, THF, rt, 56% (over 2 steps) [R1 = CH2OTIPS; R2 = Me]; (3) OsO4, NMO, acetone, pH 7 buffer; then 2,2-dimethoxypropane, CH2Cl2, PPTs; then H2O2, NaOH, THF, 66% (over 2 steps) [R1 = CH2OTIPS; R2 = Me]; (4) N-benzylmaleimide (8 eq.), PhMe, 80 °C; then H2O2, NaOH, THF; carried out on solid support [typical yields for the Diels–Alder reaction of related substrates in solution: 81–91%].](/image/article/2008/NP/b706296f/b706296f-s22.gif) | ||
| Scheme 22 Schreiber's branching pathway using boronic esters. Reagents and conditions: (1) 78 (3 eq.), Grubbs' 2nd-gen. cat. (20 mol%), CH2Cl2, reflux, then H2O; yield range: 78–92%; (2) H2O2, NaOH, THF, rt, 56% (over 2 steps) [R1 = CH2OTIPS; R2 = Me]; (3) OsO4, NMO, acetone, pH 7 buffer; then 2,2-dimethoxypropane, CH2Cl2, PPTs; then H2O2, NaOH, THF, 66% (over 2 steps) [R1 = CH2OTIPS; R2 = Me]; (4) N-benzylmaleimide (8 eq.), PhMe, 80 °C; then H2O2, NaOH, THF; carried out on solid support [typical yields for the Diels–Alder reaction of related substrates in solution: 81–91%]. | ||
A branching pathway based on the chemistry of the Michael adducts 85 was developed by Porco (Scheme 23).34 Reduction of the nitro group triggered lactamisation to yield γ-lactams such as 86a. In contrast, with appropriately positioned alkenyl and alkynyl substituents, cyclisation via ring-closing metathesis or Pauson–Khand reaction was possible. With R1 = allyl and R2 = C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2OMe, enyne metathesis yielded the cyclic diene 86b. In contrast, with R1 = CCH and R2 = allyl, a Pauson–Khand reaction allowed the remarkable bridged cyclopentenone 86c to be obtained.
CCH2OMe, enyne metathesis yielded the cyclic diene 86b. In contrast, with R1 = CCH and R2 = allyl, a Pauson–Khand reaction allowed the remarkable bridged cyclopentenone 86c to be obtained.
![Porco's branching strategy from the Michael adduct 85. Reagents and conditions: (1) Zn, AcOH–THF, then Na2CO3 (aq.) [R1 = CCMe, R2 = H], 92%; (2) Grubbs' 1st-gen. cat., ethylene, MW, 150 W, 50 °C, CH2Cl2 [R1 = allyl, R2 = CCCH2OMe], ≥86%; (3) Co2(CO)8, MW, 150 W, 80 °C, CH2Cl2 [R1 = CCH, R2 = allyl], 67%.](/image/article/2008/NP/b706296f/b706296f-s23.gif) | ||
| Scheme 23 Porco's branching strategy from the Michael adduct 85. Reagents and conditions: (1) Zn, AcOH–THF, then Na2CO3 (aq.) [R1 = CCMe, R2 = H], 92%; (2) Grubbs' 1st-gen. cat., ethylene, MW, 150 W, 50 °C, CH2Cl2 [R1 = allyl, R2 = CCCH2OMe], ≥86%; (3) Co2(CO)8, MW, 150 W, 80 °C, CH2Cl2 [R1 = CCH, R2 = allyl], 67%. | ||
Schreiber has developed a ‘branching pathway’ which exploits complementary cyclisation reactions (Scheme 24).35 A four-component Petasis condensation reaction was used to assemble flexible cyclisation precursors (e.g.87). Alternative cyclisation reactions were then used to yield products with distinct molecular skeletons: Pd-catalysed cyclisation (→ 88a); enyne metathesis (→ 88b); Ru-catalysed cycloheptatriene formation (→ 88c); Au-catalysed cyclisation of the alcohol onto the alkene (→ 88d); base-induced cyclisation (→ 88e); Pauson–Khand reaction (→ 88f); and Miesenheimer [2,3]-sigmatropic rearrangement (→ 88g). Four of these cyclisation reactions could be used again to convert the enyne 88e into molecules with four further skeletons (90a–d). In addition, Diels–Alder reactions with 4-methyl-1,2,4-triazoline-3,5-dione converted the dienes 88b, 90a and 91 into the polycyclic products 89, 92 and 93. The key to this powerful synthetic approach lay in the design of precursors (e.g.87) which were effective substrates in a wide range of efficient and diastereoselective cyclisation reactions.
