DOI:
10.1039/B801394B
(Paper)
Mol. BioSyst., 2008,
4, 746-753
A photo-electroactive surface strategy for immobilizing ligands in patterns and gradients for studies of cell polarization†‡
Received
25th January 2008
, Accepted 8th May 2008
First published on 20th May 2008
Abstract
We report a combined photochemical and electrochemical method to pattern ligands and cells in complex geometries and gradients on inert surfaces. This work demonstrates: (1) the control of density of immobilized ligands within overlapping photopatterns, and (2) the attached cell culture patterned onto ligand defined gradients for studies of directional cell polarity. Our approach is based on the photochemical activation of benzoquinonealkanethiols. Immobilization of aminooxy terminated ligands in selected region of the quinone monolayer resulted in patterns on the surface. This approach is unique in that the extent of photochemical deprotection, as well as ligand immobilization can be monitored and quantified by cyclic voltammetry in situ. Furthermore, complex photochemical patterns of single or multiple ligands can be routinely generated using photolithographic masks. Finally, this methodology is completely compatible with attached cell culture and we show how the subtle interplay between cell–cell interactions and underlying peptide gradient influences cell polarization. The combined use of photochemistry, electrochemistry and well defined surface chemistry provides molecular level control of patterned ligands and gradients on surfaces.
Introduction
The ability to spatially immobilize and organize biomolecules on solid supports is important for a wide variety of applications ranging from the development of biochips for high throughput assays to the generation of model substrates for studies of cell behavior.1,2 Most of the current methods to pattern ligands on materials rely on fabrication techniques including photo-patterning, laminar flow, and soft lithography.3–9 While these strategies can routinely immobilize ligands on surfaces in various geometries, the precise control of ligand density and the ability to immobilize multiple ligands remains difficult. In order to extend these methods to generate model substrates for detailed mechanistic studies of cell adhesion and cell migration, cell culture compatible surfaces that can present multiple ligands at defined densities in patterns and in gradients are necessary but remain technically challenging. The key criteria to generate model substrates for studies and applications in cell biology are: (1) a sophisticated surface chemistry reaction to immobilize multiple ligands with the ability to precisely modulate the density of each immobilized ligand within patterns and gradients. (2) Compatibility with cell culture conditions where the only interaction between attached cell and material is a ligand–receptor interaction. (3) A robust fabrication technique to generate complex patterns and gradients. Herein we report a new general strategy that combines surface chemistry and photochemistry to present biologically active ligands with excellent control of ligand density in patterns and gradients on electroactive self-assembled monolayers. We also show single cell polarity on small gradient patterns is directed by cell–cell interactions and the underlying peptide presenting gradient surface.
This strategy is based on the photochemical unveiling of the electroactive benzoquinone terminated alkanethiolates. Subsequent chemo-selective immobilization of aminooxy tethered ligands in selected regions of the quinone monolayer results in patterned ligands and gradients on the surface. To present a surface that can be simultaneously photochemically activated and characterized by electrochemistry we synthesized a nitroveratryloxycarbonyl (NVOC) protected hydroquinoneethylene glycol terminated alkanethiol. Previous work has shown that monolayers presenting NVOC hydroquinone groups can undergo photochemical deprotection upon UV illumination to reveal the hydroquinone.10 Subsequent electrochemical oxidation of the hydroquinone reveals the reactive quinone. The quinone form of the monolayer can then react with soluble aminooxy tagged ligands to form a stable oxime conjugate via chemoselective ligation (Fig. 1). We have shown this immobilization reaction to be chemoselective, kinetically well behaved and stable in cell culture conditions.11–14 This photochemical strategy possesses several unique features. (1) The NVOC protected hydroquinone is completely redox inactive and only upon UV illumination does the resulting monolayer become redox active, which then permits the use of cyclic voltammetry to quantitatively monitor the extent of photodeprotection. (2) The newly formed quinone oxime conjugate has a diagnostic peak determined by cyclic voltammetry that is critical for quantifying the yield and therefore the density of immobilized ligands on the surface. (3) Complex patterns of immobilized ligands can be easily generated by using patterned photolithographic masks and by using multiple masks in series (Fig. 2).
