“Naked” fluoride binding sites for physisorptive hydrogen storage
Abbie
Trewin
a,
George R.
Darling
*ab and
Andrew I.
Cooper
*a
aDepartment of Chemistry and Centre for Materials Discovery, University of Liverpool, Crown Street, Liverpool, UK L69 3BX. E-mail: aicooper@liv.ac.uk; Fax: +44 (0)151 7942304; Tel: +44 (0)151 7943548
bSurface Science Research Centre, University of Liverpool, Liverpool, UK L69 3BX. E-mail: darling@liv.ac.uk; Fax: +44 (0)151 7943870; Tel: +44 (0)151 7943888
First published on 21st November 2007
Abstract
Charge separated ammonium fluorides are calculated to have enhanced binding affinities with molecular hydrogen; a model is proposed for a hypothetical porous polymer where “naked” fluoride moieties are site isolated and available for H2physisorption .
The widespread use of hydrogen as a fuel is limited by the lack of a convenient method of H2 storage.1 Molecular H2 storage by physisorption is appealing because it is reversible, cyclable, and sorbents exist that are tolerant to minor impurities such as water. Many porous sorbents have been investigated including carbon,2 metal–organic frameworks (MOFs) and zeolites,3 and organic polymers.4 A significant drawback to H2physisorption , however, is the low temperature required and the associated thermodynamic and system weight implications. To date, most H2physisorption experiments have been conducted at 77.3 K because of the low average isosteric heat of sorption (4–7 kJ mol−1) of H2 with most porous materials.5 Hence, there is a need to consider new H2 sorbents with substantially higher average isosteric heats which might store H2 at more practicable temperatures—for example, metallation of porous frameworks has been suggested and attempted.6,7
Molecular H2 can interact with a porous substrate via weak dispersive interactions, electrostatic interactions, orbital interactions, or in non-classical dihydrogen complexes.8–10 Dispersive forces dominate in substrates such as carbon2 and porous organic polymers.4 Electrostatic interactions between H2 and charged sites are typically stronger; for example, a recent report refers to a maximum isosteric heat range of sorption (at low H2 coverage) of 10.1 kJ mol−1 for exposed Mn2+ coordination sites in a microporous MOF.11 Lochan and Head-Gordon8 have calculated much higher H2 binding affinities in the broad range 10–350 kJ mol−1 for charged metals (e.g., Li+, Al3+), ligands (e.g., SO42−, F−), and metal complexes (e.g., M[(CO)x]y+). Other studies have calculated the interaction energies for F−–(H2)n (n = 1–8) anion complexes using quantum chemical calculations at the MP2/aug-cc-pVTZ level.12 It is, however, impossible to achieve isolated “bare” ions such as Al3+ or F− in real physical materials.8
Our broad aim is to design and evaluate synthetically-viable high-energy “binding sites” for hydrogen physisorption which can be incorporated into real materials. In this study we have drawn on the concept of “naked” fluoride ions, as used quite widely as strong nucleophiles and bases in organic synthesis.13 Truly naked anions, not interacting with any other chemical species, will exist only in the gas phase. However, small “naked” anions such as fluoride can be prepared and used in chemical reactions by pairing F− with a larger and delocalized cation (e.g., tetraalkylammonium, phosphazenes ) such that the coulombic interaction between cation and anion is greatly decreased.13 In principle, such species, if “site isolated”, could also act as highly polarizing, charged binding sites for H2physisorption .
Ab initio calculations employing the GAMESS-UK code14 were used to investigate the binding interactions between molecular H2 and a range of simple molecules with increasing levels of localized charge density. All systems were geometry optimized at the MP2 level. First, we calculated the binding energy between H2 and benzene using a range of different basis sets (Table 1).
| Organic molecule | −Binding energy/kJ mol−1 | Distance/Åb | |||
|---|---|---|---|---|---|
| TZVP 6-311G* | DZP | cc-pV5Z + DZP | L–Hc | H–H | |
| a Calculated interaction energies of H2 with benzene for basis sets TZVP-dTZVP, 6-311G* and TZVPP were 3.10, 3.25 and 3.86 kJ mol−1, respectively. b Bond lengths calculated using TZVP 6-311G*. c Shortest distance between H2 molecule and binding species. | |||||
| Benzene a | 3.24 (1.71) | 2.90 (1.38) | — | 3.202 | 0.739 |
| Et2NH | 7.42 (2.25) | 5.74 (1.98) | — | 2.418 | 0.743 |
| Et3N | 7.21 (2.60) | 9.45 (5.35) | — | 2.572 | 0.743 |
| LiF | 9.03 (0.77) | 6.66 (3.60) | — | 2.539 | 0.741 |
| Et4NF | 18.16 (6.58) | 14.49 (6.51) | 13.08 (9.23) | 2.257 | 0.746 |
| Et4NF·H2O | 10.71 (0.71) | 10.04 (1.78) | 8.27 (2.49) | 1.972 | 0.746 |
| F− | 83.65 (51.31) | 49.95 (28.75) | 23.58 (18.37) | 1.050 | 1.179 |
We obtained a maximum benzene–H2 binding energy (for TZVPP) of 3.86 kJ mol−1, in agreement with a previous study,15 but somewhat lower than the 4.5–5 kJ mol−1 reported by others.8,16 At the other extreme of charge localization, we computed binding energies for a range of basis sets (with BSSE-corrected values found using the Boys–Bernardi scheme17 in parentheses) for H2 to a fluoride ion.18 The largest basis set employed was a combination of cc-pV5Z for the H2 molecule and the F− ion to which it bonds, with the remainder of the molecule represented by the DZP basis set. In this mixed basis, the binding energy of H2 to F− is in the range 18.37 to 23.58 kJ mol−1, which compares favourably with other literature values (24.1 kJ mol−1,12a 26.1 kJ mol−112b).