![Schreiber's branching pathway exploiting complementary cyclisation reactions. Reagents and conditions: (1) 10 mol% Pd(PPh3)2(OAc)2, benzene, 80 °C, [88a: single diastereomer, 81%; 90d: single diastereomer, 70%] (2) 10 mol% Hoveyda–Grubbs’ 2nd-gen. cat., CH2Cl2, reflux, [88b: trans:cis 85 : 15, 89%; 90a: trans:cis 85 : 15, 87%; 91 : trans:cis 75 : 25, 90%]; (3) 10 mol% CpRu(MeCN)3PF6, acetone, rt, [88c: single diasteromer, 85%; 90c: single diasteromer, 91%]; (4) 10 mol% NaAuCl4, MeOH, rt, single diasteromer, 80%; (5) NaH, toluene, rt, 88%; (6) Co2(CO)8, Et3NO, NH4Cl, benzene, rt, [88f: d.r. >90 : 10, 85%; 90b: single diastereomer, 85%]; (7) m-CPBA, THF, −78 → 0 °C, single diastereomer, 87%; (8) 4-methyl-1,2,4-triazoline-3,5-dione, CH2Cl2, rt [89: single diastereomer from trans diene, 72%; 92: single diastereomer, 65%; 93: 80% (combined yield from mixture of dienes)].](/image/article/2008/NP/b706296f/b706296f-s24.gif) | ||
| Scheme 24 Schreiber's branching pathway exploiting complementary cyclisation reactions. Reagents and conditions: (1) 10 mol% Pd(PPh3)2(OAc)2, benzene, 80 °C, [88a: single diastereomer, 81%; 90d: single diastereomer, 70%] (2) 10 mol% Hoveyda–Grubbs’ 2nd-gen. cat., CH2Cl2, reflux, [88b: trans:cis 85 : 15, 89%; 90a: trans:cis 85 : 15, 87%; 91 : trans:cis 75 : 25, 90%]; (3) 10 mol% CpRu(MeCN)3PF6, acetone, rt, [88c: single diasteromer, 85%; 90c: single diasteromer, 91%]; (4) 10 mol% NaAuCl4, MeOH, rt, single diasteromer, 80%; (5) NaH, toluene, rt, 88%; (6) Co2(CO)8, Et3NO, NH4Cl, benzene, rt, [88f: d.r. >90 : 10, 85%; 90b: single diastereomer, 85%]; (7) m-CPBA, THF, −78 → 0 °C, single diastereomer, 87%; (8) 4-methyl-1,2,4-triazoline-3,5-dione, CH2Cl2, rt [89: single diastereomer from trans diene, 72%; 92: single diastereomer, 65%; 93: 80% (combined yield from mixture of dienes)]. | ||
A ‘branching pathway’, developed by Martin,36 is conceptually similar (Scheme 25). Initially, a four-component reaction was used to assemble the cyclisation precursors 97. Hence, a condensation process yielded acyl iminium ions which could be intercepted by alternative nucleophiles. A range of nucleophiles could be used – Grignard reagents, organozincs and silyl enol ethers – and, thus, the diverse cyclisation precursors 97 were obtained. The functionality present in the intermediates 97 allowed them to participate in a variety of alternative cyclisation reactions. With 97a, ring-closing enyne metathesis, and cross-metathesis with styrene, were followed by intramolecular Claisen reaction to yield 98. Similarly, ring-closing olefin metathesis of 97b, and intramolecular Heck reaction, yielded the caged product 99. The fused product 100 was prepared by condensation of the aldehyde 97b with N-methylhydroxylamine and a subsequent [1,3]-dipolar cycloaddition. The molecular scaffolds accessible using this conceptually simple approach possess structural features which are reminiscent of alkaloids. As a testimony to the natural product-like structural features of Martin's library, the alkaloid (±)-roelactamine 101 was prepared from 97d using two consecutive intramolecular electrophilic aromatic substitutions.