 |
| | Fig. 1 Mixed monolayers presenting NVOC protected hydroquinone and tetra(ethyleneglycol) groups are illuminated with ultraviolet light (365 nm). Photochemical deprotection of the NVOC group reveals the hydroquinone. Subsequent oxidation of the hydroquinone results in the corresponding quinine which can then undergo chemoselective ligation with aminooxy terminated ligands. The reversible redox activities of the hydroquinone and the oxime adduct permit the use of cyclic voltammetry to characterize both the extent of photo deprotection and oxime formation on the surface in situ. Represents rhodamine, alexafluor 488, and the peptide GRGDS. | |
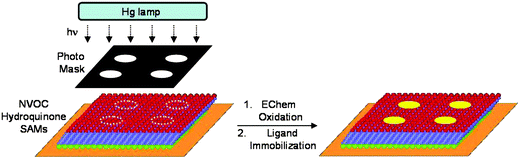 |
| | Fig. 2 A photochemical strategy for generating patterns and gradients of immobilized ligands onto an electroactive monolayer. UV illumination of the NVOC protected hydroquinone through a photomask reveals the hydroquinone in select regions on the monolayer. Electrochemical oxidation of the hydroquinone monolayer to the quinine permits selective immobilization of ligands to the patterned surface. | |
Results and discussion
We used cyclic voltammetry to characterize the rate of photochemical deprotection of the NVOC protected hydroquinone monolayer.15Fig. 3A shows cyclic voltammograms in 1 M HClO4 (50 mVs−1, Ag/AgCl reference) for the photodeprotection of the NVOC hydroquinone monolayer. The mono-protected NVOC hydroquinone monolayer is not redox active when a potential is applied to the surface. However, upon illumination of the surface with UV light an increase in the peak currents is observed which indicates the extent of photodeprotection of the NVOC groups to reveal the corresponding electroactive hydroquinone groups. Fig. 3B shows a plot of the peak currents versus time for the photodeprotection shown in Fig. 3A. The data were fit to an integrated first order rate law equation where ΓHQ is the surface density of hydroquinone, Γ0PHQ is the initial surface density of the protected hydroquinone, k is the rate constant for deprotection, and t is total time of illumination in minutes. The fit was excellent and gave a first order rate constant of 0.11 min−1.
 |
| | Fig. 3 (A) The extent of photochemical deprotection of the NVOC hydroquinone monolayer is monitored by cyclic voltammetry. Cyclic voltammograms were recorded in 8 min intervals during UV illumination of the monolayer. An increase in the peak currents indicates the extent of photo deprotection of the NVOC groups to reveal the corresponding redox active hydroquinone monolayer. (B) A plot of the normalized peak currents versus time for the photo deprotection shown in A. The data were fit to an integrated first order rate law to give a rate constant of 0.11 min−1. | |
We next demonstrate the utility of this strategy to generate complex patterns of immobilized ligands where the ligand density on the monolayer surface can be precisely controlled. For this study, we prepared a mixed monolayer presenting NVOC hydroquinone and tetra(ethylene glycol) groups (1 : 1). A photo mask consisting of vertical solid lines was placed directly in contact with the monolayer. UV illumination of the monolayer through the photo mask for 10 min reveals the hydroquinone in select regions on the surface. We expected approximately 50% of the NVOC hydroquinone monolayers to be deprotected within the pattern (based on the half-life of photodeprotection). The substrate was then electrochemically oxidized at 750 mV for 10 s and treated with soluble rhodamine oxyamine (50 mM in MeOH, 2 h). A second photo mask consisting of horizontal gradient lines was then placed in direct contact with the same substrate for subsequent UV illumination for an additional 10 min. The substrate was then again electrochemically oxidized and treated with soluble rhodamine oxyamine. Direct visualization of the monolayer by fluorescent microscopy was prohibited due to quenching of the gold film.16 In order to visualize