Table 1 shows a general increase in H2 binding energy for molecules with more localized charge density for all basis sets used. Significantly higher binding energies were calculated for simple aliphatic amines (Et2NH and Et3N) compared to benzene. The H2 molecule interacted preferentially with the lone pair of the nitrogen atom in both cases. The binding energy of H2 with the fluoride in tetraethylammonium fluoride (TEAF) was found to be the highest of all the molecules we have considered—in the range of 9–13 kJ mol−1 for the largest basis set. This is significantly higher than energies calculated for a close ion-pair such as Li+F−. A similar trend was recently reported for the binding energies of H2 and Li+ in which the binding energy of H2 to Li+ was twice that of H2 to the close ion-pair Li+OH−.19 Binding of H2 to TEAF led to notable shortening of the L–H bond distance (with respect to benzene) and lengthening of the H–H bond.
Binding energies of the order of 9–13 kJ mol−1 would facilitate physisorption at unprecedented temperatures, if reflected in an average isosteric heat of sorption.20 One practical difficulty is that tetraalkylammonium fluorides are often unstable in the anhydrous form due to Hofmann elimination reactions, unless prepared under special conditions.21Tetraethylammonium fluoride monohydrate (TEAF·H2O), by contrast, is readily prepared by heating higher hydrates of TEAF at 65 °C under vacuum. The binding energy of H2 with TEAF·H2O was calculated to be 10.04 kJ mol−1 and 10.71 kJ mol−1 using the DZP and TZVP 6-311G* basis sets, respectively, and 8.27 kJ mol−1 using cc-pV5Z + DZP. The BSSE corrected values are somewhat lower; however, it has been noted that for similar systems the BSSE over-corrects for relatively small basis sets,22,23 and that the uncorrected results for the DZP level are fortuitously accurate due to a cancellation of errors. More detailed calculations are required but we contend that the binding energy for the TEAF monohydrate is significant in comparison with the other molecules studied here. The reduction in H2 binding energy for the monohydrate with respect to anhydrous TEAF is consistent with increased delocalization of the negative fluoride charge upon introduction of the hydrogen-bonded water molecule. The close proximity of the water molecule (the distance between the bonding water hydrogen and H2 is 2.85 Å) perturbs the bonding interaction between the H2 and the F−, resulting in a shortening of the bond H2–F− distance in comparison to anhydrous TEAF.
Table 1 shows that the qualitative trends in binding energy are broadly similar for TZVP 6-311G*, DZP, and cc-pV5Z + DZP basis sets, allowing us to explore the qualitative variance in binding energies for larger assemblies using the less computationally expensive DZP basis set. The incremental binding energies for seven successive additions of H2 to TEAF were calculated (Table 2).