![Martin's branching pathway for heterocycle synthesis following a multicomponent reaction. Reagents and conditions: (1) propargyl amine, AcCl, allylZnBr, CH2Cl2, 63%; (2) allyl amine, BnCOCl, allylZnBr 4 Å MS, THF, 91%; (3) allylN(TMS)2, 10% TMSOTf, CH2Cl2 then CH2CHOTBS, AcCl, 78%; (4) MeNH2, 95, THF, Δ, then 94, −78 °C, 61%; (5) Hoveyda–Grubbs' 2nd-gen. cat., CH2Cl2, styrene, then NaHMDS, 55%; (6) Grubbs' 2nd-gen. cat., CH2Cl2, then Pd[P(tBu)3]2, DIPEA, Bu4NCl, MeCN, MW, 120 °C, 65%; (7) MeNHOH·HCl, Et3N, PhMe, Δ, 87%; (8) conc. HCl–MeOH (2 : 1), 71%.](/image/article/2008/NP/b706296f/b706296f-s25.gif) | ||
| Scheme 25 Martin's branching pathway for heterocycle synthesis following a multicomponent reaction. Reagents and conditions: (1) propargyl amine, AcCl, allylZnBr, CH2Cl2, 63%; (2) allyl amine, BnCOCl, allylZnBr 4 Å MS, THF, 91%; (3) allylN(TMS)2, 10% TMSOTf, CH2Cl2 then CH2CHOTBS, AcCl, 78%; (4) MeNH2, 95, THF, Δ, then 94, −78 °C, 61%; (5) Hoveyda–Grubbs' 2nd-gen. cat., CH2Cl2, styrene, then NaHMDS, 55%; (6) Grubbs' 2nd-gen. cat., CH2Cl2, then Pd[P(tBu)3]2, DIPEA, Bu4NCl, MeCN, MW, 120 °C, 65%; (7) MeNHOH·HCl, Et3N, PhMe, Δ, 87%; (8) conc. HCl–MeOH (2 : 1), 71%. | ||
Tan has used a complex branching-pathway strategy to prepare a stereochemically diverse library of 74 compounds with polyketide-like structural motifs (Scheme 26).37 The approach relies on the judicious use of combinations of highly stereoselective reactions. Hence, the propargylic alcohols were reduced stereoselectively to yield the E- and Z-configured allylic alcohols 103. Stereoselective epoxidation of the Z allylic alcohols was possible under substrate control using m-CPBA (→ 104a); with the E allylic alcohols, high stereoselectivity was only possible using the appropriate (matched) enantiomer of Sharpless' epoxidation catalyst (→ 104b). The allylic alcohols, 103, and the epoxy alcohols, 104, could be converted into the corresponding ketones 106 and 107 using Dess–Martin periodinane. The epoxides 104 and 107 were opened regioselectively to yield the 1,3-diols 105 and the β-hydroxy ketones 108. Finally, highly 1,3syn- or 1,3anti-selective reduction of the β-hydroxy ketones 108 gave the 1,3-diols 109. According to an established method for classifying molecular scaffolds,¶,38 six distinct scaffolds are represented in the structures 103–109. The compounds exhibited the 1,3-oxygenation and the diverse stereochemistry of polyketide natural products. In addition, Tan and Ley have prepared libraries of natural-product-like acetals.39,40
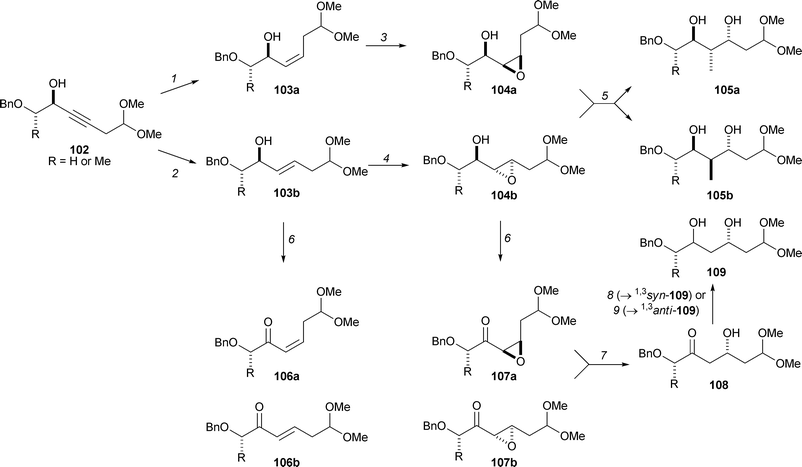 | ||
| Scheme 26 Tan's branching pathways which yield stereochemically and substitutionally diverse polyketide-like molecules. Reagents and conditions: (1) Cp2TiCl2, iBuMgBr, Et2O, 0 °C → rt, 81–86%, >88 : 12 Z:E; (2) Red-Al, Et2O, 0 °C → rt, 99%, >98 : 2 E:Z; (3) m-CPBA, NaHCO3, CH2Cl2, 0 °C → rt (R = H) or −4 °C (R = Me), 91–99%, d.r. >90 : 10; (4) diethyl tartrate (matched enantiomer), Ti(OiPr)4, tBuOOH, 4 Å sieves, CH2Cl2, −20 °C, (R = H) or −20 °C, (R = Me), 85–93%, d.r. 94 : 6; (5) (i) nBuLi, Me3Al, 1,2-dichloroethane, 0 °C→ rt; (ii) NaIO4, MeCN–H2O, 0 °C→ rt, 68–69% (over 2 steps); (6) Dess–Martin periodinane, CH2Cl2, 0 °C→ rt, 85–100%; (7) SmI2, THF, −90→ −78 °C, 65–98%; (8) NaBH(OAc)3, AcOH, MeCN, −20 °C, 87–95%, d.r. >93 : 7; (9) Et2BOMe, NaBH4. | ||
Spring41 has harnessed the diverse reactivity of diazo compounds in diversity-oriented synthesis (Scheme 27). Thus, the fluorous-tagged diazoacetate 110 was reacted with a reactive dipolarophile, DMAD, to give 111b, and, under rhodium catalysis, to yield the cyclic compounds 111a and 111c and the β-keto esters 111d–e. The cyclopropane 111a is in dynamic equilibrium (by electrocyclic ring-opening) with a cycloheptatriene. The cyclopropane was, therefore, reacted directly with DMAD to yield the Diels–Alder adduct 112a. Alternatively, methylamine was condensed with the cycloheptatriene isomer to give the alkaloid-like bicyclic compound 112b. In addition, the β-keto esters 111d-e were converted into heterocyclic compounds such as 113a and 113b. Spring's branching pathway is extremely powerful because the rich reactivity of diazo compounds allowed skeletally diverse products to be prepared by simply varying the reacting partner used. In total, a library of 223 small molecules was prepared in which 30 distinct molecular skeletons were represented.
![Spring's branching pathways strategy using the fluorous-tagged diazoacetate 110. Reagents and conditions: (1) C6H6, Rh2(O2CCF3)4, 70%; (2) dimethyl acetylenedicarboxylate [DMAD], 84% (88%); (3) PhCHO, PhNH2, then DMAD, Rh2(OAc)4, d.r. 95 : 5, 51% (80%); (4) LDA, R1COR2, then Rh2(OAc)4; 111d: 49% (90%); 111e: 68% (97%); (5) DMAD, 59%; (6) MeNH2, NaOH, then MeOH, H2SO4, 35%; (7) resorcinol, H2SO4, 74% (95%); (8) thiophene-2-carboxaldehyde, guanidine carbonate, then 3-formylchromone, 43% (98%). RF = C6F13CH2CH2–. Numbers in parentheses are purities determined by HPLC, LC-MS or 1H NMR spectroscopy.](/image/article/2008/NP/b706296f/b706296f-s27.gif) | ||
| Scheme 27 Spring's branching pathways strategy using the fluorous-tagged diazoacetate 110. Reagents and conditions: (1) C6H6, Rh2(O2CCF3)4, 70%; (2) dimethyl acetylenedicarboxylate [DMAD], 84% (88%); (3) PhCHO, PhNH2, then DMAD, Rh2(OAc)4, d.r. 95 : 5, 51% (80%); (4) LDA, R1COR2, then Rh2(OAc)4; 111d: 49% (90%); 111e: 68% (97%); (5) DMAD, 59%; (6) MeNH2, NaOH, then MeOH, H2SO4, 35%; (7) resorcinol, H2SO4, 74% (95%); (8) thiophene-2-carboxaldehyde, guanidine carbonate, then 3-formylchromone, 43% (98%). RF = C6F13CH2CH2–. Numbers in parentheses are purities determined by HPLC, LC-MS or 1H NMR spectroscopy. | ||
‘Branching pathways’ are probably the most powerful strategy devised to date for diversity-oriented synthesis. Provided that the substrates are designed carefully, many molecular skeletons can emerge through the use of a limited range of powerful transformations in combination. The key to the approach is the identification of starting materials, such as the alkynyl-substituted vinyl cyclopropanes 87 or the diazoacetates 110, which are able to participate in a wide range of complexity-generating reactions.
3 Summary and outlook
In this review, we have described some of the remarkable innovations which have been devised to address the challenges posed by diversity-oriented synthesis. We have concentrated on the diversity-oriented synthesis of compound libraries which were – in some sense – inspired by the structures of natural products.In diversity-oriented synthesis, the most challenging issue is the synthesis of skeletally diverse molecules. A range of ‘folding pathways’ have been developed in which molecules with distinct scaffolds are prepared under common reaction conditions: this approach has allowed the synthesis of libraries in which a handful of scaffolds are represented. Multicomponent reactions are extremely valuable in diversity-oriented synthesis: such reactions are particularly valuable when they can be manipulated to yield alternative scaffolds, or when they can be used in combination. In diversity-oriented synthesis, there is still a heavy reliance on the Ugi reaction to prepare substrates for other complexity-generating processes. There is, thus, still an urgent need for new multicomponent reactions,42 particularly those that yield molecules with alternative scaffolds. The ‘branching pathway’ strategy can allow libraries to be prepared in which tens of scaffolds are represented. A major challenge associated with developing branching pathways is to retain the ethos of diversity-oriented synthesis: that is, that diverse compound libraries are prepared using a limited repertoire of highly optimised reactions.
The success of using natural products to inspire diversity-oriented synthesis can ultimately only be gauged by the discovery of new biologically active molecules: not close derivatives of natural products with related functions, but molecules, which through populating productive regions of biologically-relevant chemical space, have novel biological functions. In this regard, the strategy of appending natural product scaffolds with diverse substituents has performed remarkably well, yielding chemical probes such as secramine (6), uretupamine (11), and haptamide B (14b). It is not yet possible to assess the success of the more recent innovations in diversity-oriented synthesis: in a few years' time, with the benefit of hindsight, it will be possible to assess critically the ability of these new approaches to deliver new small molecule tools for use in chemical genetic studies. It is not the structural similarity of small molecules to natural products that is ultimately important: it is discovery of valuable small molecule tools with biological functions which known natural products do not possess.
4 Acknowledgements
We thank EPSRC, the Wellcome Trust, GSK and AstraZeneca for funding, and Stuart Warriner and Stuart Leach for numerous helpful discussions.5 References
- For reviews of the chemical genetic approach, see: (a) D. P. Walsh and Y. T. Chang, Chem. Rev., 2006, 106, 2476 CrossRef CAS; (b) D. S. Bellows and M. Tyers, Science, 2004, 306, 67 CrossRef CAS; (c) T. U. Mayer, Trends Cell Biol., 2003, 13, 270 CrossRef CAS; (d) R. Breinbauer, Angew. Chem., Int. Ed., 2003, 42, 1086 CrossRef CAS; (e) B. R. Stockwell, Nat. Rev. Genet., 2000, 1, 116 CrossRef CAS; (f) S. L. Schreiber, Nat. Chem. Biol., 2005, 1, 64 CrossRef CAS; (g) D. R. Spring, Chem. Soc. Rev., 2005, 34, 472 RSC.
- (a) F. G. Kuruvilla, A. F. Shamji, S. M. Sternson, P. J. Hergenrother and S. L. Schreiber, Nature, 2002, 416, 653 CrossRef CAS; (b) A. C. Bishop, J. A. Ubersax, D. T. Petsch, D. P. Matheos, N. S. Gray, J. Blethrow, E. Shimizu, J. Z. Tsien, P. G. Schultz, M. D. Rose, J. L. Wood, D. O. Morgan and K. M. Shokat, Nature, 2000, 407, 395 CrossRef CAS.
- (a) M. Frank-Kamenetsky, X. M. Zhang, S. Bottega, O. Guicherit, H. Wichterle, H. Dudek, D. Bumcrot, F. Y. Wang, S. Jones, J. Shulok, L. L. Rubin and J. A. Porter, J. Biol., 2002, 1, 10 Search PubMed; (b) R. Weigert, A. Colanzi, A. Mironov, R. Buccione, C. Cericola, M. G. Sciulli, G. Santini, S. Flati, A. Fusella, J. G. Donaldson, M. D. Girolamo, D. Corda, M. A. De Matteis and A. Luini, J. Biol. Chem., 1997, 272, 14200 CrossRef CAS.
- (a) T. U. Mayer, T. M. Kapoor, S. J. Haggarty, R. W. King, S. L. Schreiber and T. J. Mitchison, Science, 1999, 286, 971 CrossRef CAS; (b) A. F. Straight, A. Cheung, J. Limouze, I. Chen, N. J. Westwood, J. R. Sellers and T. J. Mitchison, Science, 2003, 299, 1743 CrossRef CAS.
- (a) M. A. Koch, A. Schuffenhauer, M. Scheck, S. Wetzel, M. Casaulta, A. Odermatt, P. Ertl and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 17272 CrossRef CAS; (b) M. A. Koch, L. O. Wittenberg, S. Basu, D. A. Jeyaraj, E. Gourzoulidou, K. Reinecke, A. Odermatt and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16721 CrossRef CAS.
- For other reviews of aspects of diversity-oriented synthesis, see: (a) M. D. Burke and S. L. Schreiber, Angew. Chem., Int. Ed., 2004, 43, 46 CrossRef; (b) D. S. Tan, Nat. Chem. Biol., 2005, 1, 74 CrossRef CAS; (c) D. R. Spring, Org. Biomol. Chem., 2003, 1, 3867 RSC; (d) R. J. Spandl, A. Bender and D. R. Spring, Org. Biomol. Chem., 2008, 6, 1149 RSC.
- (a) B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPardo, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber, P. S. Anderson, R. S. L. Chang, V. J. Lotti, D. J. Cerino, T. B. Chen, P. J. Kiling and K. A. Kunkel, J. Med. Chem., 1988, 31, 2235 CrossRef CAS; (b) K. C. Nicolaou, J. A. Pfefferkorn, H. J. Mitchell, A. J. Roecker, S. Barluenga, G. Q. Cao, R. L. Affleck and J. E. Lillig, J. Am. Chem. Soc., 2000, 122, 9954 CrossRef.
- B. E. Bernstein, J. K. Tong and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13708 CrossRef CAS.
- A. Zewail, M. W. Xie, Y. Xing, L. Lin, P. F. Zang, W. Zou, J. P. Saxe and J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3345 CrossRef CAS.
- K. F. Xu, X. Shen, H. Li, G. Pacheco-Rodriguez, J. Moss and M. Vaughan, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2784 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feenby, Adv. Drug Delivery Res., 1997, 23, 3 Search PubMed.
- (a) K. Lu, M. Huang, Z. Xiang, Y. Liu, J. Chen and Z. Yang, Org. Lett., 2006, 8, 1193 CrossRef CAS; (b) A. B. Smith III, S. P. Walsh, M. Frohn and M. O. Duffey, Org. Lett., 2005, 7, 139 CrossRef.
- H. E. Pelish, N. J. Westwood, Y. Feng, T. Kirchhausen and M. D. Shair, J. Am. Chem. Soc., 2001, 123, 6740 CrossRef CAS.
- S. M. Sternson, J. B. Louca, J. C. Wong and S. L. Schreiber, J. Am. Chem. Soc., 2001, 123, 1740 CrossRef CAS.
- (a) H. E. Blackwell, L. Pérez, R. A. Stavenger, J. A. Tallarico, E. C. Eatough, M. A. Foley and S. L. Schreiber, Chem. Biol., 2001, 8, 1167 CrossRef CAS; (b) P. A. Clemons, A. N. Koehler, B. K. Wagner, T. G. Sprigings, D. R. Spring, R. W. King, S. L. Schreiber and M. A. Foley, Chem. Biol., 2001, 8, 1183 CrossRef CAS.
- A. N. Koehler, A. F. Shamji and S. L. Schreiber, J. Am. Chem. Soc., 2003, 125, 8420 CrossRef CAS.
- D. R. Spring, S. Krishnan, H. E. Blackwell and S. L. Schreiber, J. Am. Chem. Soc., 2002, 124, 1354 CrossRef CAS.
- M. D. Burke, E. M. Berger and S. L. Schreiber, J. Am. Chem. Soc., 2004, 126, 14095 CrossRef CAS.
- H. Oguri and S. L. Schreiber, Org. Lett., 2005, 7, 47 CrossRef CAS.
- S. Dandapani, M. Duduta, J. S. Panek and J. A. Porco, Jr, Org. Lett., 2007, 9, 3849 CrossRef CAS.
- D. A. Spiegel, F. C. Schroeder, J. R. Duvall and S. L. Schreiber, J. Am. Chem. Soc., 2006, 128, 14766 CrossRef CAS.
- T. Yang, L. Campbell and D. J. Dixon, J. Am. Chem. Soc., 2007, 129, 12070 CrossRef CAS.
- O. Jiménez, G. de la Rosa and R. Lavilla, Angew. Chem., Int. Ed., 2005, 44, 6521 CrossRef CAS.
- (a) W. J. Bromley, M. Gibson, S. Lang, S. A. Raw, A. C. Whitwood and R. J. K. Taylor, Tetrahedron, 2007, 63, 6004 CrossRef CAS; (b) Y. Fernández Sainz, S. A. Raw and R. J. K. Taylor, J. Org. Chem., 2005, 70, 10086 CrossRef; (c) S. A. Raw and R. J. K. Taylor, Chem. Commun., 2004, 508 RSC; (d) S. A. Raw and R. J. K. Taylor, Tetrahedron Lett., 2004, 45, 8607 CrossRef CAS; (e) S. A. Raw and R. J. K. Taylor, J. Am. Chem. Soc., 2004, 126, 12260 CrossRef CAS.
- O. Kwon, S. Bum Park and S. L. Schreiber, J. Am. Chem. Soc., 2002, 124, 13402 CrossRef CAS.
- N. Kumar, M. Kiuchi, J. A. Tallarico and S. L. Schreiber, Org. Lett., 2005, 7, 2535 CrossRef CAS.
- H. Kubota, J. Lim, K. M. Depew and S. L. Schreiber, Chem. Biol., 2002, 9, 265 CrossRef CAS.
- D. Lee, J. K. Sello and S. L. Schreiber, Org. Lett., 2000, 2, 709 CrossRef CAS.
- J. K. Sello, P. R. Andreana, D. Lee and S. L. Schreiber, Org. Lett., 2003, 5, 4125 CrossRef CAS.
- K. Lu, T. Luo, Z. Xiang, Z. You, R. Fathi, J. Chen and Z. Yang, J. Comb. Chem., 2005, 7, 958 CrossRef CAS.
- R. Gámez-Montano, E. González-Zamora, P. Potier and J. Zhu, Tetrahedron, 2002, 58, 6351 CrossRef CAS.
- C. T. Calderone and D. R. Liu, Angew. Chem., Int. Ed., 2005, 44, 7383 CrossRef CAS.
- G. C. Micalizio and S. L. Schreiber, Angew. Chem., Int. Ed., 2002, 41, 152 CrossRef CAS.
- E. Comer, E. Rohan, L. Deng and J. A. Porco, Org. Lett., 2007, 9, 2123 CrossRef CAS.
- N. Kumagai, G. Muncipinto and S. L. Schreiber, Angew. Chem., Int. Ed., 2006, 45, 3635 CrossRef CAS.
- J. D. Sunderhaus, C. Dockendorff and S. F. Martin, Org. Lett., 2007, 9, 4223 CrossRef CAS.
- S. Shang, H. Iwadare, D. E. Macks, L. M. Ambrosini and D. S. Tan, Org. Lett., 2007, 9, 1895 CrossRef CAS.
- A. Schuffenhauer, P. Ertl, S. Roggo, S. Wetzel, M. A. Koch and H. Waldmann, J. Chem. Inf. Model., 2007, 47, 47 Search PubMed.
- (a) J. S. Potuzak, S. B. Moilanen and D. S. Tan, J. Am. Chem. Soc., 2005, 127, 13796 CrossRef CAS; (b) S. B. Moilanen, J. S. Potuzak and D. S. Tan, J. Am. Chem. Soc., 2006, 128, 1792 CrossRef CAS.
- L.-G. Milroy, G. Zinzalla, G. Prencipe, P. Michel, S. V. Ley, M. Gunaratnam, N. Beltran and S. Neidle, Angew. Chem., Int. Ed., 2007, 46, 2493 CrossRef CAS.
- E. E. Wyatt, S. Fergus, W. R. J. D. Galloway, A. Bender, D. J. Fox, A. T. Plowright, A. S. Jessiman, M. Welch and D. R. Spring, Chem. Commun., 2006, 3296 RSC.
- For example, see: B. B. Touré, H. R. Hoveyda, J. Tailor, A. Ulaczyk-Lesanko and D. G. Hall, Chem. Eur. J., 2003, 9, 466 Search PubMed.
Footnotes |
| † Substitutional diversity has also been described as ‘appendage diversity’. |
| ‡ The forward chemical genetic approach involves the discovery of chemical probes through their phenotypic effect. In contrast, the reverse chemical genetic approach involves the discovery of chemical probes which modulate a predetermined macromolecular target. |
| § In the context of this review, a multi-component reaction is simply considered to involve the assembly of two or more components. |
| ¶ In this classification scheme, scaffolds with different unsaturation patterns are classified separately. This classification reflects the important influence of sp2-hybridised atoms on conformation (ref. 38). |
| This journal is © The Royal Society of Chemistry 2008 |