the surface we transferred the monolayer to a transparent medium where we could then characterize the photopatterning by fluorescent microscopy.17Fig. 4A shows a fluorescent image with overlapping patterns consisting of solid and gradient lines of surface immobilized rhodamine–oxyamine. The intensity profile for the line drawn across the image in Fig. 4A shows the relative fluorescence of the patterns on the surface (Fig. 4B). The fluorescent intensity for the region where the vertical and horizontal lines overlap is approximately double in magnitude to that of the nonoverlapped patterns, which is consistent with the conditions used for photodeprotection and immobilization. This result confirms that aminooxy ligands react only to photoactivated benzoquinone groups and in high yield. More importantly, the density of immobilized ligands within the pattern is directly proportional to the density of photoactivated benzoquinone and subsequent reaction of rhodamine–oxyamine on the surface. We also show that it is straight forward to extend this methodology for the preparation of patterned substrates with multiple ligands. Fig. 4C shows a fluorescent image for the photopatterning of two immobilized fluorescent ligands (Alexafluor 488 and rhodamine oxyamine) in dumbbell gradients on the same substrate.18
 |
| | Fig. 4 (A) A fluorescent image of a surface that was serially photo deprotected and reacted with rhodamine oxyamine. The first photo deprotection and immobilization was of two rectangles and the second was of two gradients. The resulting image is over lapping patterns that consist of solid and gradient lines of surface immobilized rhodamine. (B) An intensity profile that describes the relative fluorescence for the line drawn across the image shown in A. (C) A fluorescent image for the photo patterning of two immobilized ligands (Alexafluor 488 and rhodamine oxyamine) in dumb bell gradients on the same substrate. The fluorescent images for the two fluorophores were taken separately, and then superimposed to show their relative positions with each other on the surface. | |
In order to extend this methodology for studies of cell adhesion and cell migration, we immobilized adhesive peptide ligands in patterns and gradients on a surface that is otherwise inert to nonspecific protein adsorption.19 We used solid phase peptide synthesis to generate an RGD-oxyamine ligand.11 The RGD peptide is found in the extracellular matrixprotein fibronectin and is known to facilitate cell adhesion through cell surface integrin receptors.20 We prepared a mixed monolayer containing 1% NVOC protected hydroquinone groups and 99% tetra(ethylene glycol) groups for the photopatterning of cells. The background ethylene glycol groups are critical for the surface to resist nonspecific protein adsorption.21After UV illumination through a photo mask with various patterns for 20 min, soluble aminooxy tagged RGD peptide (20 mM in PBS for four hours) was installed onto the surface. Swiss 3T3 Fibroblasts were then added to the resulting substrate for 12 h. Cells attached and became confluent within the patterns. To show the patterned cell attachment was biospecific and only mediated by the immobilized RGD ligands and not due to photo-damage of the monolayer, soluble RGD was added to the media (1 mM for 1 h) and all the adhered cells detached. Fig. 5 shows representative fluorescent images of fixed cells on various RGD immobilized photo (pattern geometries and gradients.22 The cells are completely confined within the patterns demonstrating that the use of UV light and electrochemistry to unveil and oxidize the reactive quinone groups which then undergo chemoselective reaction with RGD oxymine groups leads to specific attachment. Analysis of focal adhesions within the cells by anti-paxillin antibody (a protein found in focal adhesions) showed more and larger focal adhesions at the higher RGD ligand density region of the gradient (Fig. 5G).
 |
| | Fig. 5 (A–F) Representative fluorescent images of Swiss 3T3 Fibroblasts on various photo patterns of RGD ligands. Patterned cells were stained for actin (red), microtubules (green), and nuclei (blue). (G) Fluorescent images of immobilized ligands and cells on a gradient. A phase contrast image shows a hexagonal photomask with a gradient used in the preparation of the photo patterning of rhodamine and RGD peptide ligands (top). A fluorescent image shows the photo patterning of rhodamine on a gradient (middle). A fluorescent image of patterned cells on a gradient of immobilized RGD ligands (bottom). Cells were stained for paxillan, actin, and nuclei to show the formation of focal adhesions (greenspots) on RGD ligands presented on molecularly defined gradient surfaces. | |
We next extend this gradient methodology to study single cell polarization and the effects of cell–cell interactions on polarization. The ability for a cell to polarize and therefore generate asymmetry within itself due to external factors is critical for a range of biological processes.23 The role of the underlying adhesive environment and cell–cell interactions are critical for establishing cell polarity but are not well understood due to a lack of studies on molecularly well-defined surfaces. In order to study the underlying surface chemistry requirements for cell polarity, we confined cells to small patterns and gradients where the cells attach and become polarized due to the combination of cell–cell interactions and cell-peptide immobilized gradient but are not able to migrate. The most conclusive method to determine cell polarity is to measure the vector between the cell nucleus, concentrated Golgi and the centrosome (Fig. 6).24
 |
| | Fig. 6 Polarity vector defining the distance and direction between nucleus center, centrosome center and Golgi center. The vector between the concentrated Golgi with respect to the nucleus center and centrosome was used to define internal polarity on RGD-peptide patterns and gradients. Fluorescent images of the cells stained for centrosome (red) nuclei (blue) and Golgi apparatus (green). | |
The polarity of a cell can be experimentally observed and measured through the systematic reorientation and alignment of these organelles, which can be visualized using fluorescent dyes to map the direction of polarity. Giantin, a protein found in the membrane of the Golgi apparatus cisternae was chosen as a marker for that organelle.25,26 As can be seen in the series of fluorescent micrographs in Fig. 7, the adherent cells adopt a morphology in which the nucleus is located approximately in the center of the cell. By determining the alignment vector of the Golgi apparatus relative to the nucleus and centrosome, we found no consistent directional polarity on the non-gradient square patterns for many cells (n = 24, Fig. 7A). In fact, almost all cells had a diffuse Golgi around the nucleus, a strong indicator of no polarity. Interestingly, by the simple addition of another cell within the pattern, both cells sense each other and generate polarity opposite to each other (Fig. 7B and C). By seeding cells on a gradient pattern, the cells attach only to the higher density region but the polarity is still determined by the cell–cell interactions and not the underlying peptide gradient. Clearly, the interplay between cell–cell interactions and the surface presentation of ligands influences cell polarity. In order to separate the influences of cell–cell interactions, pattern edge effects and the underlying gradient on cell polarity we generated very large RGD peptide gradient patterns (Fig. 8). Cells were seeded on the large gradients at low density in order to reduce cell–cell interactions (Fig. 8D). By analyzing the individual cells’ polarity vectors it became clear that the cells polarized overall to the higher density region of the gradient, independent of their position on the gradient. When the cells were allowed to proliferate and become contact inhibited on the large gradient, analysis of the cells showed no directional polarity due to the many cell–cell interactions.
 |
| | Fig. 7 Phase contrast and fluorescent images of polarized cells on RGD immobilized patterns and gradients. (A) A single cell on a symmetric square pattern has no net polarity. (B) Introduction of a second cell to the symmetric square pattern causes the cells to interact and to polarize in opposite directions from each other. (C) Two cells on a circular pattern sense each other and become polarized in opposite directions. (D) A gradient pattern of RGD peptide provides adhesion to multiple cells. The cells attach to the higher density region but their polarity is directed by cell–cell interactions. The cells each polarize away from each other. Fluorescent images of the cells stained for actin (red) nuclei (blue) and Golgi apparatus (green). | |
 |
| | Fig. 8 Determining directional cell polarity on a RGD peptide gradient without edge or cell–cell interactions. (A) Micrograph of the microfiche gradient pattern. (B) Fluorescent image of a rhodamine–oxyamine immobilized gradient. (C) Confluent cells attached to the RGD peptide gradient. Determining single cell polarity is complicated by the numerous cell–cell interactions. (D) Low cell seeding density on the gradient allows single cells to adhere and polarize without edge or cell–cell interactions. Analysis of the fluorescent staining for polarity vector shows the overall net polarity vector for the cells is towards the higher density region of the gradient. Scale bar represents 600 μm.‡ | |
In order to determine the peptide density for cell attachment and polarity along the gradient, we used an image analyzing software (ImageJ) to first obtain a density profile of the gradient based on the pixel intensity of the fluorescent gradient. Fig. 9A shows fluorescent micrographs of rhodamine immobilized ligands generated from the gradient photomasks. Fig. 9B shows the fluorescent images of attached cell culture on RGD immobilized gradients generated by the photo-mask shown in 9A. Fig. 9C shows a plot for the relative density with respect to the distance along the gradient generated from ImageJ. To extrapolate the density along the gradient from this plot, we assume that the maximum peptide density is 2.7 × 104 molecules μm−2 based on 1% NVOC hydroquinone monolayer that has been completely modified to the peptide ligand on the surface. The ligand density at 1% NVOC hydroquinone was determined based on integration of the area under the oxidative or reductive waves corresponding to the hydroquinone monolayer after the photo-deprotection of monolayer presenting only the NVOC hydroquinone groups. The value obtained was 4.5 × 10−20 mol μm2. By aligning the density plot with the attached cells on the large gradient, we determine precisely the ligand density for supporting cell adhesion along the gradient. For high cell seeding, cells attach, proliferate and migrate but are able to alter the minimum RGD density required for supporting cell adhesion that is dependent on the slope of the gradient. Higher ligand density is required to support cell attachment on steep slopes and lower ligand density on more shallow gradients. Clearly, the slope and density of the ligands modulate the ability of the cells to attach to the surface and proliferate. Interestingly, our data show that individual cells polarize consistently towards the higher density of the gradient irrespective of the slope of the gradient in the absence of cell–cell interactions upon low cell seeding. This result suggests that cell polarity is dependent on the slope of surface-bound ligands. Further studies are being pursued to determine the role of slope versus ligand density on cell polarity and cell migration rate with these gradient surfaces.
 |
| | Fig. 9 Determining the role of slope and density for cell adhesion and cell polarization on a RGD peptide gradient. (A) Fluorescent image of a rhodamine oxyamine immobilized gradient. (B) At high cell seeding density, cell attachment and survival is dependent upon the slope and density of RGD peptides. (C) A plot of the slope was generated from the data in (A) by ImageJ software and shows how the greater the slope the higher the ligand density required for adhesion (D represents distance in μm and Γ represents ligand density. For low cell seeding on these gradients, individual cells in the absence of cell–cell interactions consistently polarize to the higher density region of the gradient. | |
These are the first results to show the underlying gradient directly determines cell polarization using the acceptable polarity measure of the vector between the nucleus, Golgi and centrosome. Furthermore, we showed that focal adhesionkinase deficient fibroblast cells (FAK−/−) were unable to polarize on the large gradient surfaces due to a severe defect in forming focal adhesions, which are crucial for relaying information from the adhesive surface to the polarity signaling machinery within cells. This combined photochemical and electroactive surface strategy may be used to determine single cell polarity on a variety of ligand immobilized surfaces and how cell geometry may influence the requirements for adhesion and migration on molecularly defined gradients.
Conclusion
We have developed a photochemical strategy to present immobilized ligands in patterns and gradients on photoactivated electroactive SAMs surfaces. We have used this methodology to demonstrate control of ligand density on complex patterned surfaces, and cell attachment on molecularly well-defined gradients. This methodology allows for the immobilization of multiple ligands in patterns and gradients onto a surface and is compatible with cell culture. We also show how this strategy may be used to study the factors that influence and regulate cell polarity. The methodology presented here can be used with various cell lines and various ligands to study the interplay of gradient and ligand composition on adhesion, migration and cell–cell communication.27,28 The synthetic flexibility of the strategy also allows for the straightforward immobilization of a variety of biomolecules, different affinity ligands and small molecules that contain the oxyamine group. The method can be used with high-throughput microarray technologies, microelectrode arrays,12 microfluidics and several surface spectroscopy techniques for massively parallel assays to generate novel biosensors for biotechnological applications. The gradient surfaces are designed at the molecular level and therefore provide exquisite control of the density and presentation of ligands for numerous cell based studies and assays. This methodology combined with live cell fluorescence microscopy,29 nano-patterned surfaces30 and dynamic substrates31 will provide the generation of dynamic gradient surfaces where the spatial and temporal control of cell migration may allow for internal and external insights into the subcellular nanoarchitecture that governs cell movement and cell–cell communication.
Experimental section
All the solvents for the synthesis were HPLC grade. THF was distilled from sodium benzophenone under nitrogen before use. Absolute ethanol was purchased from Aaper Alcohol Chemical Company. Flash chromatography was carried out using silica gel (230–400 mesh). All amino acids and resin were purchased from Anaspec, Inc. (La Jolla, CA). All reagents were purchased from Aldrich and used as received. All reagents used in cell culture were obtained from Gibco BRL. 3T3-Swiss albino fibroblasts and FAK−/− mouse embryonic fibroblasts (MEF) were purchased from ATCC.
Synthesis of alkanethiolates
NVOC protected hydroquinone tetra(ethylene glycol) alkanethiol, tetra(ethylene glycol) alkanethiol, and rhodamine oxyamine were prepared as previously described.10,11
Solid-phase peptide synthesis
All peptides were synthesized by automated solid phase peptide synthesis using the CS136XT Peptide Synthesizer (CS Bio Co., Menlo Park, CA). Fmoc (9-fluorenylmethoxycarbonyl)-protected amino acids were used on Fmoc-Ser(tBu)-Rink Amide-MBHA resin. Synthesized peptide was cleaved from the resin by agitating in a solution of trifluoroacetic acid (TFA) : water : triisopropylsilane (95 : 2.5 : 2.5) for 3 h. TFA was evaporated and the cleaved peptide was precipitated in cold diethyl ether. The water-soluble peptide was extracted with water and lyophilized. Mass spectral data confirmed the peptide product. MS (ESI) (m/z): [M+H+] calculated for linear RGD-oxyamine (C25H45N11O11), 676.69; found, 676.5. [M+H+] calculated for control scrambled peptide, GRD-oxyamine (C25H45N11O11), 676.69; found, 676.4. [M+H+] calculated for control soluble peptide RGD (C17H31N9O8), 490.48; found, 490.3.
Microscopy of surface immobilized dyes
Scotch tape (3M) was adhered to the monolayer. The resulting substrate was then cured at 85 °C for 20 min. The tape was peeled from the substrate, resulting in transfer of the monolayer from the gold substrate to the tape.
Microscopy of attached cell culture
Adherent cells were fixed in 3.7% paraformaldehyde in phosphate buffer saline (PBS) for ten minutes and then permeabilized with 0.1% Triton X in PBS (PBST) for ten minutes. Cells were then stained with anti-tubulin (1 : 1000) in PBS containing 10% goat serum for one hour, followed by Alexa 488-conjugated goat anti-mouse IgG (1 : 100 in PBST), phalloidin-tetramethylrhodamine B isothiocyanate (1 : 50 in PBS), and DAPI (1 : 300 in PBST) for one hour. Substrates were rinsed with deionized water before being mounted onto glass cover slips for microscopy. For single cell polarity studies on surface gradients, a combination of fluorescent dyes was used to visualize the fibroblasts: DAPI (4′,6-diamidino-2-phenylindole dihydrochloride for the nucleus, Sigma, St. Louis, MO), phalloidin-tetramethylrhodamine B isothiocyanate (Sigma, St. Louis, MO) for F-actin cytoskeleton, anti-giantin (Covance Research Products, Berkeley, CA) with a fluorescent tagged secondary antibody (fluorescein conjugated goat anti-rabbit IgG, Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) targeting the Golgi apparatus, and mouse monoclonal anti-gamma tubulin (Sigma) to track centrosome position. All optical and fluorescent micrographs were imaged using a Nikon inverted microscope (model TE2000–E). All images were captured and processed by MetaMorph.
Preparation of monolayers
All gold substrates were prepared by electron-beam deposition of titanium (5 nm) and then gold (15 nm) on glass cover slips (75 mm × 25 mm). All gold coated glass substrates were cut into 1 cm2 pieces and washed with absolute ethanol. The substrates were immersed in an ethanolic solution containing the alkanethiolates (1 mM) for 12 h, and then cleaned with ethanol prior to each experiment.
Electrochemical measurements
All electrochemical experiments were performed using a Bioanalytical Systems CV–100 W potentiostat. Electrochemical oxidation of the monolayer was performed by applying an oxidative potential at 750 mV for 10 s in 1M HClO4, using a platinum wire as the counter electrode, Ag/AgCl as reference, and the gold/SAM substrate as the working electrode.
Fabrication of photomasks
The photopatterns were designed and drawn in PowerPoint. The patterns were then reduced 25 times and printed onto microfiches.
Photochemical deprotection of substrates
A substrate presenting NVOC protected hydroquinone and tetra(ethylene glycol) groups (1 : 1) was illuminated with ultraviolet light (100 W Hg lamp, Nikon) filtered through a band-pass filter (365 nm) for 30 minutes to ensure complete deprotection of the NVOC groups.
Photopatterning of peptide ligands
UV illumination of a substrate presenting NVOC protected hydroquinone and tetra(ethylene glycol) groups (1:99) through a photomask for 30 minutes removed the NVOC groups. The substrate was then oxidized electrochemically at 750 mV for 10 s to convert the hydroquinone to the quinone. A RGD oxyamine solution (50 mM in PBS) was added to the substrate for four hours to ensure complete immobilization of the peptide ligands. The substrate was then clean with water and dried before using for cell culture.
Cell culture
Swiss 3T3 fibroblasts and FAK−/− Mouse Embryonic Fibroblasts (MEFs) were cultured in Dulbecco’s Modified Eagle Medium (Gibco) supplemented with 10% calf bovine serum and penicillin/streptomycin. Cells were removed with a solution of 0.05% trypsin/0.53 mM EDTA, resuspended in serum-free culture medium (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells/mL), and plated onto the SAM substrates. After 2 hours, the substrates were placed in serum containing media and maintained at 37 °C in a humidified 5% CO2 atmosphere.
000 cells/mL), and plated onto the SAM substrates. After 2 hours, the substrates were placed in serum containing media and maintained at 37 °C in a humidified 5% CO2 atmosphere.
Acknowledgements
This work was supported by the Carolina Center for Cancer Nanotechnology Excellence and grants from the NIH and the Burroughs Wellcome Foundation (Interface Career Award). We thank Professors Keith Burridge, Rudy Juliano, James Bear and Klaus Hahn for their comments and suggestions.
References
- T. H. Park and M. L. Shuler, Biotechnol. Prog., 2003, 19, 243–253 CrossRef CAS.
- S. Panda, T. K. Sato, G. M. Hampton and J. B. Hogenesch, Trends Cell Biol., 2003, 13, 151–156 CrossRef CAS.
- M. S. Hahn, L. J. Taite, J. J. Moon, M. C. Rowland, K. A. I. Ruffino and J. L. West, Biomaterials, 2006, 27, 2519–2905 CrossRef CAS.
- G. T. Carroll, D. N. Wang, N. J. Turro and J. T. Koberstein, Langmuir, 2006, 22, 2899–2905 CrossRef CAS.
- D. Ryan, B. A. Parviz, V. Linder, V. Semetey, S. K. Sia, J. Su, M. Mrksich and G. M. Whitesides, Langmuir, 2004, 20, 9080–9088 CrossRef CAS.
- R. Michel, J. W. Lussi, G. Csucs, I. Reviakine, G. Danuser, B. Ketterer, J. A. Hubbell, M. Textor and N. D. Spencer, Langmuir, 2002, 18, 3281–3287 CrossRef CAS.
- C. B. Herbert, T. L. McLernon, C. L. Hypolite, D. N. Adams, L. Pikus, C. C. Huang, G. B. Fields, P. C. Letourneau, M. D. Distefano and W.-S. Hu, Chem. Biol., 1997, 4, 731–737 CrossRef CAS.
- X. Y. Jiang, Q. B. Xu, S. K. W. Dertinger, A. D Stroock, T. M. Fu and G. M. Whitesides, Anal. Chem., 2005, 77, 2338–2347 CrossRef CAS.
- M. N. Yousaf, E. W. L. Chan and M. Mrksich, Angew. Chem., Int. Ed., 2000, 30, 1943–1946 CrossRef.
- W. S. Dillmore, M. N. Yousaf and M. Mrksich, Langmuir, 2004, 20, 7223–7231 CrossRef CAS.
- E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2006, 128, 15542–15546 CrossRef CAS.
- E. W. L. Chan and M. N. Yousaf, ChemPhysChem, 2007, 8, 1469–1472 CrossRef CAS.
- D. K. Hoover, E.-J. Lee, E. W. L. Chan and M. N. Yousaf, ChemBioChem, 2007, 8, 1920–1923 CrossRef CAS.
- N. P. Westcott and M. N. Yousaf, Langmuir, 2008, 24, 2261–2265 CrossRef CAS.
- Substrates were illuminated with ultraviolet light (100 W Hg lamp, Nikon) filtered through a band-pass filter (365 nm).
- D. H. Waldeck, A. P. Alivisatos and C. B. Harris, Surf. Sci., 1985, 158, 103–125.
- Scotch tape (3M) was adhered to the monolayer. The resulting substrate was then cured at 85 °C for 20 min. The tape was peeled from the substrate, resulting in transfer of the monolayer from the gold substrate to the tape.
- Mixed monolayers presenting NVOC protected hydroquinone and tetra(ethylene glycol) groups (1 : 1) were exposed to UV light through a mask containing vertical dumbbell patterns for 10 min. Electrochemical oxidation, followed by Rhodamine immobilization (50 mM in MeOH, 2 h) resulted in the first ligand immobilized photo pattern. The substrate was rinsed with methanol and water, and then resubjected to UV illumination through a photomask with horizontal dumbbell patterns. Subsequent electrochemical oxidation and immobilization of Alexafluor 488 oxyamine (Molecular Probes, 50 mM in H2O, 2 h) gave the second photo pattern.
-
M. N. Yousaf and M. Mrksich, in New Technologies for Life Sciences: A Trends Guide, ed. E. Wilson, Elsevier, 1999, pp. 28–35 Search PubMed.
- E. Ruoslahti, Annu. Rev. Cell Dev. Biol., 1996, 12, 697–748 CrossRef CAS.
- K. L. Prime and G. M. Whitesides, Science, 1993, 252, 1164–1167.
- Patterned cells were fixed with 3.7% paraformaldehyde in PBS for 10 min, washed with PBS twice, permeablized with 0.1% Trinton X in PBS for 30 min before staining was proceeded.
- S. K. Shastri and K. Burridge, Exp. Cell Res., 2000, 261, 25–36 CrossRef CAS.
- W. J. Nelson, Nature, 2003, 422, 766–774 CrossRef CAS.
- M. Thery, V. Racine, M. Piel, A. Pepin, A. Dimirov, Y. Chen, J. Sibarita and M. Bornens, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19771–19776 CrossRef CAS.
- A. D. Linstedt and H.-P. Hauri, Mol. Biol. Cell, 1993, 4, 679–693 CAS.
- D. G. Barrett and M. N. Yousaf, Angew. Chem., Int. Ed., 2007, 39, 7581–7583 CrossRef.
- E. W. L. Chan and M. N. Yousaf, Angew. Chem., Int. Ed., 2007, 46, 3881–3884 CrossRef CAS.
- L. Hodgson, E. W. L. Chan, K. M. Hahn and M. N. Yousaf, J. Am. Chem. Soc., 2007, 129, 9264–9265 CrossRef CAS.
- D. K. Hoover, E. W. L. Chan and M. N. Yousaf, J. Am. Chem. Soc., 2008, 130, 3280–3281 CrossRef CAS.
- D. G. Barrett and M. N. Yousaf, ChemBioChem, 2008, 9, 62–66 CrossRef CAS.
Footnotes |
| † This article was originally submitted for the Molecular BioSystems ‘Emerging Investigators’ issue highlighting the work of outstanding young scientists at the chemical- and systems-biology interfaces. For more papers from this issue please see the table of contents of Issue 6, 2008 at http://www.molecularbiosystems.org/ei. |
| ‡ Electronic supplementary information (ESI) available: Cyclic voltammograms detailing the photo-deprotection and subsequent immobilization of ligands to the electroactive surface. See DOI: 10.1039/b801394b |
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.