Table 2 shows that it is energetically favorable to bind up to six H2 molecules to the hypothetical anhydrous TEAF species. This suggests that a suitably isolated TEAF moiety might physisorb up to six H2 molecules at low temperatures (e.g., 77.3 K). The successive BE depends strongly on the number of dihydrogens coordinated, as found for models of isolated F−.12a Even allowing for the significant overestimation of the H2 binding energy using DZP, comparison with the larger basis sets (Tables 1 and 2) suggests that the first three (or possibly more) H2 molecules could bind strongly enough to display persistent physisorption up to higher temperatures (Fig. 1).20
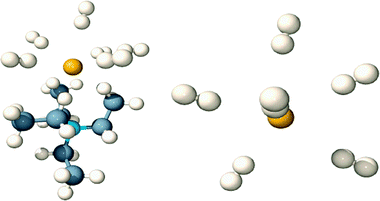 | ||
| Fig. 1 Left: Side view of molecular model showing six H2 molecules binding to Et4N+F−. Right: Top view showing distribution of H2 molecules around the fluoride anion (tetraethylammonium cation removed for clarity). | ||
The calculations above refer to isolated TEAF molecules. Clearly, TEAF and its hydrate are non-porous solids and the scope for interaction with H2 is limited. Even if one could increase the surface area of TEAF—for example, by the production of very small nanoparticles—the “naked” F− character discussed above would tend to be dissipated by multiple intermolecular cation–anion interactions which occur in the extended solid. Likewise, TEAF or its hydrate could in principle be supported on a high surface area porous substrate—such as activated carbon2—but this may also lead to TEAF–TEAF interactions, especially at significant TEAF loadings, or substantial interactions between the fluoride ion and the porous support. In all of these cases one might expect the strength of these idealized TEAF–H2 interactions to be greatly diminished, even if the coordination sphere around the fluoride ion remains somewhat accessible to H2. It is possible however to conceive of extended porous solids where the fluoride anion is fixed in space in such a way that intermolecular self-interactions are minimized and that the coordination sphere of the anion is left partially vacant to facilitate H2 coordination. A hypothetical extended structure consisting of a biphenyl-linked tetrabiphenylammonium fluoride subunit is shown in Scheme 1.
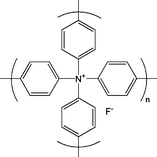 | ||
| Scheme 1 Repeat unit structure for hypothetical biphenyl-linked tetrabiphenylammonium fluoride network. | ||
To assess the viability of a structure composed of such units, a model was constructed using the Materials Studio Modeling 4.0 package (Accelrys Inc., San Diego, CA, 2005) and fully optimized (both cell parameters and the atomic positions) using the density functional theory code VASP24 (with the standard ultrasoft pseudopotentials) treating exchange and correlation with the PW91 generalized gradient approximation,25 using a 450 eV cutoff for the plane wave basis set sampled at the Γ-point of the Brillouin zone (Fig. 2).
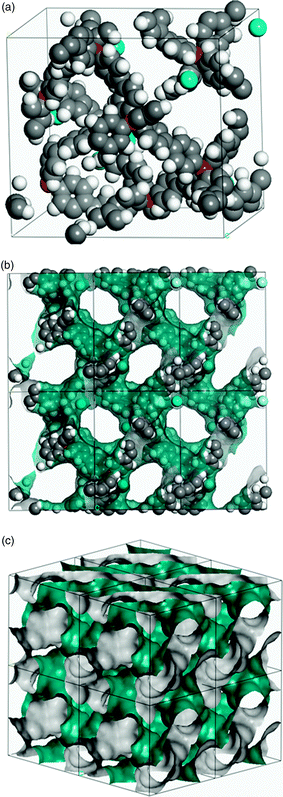 | ||
| Fig. 2 Molecular simulation of biphenyl-linked tetrabiphenylammonium fluoride (a) unit cell with dimensions 2.2 × 2.2 × 2.4 nm, nitrogen atoms are shown as red spheres, and fluorine as blue. (b) 3 × 2 lattice with a Connolly surface shown in green/grey. (c) A 2 × 2 × 2 lattice showing only the Connolly surface, omitting atoms for clarity. | ||
After minimization, this hypothetical structure has a relatively low calculated bulk density of 0.40 g cm−3. The micropore structure in the model is fully interconnected and the network has a distorted diamondoid structure. The fluoride ions are situated adjacent to micropore cavities; as such, each “naked” fluoride has a substantial vacant coordination space. The fluoride ions are well separated with an average F−–F− distance of 11.03 Å. This separation is a function of the specific polymer structure and will not necessarily be realized in other porous materials incorporating fluorine.3b The calculated surface area for this modelled structure was 4464 m2 g−1 with a calculated pore volume of 1.89 cm3 g−1. These values were obtained by creating a Connolly surface using the Atom, Volumes and Surface module in Materials Studio using a coarse grid resolution and a Connolly radius set to 1.82 Å (the kinetic radius of N2).4d While this is a hypothetical structure and we have no synthetic route to this hypothetical tetraphenyl-onium polymer, real materials do exist that have micropore properties similar to those calculated here. For example, IRMOF-20 was reported to have a Langmuir surface area of 4346 m2 g−1 (calculated Connolly surface area = 3275 m2 g−1) and a measured pore volume of 1.53 cm3 g−1.26 We are currently exploring the synthetic potential of amorphous conjugated microporous polymers as substrates to incorporate “site isolated” fluoride groups for gas sorption applications.27 We also believe that this general strategy might be applicable to other microporous materials such as MOFs3 and polymers of intrinsic microporosity.4a
Acknowledgements
We thank the EPSRC (EP/C511794/1) for financial support and Matt Rosseinsky (Liverpool), Dave Adams (Unilever, Colworth) and Jens Thomas (CFS, CCLRC, Daresbury Laboratory) for helpful discussions.References
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef.
- Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673 CrossRef CAS.
- See for example: (a) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS and references therein; (b) G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS; (c) A. Zecchina, S. Bordiga, J. G. Vitillo, G. Ricchiardi, C. Lamberti, G. Spoto, M. Bjorgen and K. P. Lillerud, J. Am. Chem. Soc., 2005, 127, 6361 CrossRef CAS.
- (a) N. B. McKeown, B. Gahnem, K. J. Msayib, P. M. Budd, C. E. Tattershall, K. Mahmood, S. Tan, D. Book, H. W. Langmi and A. Walton, Angew. Chem., Int. Ed., 2006, 45, 1804 CrossRef; (b) J. Y. Lee, C. D. Wood, D. Bradshaw, M. J. Rosseinsky and A. I. Cooper, Chem. Commun., 2006, 2670 RSC; (c) J. Germain, J. Hradil, J. M. J. Fréchet and F. Svec, Chem. Mater., 2006, 18, 4430 CrossRef CAS; (d) C. D. Wood, B. Tan, A. Trewin, H. Niu, D. Bradshaw, M. J. Rosseinsky, Y. Z. Khimyak, N. L. Campbell, R. Kirk, E. Stöckel and A. I. Cooper, Chem. Mater., 2007, 19, 2034 CrossRef CAS.
- S. K. Bhatia and A. L. Myers, Langmuir, 2006, 22, 1688 CrossRef CAS.
- S. S. Han and W. A. Goddard, J. Am. Chem. Soc., 2007, 129, 8422 CrossRef CAS.
- K. L. Mulfort and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 9604 CrossRef CAS.
- R. C. Lochan and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 1357 RSC.
- L. Gagliardi and P. Pyykko, J. Am. Chem. Soc., 2004, 126, 15014 CrossRef CAS.
- J. Niu, B. K. Rao and P. Jena, Phys. Rev. Lett., 1992, 68, 2277 CrossRef CAS.
- M. Dincă, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS.
- (a) B. Nyulasi and A. Kovács, Chem. Phys. Lett., 2006, 426, 26 CrossRef CAS; (b) J. G. Vitillo, A. Damin, A. Zecchina and G. Ricchiardi, J. Chem. Phys., 2006, 124, 12 , Art. No. 224308.
- (a) K. O. Christe and H. D. B. Jenkins, J. Am. Chem. Soc., 2003, 125, 9457 CrossRef CAS; (b) T. Borrmann, E. Lork, M. Mews and W. D. Stohrer, J. Fluorine Chem., 2004, 125, 903 CrossRef CAS.
- M. F. Guest, I. J. Bush, H. J. J. Van Dam, P. Sherwood, J. M. H. Thomas, J. H. Van Lenthe, R. W. A. Havenith and J. Kendrick, Mol. Phys., 2005, 103, 719 CrossRef CAS.
- S. Patchkosvkii, J. S. Tse, S. N. Yurchenko, L. Zhechkov, T. Heine and G. Seifert, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10439 CrossRef CAS.
- O. Hubner, A. Gloss, M. Fichtner and W. Klopper, J. Phys. Chem. A, 2004, 108, 3019 CrossRef.
- S. B. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553.
- B. Hartke and H.-J. Werner, Chem. Phys. Lett., 1997, 280, 430 CrossRef CAS.
- O. Hubner and W. Klopper, J. Phys. Chem. A, 2007, 111, 2426 CrossRef.
- Following the method of Bhatia and Myers (ref. 5), the optimal H2 delivery temperature, Topt, for a real isosteric heat of −13.08 kJ mol−1 would be 258.1 K (P1 = 30 bar, P2 = 1.5 bar; ΔS° = −8R). In most real materials, however, high-energy binding sites would contribute to a lower average isosteric heat of sorption.
- (a) H. R. Sun and S. G. DiMagno, J. Am. Chem. Soc., 2005, 127, 2050 CrossRef CAS; (b) H. R. Sun and S. G. DiMagno, Chem. Commun., 2007, 528 RSC.
- C. J. Cramer, Essentials of Computational Chemistry, Theories and Models, Wiley, West Sussex, UK, 2nd edn, 2004 Search PubMed.
- M. Masamura, Theor. Chem. Acc., 2001, 106, 301 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B, 1993, 48, 13115 CrossRef CAS.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B, 1992, 46, 6671 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 1304 CrossRef CAS.
- (a) J.-X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky, Y. Z. Khimyak and A. I. Cooper, Angew. Chem., Int. Ed., 2007, 46, 8574 CrossRef; (b) J.-X. Jiang, F. Su, H. Niu, C. D. Wood, N. L. Campbell, Y. Z. Khimyak and A. I. Cooper, Chem. Commun., 2007 10.1039/b715563h.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |