Engineering stochasticity in gene expression†
Jeffrey J.
Tabor‡
a,
Travis S.
Bayer
b,
Zachary B.
Simpson
a,
Matthew
Levy
a and
Andrew D.
Ellington
*ac
aCenter for Systems and Synthetic Biology and Institute for Cell and Molecular Biology, University of Texas at Austin, Austin. E-mail: andy.ellington@mail.utexas.edu
bDivision of Biology, California Institute of Technology, Pasadena, California 91125
cDepartment of Chemistry and Biochemistry, University of Texas at Austin, Austin, TX 78712
First published on 1st May 2008
Abstract
Stochastic fluctuations (noise) in gene expression can cause members of otherwise genetically identical populations to display drastically different phenotypes. An understanding of the sources of noise and the strategies cells employ to function reliably despite noise is proving to be increasingly important in describing the behavior of natural organisms and will be essential for the engineering of synthetic biological systems. Here we describe the design of synthetic constructs, termed ribosome competing RNAs (rcRNAs), as a means to rationally perturb noise in cellular gene expression. We find that noise in gene expression increases in a manner proportional to the ability of an rcRNA to compete for the cellular ribosome pool. We then demonstrate that operons significantly buffer noise between coexpressed genes in a natural cellular background and can even reduce the level of rcRNA enhanced noise. These results demonstrate that synthetic genetic constructs can significantly affect the noise profile of a living cell and, importantly, that operons are a facile genetic strategy for buffering against noise.
Introduction
Gene expression is inherently stochastic, due primarily to the small numbers of molecules involved in the process.1 Noise intrinsic to gene expression is thought to be dictated by fluctuations in mRNA levels, which may arise from random transitions in promoter states2–6 followed by bursts of transcription7 or the random births and deaths of mRNAs themselves.8–11Messenger RNA fluctuations are then amplified by random mRNA–ribosome interactions and concomitant bursts in protein production.8,12–16 As a consequence, the concatenation of multiple genes into a single expression unit (operon formation) has been predicted to decrease uncorrelated fluctuations in the levels of protein products.11 Noise has also been shown to result from fluctuations in factors extrinsic to the genes themselves. These factors are thought to include pathway specific modulators2,3,17–19 and global factors of gene expression such as the levels of nucleic acid polymerases or ribosomes.18,20–23Synthetic biology, or the (re)construction of gene networks with defined performance characteristics, has proven a useful paradigm for studying the principles which govern cellular function.24–26 Several synthetic studies have demonstrated that noise can drive identical gene regulatory networks to encode significant variation across cell populations.6,27–32 It is now apparent that the study and construction of genetic regulatory circuits will require strategies for understanding and controlling intracellular noise. To this end, we constructed ribosome-competing RNA constructs (rcRNAs) as a synthetic biological tool to study the impact of synthetic genetic elements on noise in living cells, and to begin to develop methods for engineering noise buffering.
Results and discussion
Ribosome competition increases noise in gene expression
To examine how the expression of exogenous genes might affect noise, we engineered a series of small mRNAs whose predicted function was to compete for ribosomes in E. coli (Fig. 1a). These RNAs, which we term ribosome competing RNAs (rcRNAs), contained hairpin stems of varying stabilities that occluded a ribosome binding site (RBS) to different degrees (Fig. 1b). If rcRNAs were expressed to high levels in cells, they would presumably compete for cellular ribosome pools based on the extent of exposure of the RBS. Natural and synthetic strategies for RBS occlusion have previously been shown to be efficient methods for blocking ribosome association and subsequent translation as well.33–36 | ||
| Fig. 1 rcRNA strategy for ribosome competition. (a) Cellular mRNAs (blue) associate with ribosomes probabilistically at a rate dependent upon the concentration of the mRNA and ribosomes in that cell. (Top) “Wild-type” gene expression scenario. (Bottom) An exogenous rcRNA competes with bulk cellular mRNAs for translational machinery. A reduction in the number of available ribosomes results in a decreased probability of a given cellular mRNA associating with a ribosome. (b) A series of rcRNAs based on rc1 (shown) were engineered to contain ribosome binding sites (green) of increasingly single-stranded nature. Mutations of 2, 3, 4 or 6 mismatches were made in the antisense stem (bracketed) such that the region in and around the RBS became destabilized. (c) In-line probing of the rcRNA structure. Each rcRNA molecule was transcribed in vitro. The 5′-most nucleotide was then radioactively labeled with a phosphate group containing a 32P atom and the RNAs were incubated under conditions which promote spontaneous hydrolysis of the phosphodiester backbone.37 Nucleotide residues which tend to be single-stranded undergo hydrolysis significantly faster than nucleotides involved in a base pair. Hydrolysis results in two truncated RNAs, a labeled 5′-fragment and the remaining 3′ fragment. The RNA population is then separated on a polyacrylamide gel matrix and runs as a pattern of degradation products of different sizes, based on the position of the hydrolysis event relative to the 5′-end of the RNA. Nucleotide residues which are more single stranded show up as more intense bands while residues which are more double stranded show less intense bands. Equivalent counts of 32P containing RNA were added to each lane of a 10% denaturing polyacrylamide gel. Schematic of the rcRNA transcript is shown at right. (d) In vitro translation competition assays. A fixed amount of gfp DNA template was added to a coupled in vitro transcription–translation reaction along with increasing molar ratios of rcRNA DNA templates. The unpaired rcRNA, rc6, is represented as grey bars while rc1 is represented as white bars. Data are normalized to a gfp only control. Error bars represent 95% confidence intervals derived from 3 experiments run in parallel. | ||
To examine whether rcRNAs could directly compete for translation in a manner dependent on the availability of the RBS, we performed two in vitro assays. First, the predicted secondary structures of rcRNAs were verified by limited hydrolysis of single-stranded regions (in-line probing).37 As predicted, an rcRNA variant engineered to have a perfectly base-paired RBS/anti-RBS stem (rc1), showed the greatest double-strandedness in both regions (Fig. 1c). Thus, rc1 was predicted to have minimal capacity for translation competition amongst the rcRNA series. As bulges were designed into the anti-RBS stem in different rcRNA variants, both the RBS and anti-RBS regions became less structured, rationally allowing for increased ribosome binding (Fig. 1c).
Second, to directly assay the functionality of the rcRNAs, we performed translation competition assays in a reconstituted E. coli lysate (Experimental). A fixed amount of reporter gene (gfp) DNA template was added to a coupled in vitro transcription–translation reaction and increasing amounts of DNA templates for either a highly structured (rc1) or a largely unstructured (rc6) rcRNA were added to the reaction. Interestingly, addition of either rcRNA template at low levels resulted in an increase in GFP protein abundance. This effect is likely due to stabilization of the gfpmRNAtranscript through RNAse competition. The rc1 template continued to increase GFP abundance when added at a 5 : 1 ratio relative to the gfp template, but resulted in no further increase in GFP production when added at a 10 : 1 ratio. In sharp contrast, the addition of increasing amounts of the relatively unstructured rc6 template resulted in a strong inhibition of translational capacity (Fig. 1d). These results demonstrated that rcRNAs are capable of reducing translation rate in a manner proportional to the availability of the RBS sequence within the engineered helix.
To measure the effect of ribosome competition on noise, we then followed the expression of a gfp gene in individual E. coli cells using flow cytometry. Individual rcRNA constructs that could compete for ribosomes to different extents were introduced into E. coli, and rcRNA expression was driven to high levels by T7 RNA polymerase (see Experimental). Noise was quantified as the standard deviation in GFP abundance divided by the mean over the cell populations, a metric also known as the coefficient of variation (CV). As predicted, the availability of the rcRNA RBS showed a strong positive correlation with noise, and a strong negative correlation with GFP abundance (Fig. 2a, Table 1). It is worthy to note that though the availability of the RBS from the in-line probing experiments had a largely predictable effect on in vivo expression data, the correlation between the two experiments was not perfect. For example, though rc1 appears to have a less available RBS than rc2 and rc3 (Fig. 1c), rc1 expression results in lower GFP abundance and a slightly higher noise profile than does the expression of rc2 and rc3 (Table 1). The RBS of rc4, however, was shown to be the most available in the in-line assays, and rc4 indeed resulted in the lowest GFP abundance and the greatest amount of noise in vivo. As was the case with the in vitro translation reactions above, expression of the highly structured rcRNAs (rc1–3) generally led to increased GFP abundance, while expression of the mostly unstructured rcRNAs (rc4, 5 and 6) decreased GFP.
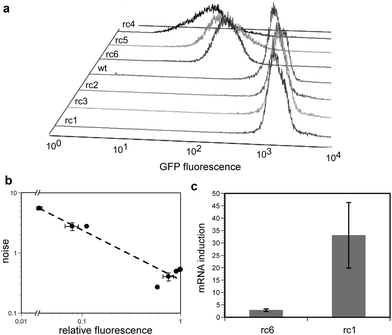 | ||
| Fig. 2 The effect of ribosome competing RNAs (rcRNAs) on gene expression. (a) Flow cytometric histograms of E. coli populations expressing GFP as well a single rcRNA variant. rcRNA variants expressed (near to far): rc1, rc3, rc2, wild-type (uninduced cells carrying the plasmid for rc1), rc6, rc5, rc4. (b) relative GFP fluorescence versus noise (standard deviation divided by mean protein abundance) from the flow cytometry data in panel ‘a’. The data were fit to an equation of the form y = axb where the value of b is −0.83 (dashed line). Three cultures of each sample were grown in parallel and assayed under the conditions described in the Experimental. Error bars represent 95% confidence intervals. (c) Relative abundance of gfpmRNA in E. coli treated with 200 ng mL−1 anhydrotetracycline (aTc) as compared to cells treated with no inducer in the presence of a high affinity (rc6) or low affinity (rc1) rcRNA. RNA was prepared and quantitated from three cultures grown in parallel under the conditions described in Experimental. Error bars represent 95% confidence intervals. | ||
| Construct | Designed bulges | Relative GFP (×10−1) | Noise (×10−1) |
|---|---|---|---|
| Wild-type | — | 5.8 ± 0.1 | 2.7 ± 0.1 |
| rc1 | 0 | 10 ± 0.1 | 5.3 ± 0.04 |
| rc2 | 2 | 7.6 ± 0.9 | 4.1 ± 0.7 |
| rc3 | 4 | 9.1 ± 0.1 | 4.9 ± 0.1 |
| rc4 | 4 | 0.4 ± 0.1 | 55 ± 3.5 |
| rc5 | 6 | 0.8 ± 0.1 | 28 ± 4.2 |
| rc6 | 6 | 1.2 ± 0.007 | 28 ± 0.2 |
The in vivo rcRNA expression data showed that noise and GFP abundance obeyed an inverse power law relationship (Fig. 2b), a scaling previously observed in studies in which the rate of transcription was modulated.17,18 Given that mRNAs are known to compete for cellular ribosome pools38 and that highly expressed mRNAs are capable of sequestering nearly all cellular ribosomes,39 one interpretation of these results is that rcRNA-induced noise results from probabilistic mRNA–ribosome interactions which become infrequent as ribosomes become rare. It is very important to note, however, that there are many downstream effects resulting from the initial competition for ribosomes which likely contribute to the noise profiles observed in our experiments. For example, competition for ribosome pools would reduce the expression level of all cellular proteins , including RNA polymerases. A reduction in RNA polymerase abundance should itself increase total noise in gene expression, due to a reduction in transcription rate. Though it has previously been shown that noise scales inversely with net expression level,8,40 the results of these experiments demonstrate that the same scaling can arise from ribosome competition as a result of the expression of exogenous genes.
Our results are also consistent with the hypothesis that inefficiently translated rcRNAs function as RNAse competitors, stabilizing the gfpmRNA, increasing GFP abundance and in turn decreasing noise in GFP levels. The intracellular stability of mRNAs is thought to be governed in part by a competition between ribosome binding and RNAseE-induced degradation.41 We therefore expected that when ribosome competition increases, mRNA levels should decline, increasing the significance of mRNA fluctuations and therefore noise in protein levels. To examine this effect, we quantitated gfpmRNA levels in strains expressing different rcRNAs. Consistent with the mRNA stability model, the magnitude of induction of gfpmRNA (see Experimental) was approximately 12-fold lower in E. coli expressing an rcRNA that could effectively compete for ribosomes as compared with a control rcRNA with an occluded RBS (Fig. 2c). This is likely a combined effect of ribosome competition and lower mRNA half-life. These two factors can feedback on one another, as the observed decline in mRNA levels should further reduce the frequency of mRNA–ribosome interactions, which could in turn reduce transcriptional or metabolic capability and thus mRNA production. The possibility that such a noise amplification cascade exists would argue strongly that there should be mechanisms for the reduction or regulation of noise in cells.
Operons buffer gene expression noise
To determine if cells have natural ‘noise abatement’ mechanisms, we used our synthetic noise generators to investigate a standing mathematical model which predicts that the concatenation of multiple genes onto a single mRNA (operon formation) buffers relative fluctuations in the levels of coexpressed proteins .11 This buffering is predicted to occur because mRNA fluctuations contribute strongly to the noise profile of a given gene (as we have also shown) and operons virtually eliminate relative mRNA fluctuations between genes. To examine the model, we constructed a synthetic operon based on the dual fluorescent protein reporter system.17 In the engineered operon, two fluorescent genes (cfp and yfp) were arrayed in series, each preceded by a strong RBS (Fig. 3a). The mRNA encoding these genes was transcribed from an inducible promoter (Ptet) which allowed for examination of the noise profile over a large range of expression levels. The translation of the two genes was made independent by the incorporation of two consecutive strong stop codons at the end of each open reading frame. As a control, an analogous monocistronic version was built in which the two fluorescent reporter genes were transcribed independently from identical promoters and translated from identical ribosome binding sites (Fig. 3a). | ||
| Fig. 3 Noise in monocistronic and bicistronic expression platforms. (a) Schematic for engineered monocistronic (left) or bicistronic (right) CFP/YFP expression platforms. The monocistronic plasmid, pASKJ13065, was assembled such that both genes were expressed from the same TetR-repressed promoter (Ptet), carried the same RBS (green), and were followed by the same transcription terminator (not shown after YFP). On both plasmids, both genes contained double stop codons at the end of the respective open reading frame (asterisks shown in bicistronic version). The bicistronic plasmid, pASKJ13004, was designed exactly as pASKJ13065 except that both genes were expressed from a single Ptet promoter. (b) Intrinsic noise of mono and bicistronic constructs. E. coli carrying the monocistronic (black squares) and bicistronic (red dots) plasmids were induced to different extents by the addition of increasing amounts of the inducer anhydrotetracycline (aTc; between 0 and 500 ng mL−1) to the media. Reporter gene expression and noise were calculated as described in the Experimental. Relative fluorescence indicates CFP values from which E. coli autofluorescence and YFP bleedthrough were corrected. The data used to compare low level expression from the two constructs is the point at which each produces 0.012 relative fluorescence units (2nd point from left in each data set). Lines connecting data points are guides to the eye. (c) CFP fluorescence and intrinsic noise data in E. coli populations expressing mono or bicistronic CFP and YFP as well as the noisy rcRNA, rc6. High level CFP/YFP expression was matched between cells carrying the two constructs by inducing pASKJ13004 carrying cells with 250 ng mL−1 aTc (equivalent to green arrow in Fig. 3b) and pASKJ13065 carrying cells with 37.5 ng mL−1 aTc. Noise was quantitated from three cultures grown in parallel. Error values represent 95% confidence intervals. | ||
E. coli populations carrying the monocistronic and bicistronic constructs were grown with increasing concentrations of the transcriptional inducer anhydrotetracycline (aTc) and populations were analyzed by multi-channel flow cytometry (see Experimental). In agreement with the model,11 the bicistronic expression platform resulted in significantly less noise than the monocistronic version over a large range of low protein expression levels (Fig. 3b). To directly compare the noise profiles of two platforms, the mono and bicistronic constructs were induced to the same level of protein expression (0.012 relative fluorescence units) and the noise was quantified. At this low expression level, the operon showed ∼70% less noise than the monocistronic construct (Fig. 3b).
The mathematical model,11 predicts that the noise buffering properties of the operon will be greatest at low protein expression levels, and that noise in mono and bicistronic systems will converge as protein abundance increases. We investigated this prediction by varying protein expression from each construct from very low to very high levels. In agreement with the model, the noise output of the two constructs converged as protein abundance increased, and that at the highest expression levels the operon had no noise buffering effect (Fig. 3b). The contribution of mRNA fluctuations to noise is expected to be greatest at low expression levels, and this was also the regime under which the operon served to most efficiently buffer noise. At higher expression levels, mRNA fluctuations become less significant, diminishing the noise buffering effect of bicistronic encoding. Taken together, the noise trends of the mono and bicistronic constructs are in strong agreement with the predictions of the model over the protein abundances in our experiments.
We then introduced rcRNAs with the mono and bicistronic reporter constructs in order to assay the tolerance of the different expression systems to genetically-encoded noise. To better compare the mono and bicistronic systems, we induced both to the same high expression level, where their respective noise levels were low. This was achieved by inducing cells carrying the bicistronic plasmid to the maximal expression level (Fig. 3b) and varying the aTc concentration added to cells carrying the monocistronic plasmid to achieve the same level of fluorescence. In this high expression regime, noise in the monocistronic construct is naturally lower than the bicistronic version (Fig. 3b, green arrow). Even so, the introduction of a noisy rcRNA, rc6, resulted in 20% more noise in the monocistronic construct than in the bicistronic version (Fig. 3c). That is, the operon was significantly more tolerant to rcRNA induced noise than the monocistronic construct. These results bolster the hypothesis that operons function as genetic noise insulators, and demonstrate their efficacy in buffering artificially enhanced noise resulting from the expression of foreign genes.
Conclusions
The origins of stochasticity in gene expression vary widely between different organisms and even between genes within individual organisms.2,8,9 Random fluctuations in promoter states, transcription events and mRNA deaths are amplified by translation and lead to noise in protein levels across genetically identical cell populations. This noise in turn, can lead to dramatically different phenotypes between individual cells.6,30–32,42–46Here we show that noise in gene expression can be rationally engineered and that noise is strongly affected by the introduction of exogenous mRNAs which compete for ribosomes. Previous studies have shown that bulk cellular mRNAs compete for access to the ribosome,38 and that highly expressed mRNAs are capable of occupying nearly all cellular ribosomes.39 Our artificial ribosome-depleted states may mimic naturally occurring cellular states that arise when certain genes, such as those involved in stress response, are expressed to high levels or translated preferentially.47–54 The relationship between competition for translation and noise may prove to be universal for any of a number of factors required for gene expression (e.g., transcription factors), a hypothesis that is directly amenable to experimental evaluation. Indeed, competition for ribosomes may affect the translation and availability of other gene expression machinery and thereby initiate feedback cascades that further exacerbate noise in gene expression.
We also demonstrate that operons strongly buffer against noise in the coexpression of multiple genes, reducing noise ∼70% (nearly 4-fold) at low expression levels. While the cotranscription of multiple genes on a single mRNA undoubtedly reduces against noise generated by random promoter transitions and mRNA birth and death events,3–5,10 we demonstrate here that operons also buffer against noise arising from diminished ribosome pools. The noise buffering properties of the operon were particularly strong at low expression levels in our experiments. These low expression levels are likely to be more representative of natural protein abundances than the higher expression levels, as the genes were expressed from a multicopy plasmid. This suggests that noise buffering may be a relevant property of many natural operons. It is possible, then, that operons represent a reliable strategy by which cells can stoichiometrically couple the expression of multiple genes, even in translationally compromised or stressed environments.
The finding that operons have noise buffering properties has fundamental evolutionary implications. Though operons are one of the most ubiquitous forms of gene organization in nature, they are relatively unstable over evolutionary time. Operons frequently decompose into multiple genetic loci which are regulated by the same transcription factors, but are independently transcribed and translated.55 The most stable operons share an intriguing feature: they encode genes whose products physically interact.56 When gene products interact (e.g. in multi-protein complexes), stoichiometric coupling becomes critical, as the over- or underproduction of any single product will squander cellular resources and have deleterious fitness effects, and polycistronic encoding may provide a strong selective advantage against these effects.
Our results are also relevant to many approaches in synthetic biology. The engineering of synthetic biological systems with complex behaviors is proving to be challenging, as noise in the expression of certain gene products thwarts efforts to forward engineer deterministic phenotypes.29 Moreover, as the size (in DNA base pairs) of the synthetic biological constructs introduced into living cells is increased, competition for cellular resources, including ribosomes, will become greater and noise will concomitantly increase. Our experiments suggest that operons may offer a robust design strategy for those attempting to engineer synthetic pathways and behaviors that require reliable stoichiometric coupling of multiple gene products (such as the efficient production of foreign metabolites).57
Ultimately, these results nicely emphasize the increasing crossover between systems and synthetic biology. It was a synthetic tool (and orthogonal noise generator) that allowed the rational manipulation of the noise inherent in genetic expression. Novel synthetic circuits are frequently at the mercy of the cellular backgrounds in which they are implanted, and it is thus difficult to predict and model their performance. Our synthetic tool can now also be applied to any synthetic circuit, acting as an perturbant to determine whether and to what extent the cellular machinery is taxed by a synthetic circuit (and vice versa).
Experimental
rcRNA plasmid construction
rcRNAs (Supplementary Table 1† ) were embedded between the NcoI and BlpI sites (italic) in the following 100 nucleotide DNA: 5′-GATGGCAGCTACTAATGCTAGCTAAGATACCATG-31nt_rcRNA-GCTGAGCAACGAGCTATAGCTACTACTGATAGTCC-3′. 1 pmol of the oligonucleotide was amplified by PCR using the flanking primers 44.31F: 5′-ACGAGATCTCAGATCAGACCAGGATGGCAGCTACTAATGCTAGC-3′ and 44.31R: 5′-CGGTGAACTCGTCTATGGATCTGGACTATCAGTAGTAGCTATAG-3′, the final 20 nt of which were complimentary to the 100 nt oligo. The resulting double stranded DNA was purified using the Qiaquick PCR purification kit (Qiagen, Valencia CA) and digested with NcoI and BlpI (New England Biolabs, Ipswich MA) according to manufacturer instructions. Appropriate sized DNA fragments were isolated by gel purification using 4% NuSieve GTG agarose (Cambrex, Baltimore MD) in TBE, and recovered using a Wizard SV gel purification column (Promega, Madison WI). 1 μg of the rcRNA host vector, pACYCDuet-1 (Novagen, Madison WI) was digested with NcoI and BlpI, and gel purified using 2% Seaplaque agarose (Cambrex) in TAE. 60 fmol of digested rcRNA DNA was ligated into 20 fmol digested pACYCDuet-1 with T4 DNA ligase (Invitrogen, Carlsbad CA) and transformed into Top10 cells (Invitrogen) according to manufacturer instructions. The insert regions were sequence verified and the plasmids were transformed into BL21DE3 (Novagen) along with the appropriate reporter plasmid for all assays. All DNA oligonucleotides and primers were purchased from Integrated DNA Technologies (Coralville, IA), resuspended in H2O and used directly. The pACYCDuet-1 host plasmid carries a p15A (∼10–12 copy) origin of replication, and a chloramphenicol resistance marker. rcRNAs were expressed by the IPTG inducible T7 RNA polymerase (carried in the genome of BL21DE3) through the IPTG inducible T7 promoter upstream of the first MCS of pACYCDuet-1. All pACYC-hosted rcRNA plasmids were maintained in BL21DE3 using 34 μg mL−1chloramphenicol.Reporter plasmids
The expression plasmid pASKIBA3.GFPm2 was used for all GFP flow cytometry assays. The gene gfpm2 was expressed from the multiple cloning site of the host plasmid pASKIBA3 (IBA, Göttingen Germany) under the control of a Tetracycline Repressor (TetR) controlled promoter. pASKIBA3 carries a marker for ampicillin resistance, a pBR322 (∼40 copy number) based origin of replication, and the TetR gene. pASKIBA3.GFPm2 was maintained in BL21DE3 using 50 μg mL−1ampicillin.The CFP/YFP expression plasmids were constructed using MIT’s registry of standard biological parts (http://parts.mit.edu) and DNA isolation, purification and ligation methods as described above. The host plasmid pSB4A3 (MIT’s registry of standard biological parts) was used for construction of the monocistronic (J13065) and the bicistronic constructs (J13004). pSB4A3 bears a pSC101 origin of replication (∼5 copy) and an ampicillin resistance marker. 25 μg mL−1ampicillin was used to maintain all pSB4A3-based plasmids. For noise assays, J13065 and J13004 were cloned into pASKIBA3 and expressed under the control of TetR.
To build the monocistronic construct (J13065), a DNA sequence bearing the Ptet promoter (R0040), a strong RBS (B0034), an ECFP coding sequence (E0020) and a strong transcriptional terminator (B0015) was cloned into pSB4A3 using the restriction enzymes EcoRI and PstI. A second strong transcriptional terminator (B0015) was cloned downstream of the first. Finally an EYFP expression cassette carrying the Ptet promoter, strong RBS (B0034), EYFP gene (E0030) and transcription terminator (B0015) were cloned downstream of the tandem transcription terminators using standard biobrick assembly methods (http://parts.mit.edu). The ECFP/EYFP monocistronic expression construct (J13065) was cloned into the pASKIBA3 plasmid using the restriction sites PflMI and PstI. The primers jt_pflmI_bb_prefix_F: 5′-CGTAGACTAGACCACCATCGAATGGGAATTCGCGGCCGCTTCTAG-3′ and BBa_G00101: 5′- ATTACCGCCTTTGAGTGAGC-3′ were used to add an upstream PflMI site to the beginning of J13065 while maintaining the downstream PstI site from pSB4A3. The amplified DNA was digested and cloned into pASKIBA3 using methods described above.
The bicistronic CFP/YFP expression construct, J13004 carries the same promoter, RBS, coding sequences and transcriptional terminator as J13065 (Fig. 3a). It was constructed by cloning a DNA segment containing R0040, B0034 and E0020 upstream of a segment containing B0034, E0030 and B0015 as above. J13004 was then moved to pASKIBA3 as above.
GFP Flow cytometry assays
E. coli cultures were grown overnight at 37 °C with shaking at 250 rpm from frozen stocks in LB plus appropriate antibiotics. Three parallel cultures were diluted 1 : 100 in fresh media with 100 μM IPTG as appropriate, grown to early log phase (OD600 ∼ 0.1) and treated with 200 ng mL−1 anhydrotetracycline (aTc) to induce GFP expression. 4 h later, 1–10 μL of cells were diluted into 0.5–1 mL phosphate buffered saline (PBS) and scanned for GFP fluorescence. BL21DE3 carrying no plasmid were measured for autofluorescence and subtracted from all data sets. All GFP expression data were collected on a Becton Dickinson FACSCalibur flow cytometer with a 488 nm argon excitation laser and a 515–545 emission filter. 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events were collected for each data set and the cell populations were then gated away from other objects by polygonal gates around the dominant forward scatter/side scatter profile. Non-fluorescent objects not representative of the cell population were discarded. Data were analyzed using WinMDI version 2.8 (Joseph Trotter, The Scripps Research Institute).
000 events were collected for each data set and the cell populations were then gated away from other objects by polygonal gates around the dominant forward scatter/side scatter profile. Non-fluorescent objects not representative of the cell population were discarded. Data were analyzed using WinMDI version 2.8 (Joseph Trotter, The Scripps Research Institute).
CFP/YFP flow cytometry
E. coli cultures were grown overnight from frozen stocks as above. Cultures were diluted 1 : 100 in fresh media with 100 μM IPTG when appropriate and grown to OD600 ∼ 0.1–0.2. The cultures were then induced with the appropriate amount of anhydrotetracycline (from 2 ng mL−1 to 500 ng mL−1) and grown shaking at 250 rpm and 37 °C for 3.75 h. 50 μL of each culture was then diluted into 2 mL of PBS and analyzed on a Becton-Dickinson LSRII cytometer with Trigon Violet and Octagon Blue lasers for CFP and YFP, respectively. ECFP fluorescence was measured through a 525/50 bandpass filter while EYFP expression was measured through a 530/30 bandpass filter. BL21DE3 cells carrying no fluorescent proteins were measured for autofluorescence and subtracted from all data sets. 30000 data points were collected at the LOW flow rate and cells expressing CFP or YFP alone were used to calculate filter bleedthrough, which was used to correct all data sets. Data were exported from the associated FACSDiva software as fcs files and converted to raw, ASCII format with FlowJo7.2 (Tree Star Inc., Ashland, OR) for further analysis.
Cell populations were gated away from other objects by a polygonal gate based on the dominant forward scatter/side scatter profile. Objects with fluorescence values at the lower limit of detection of the cytometer were discarded. The first and last 0.2 s of the data were then removed to eliminate cytometer flow rate variability.40
CFP/YFP Noise calculations
Intrinsic noise in CFP and YFP was quantified using the equation of Elowitz et al.17 as follows.Where η is noise, c indicates CFP fluorescence, y indicates YFP fluorescence and 〈
 〉 indicates a population averaged measurement.
〉 indicates a population averaged measurement.
mRNA quantitation assays
Cells were grown overnight as above, subcultured 1 : 100 into fresh media with 100 μM IPTG to induce rcRNA expression when appropriate. The cultures were grown to early-log phase (OD600 ∼ 0.1) at which point transcription of the gfpm2mRNA was induced by the addition of 200 ng mL−1 anhydrotetracycline (aTc) when appropriate. Cells were grown for 4 h, at which point total RNA was recovered and DNase-treated using an RNeasy kit (Qiagen) according to manufacturer’s instructions. cDNA was created for the gfpm2mRNA, and 16 s ribosomal RNA using MMLV reverse transcriptase (Ambion, Austin TX) according to manufacturer instructions. gfpm2mRNA was specifically reverse transcribed using the primer GFPm2.rt.R: 5′- TTTTCGTTGGGATCTTTCGAA-3′ and 16 s rRNA using 16s.rt.R: 5′-ACCGCTGGCAACAAAAGATAA-3′. gfpm2cDNA was quantitated on an Applied Biosystems 7900HT real-time PCR machine using the forward primer GFPm2.rt.F: 5′- GATGGCCCTGTCCTTTTACCA-3′, GFPm2.rt.R, and the Taqman probe GFPm2.rt.p: 5′- 6FAM/ACAACCATTACCTGTCCACACAATCTGCC/BHQ1 where BHQ1 indicates Black Hole Quencher 1 (Integrated DNA technologies). Cycle threshold (C(T)) values were compared to 16 s ribosomal RNA C(T) values. 16 s cDNA was amplified using the forward primer 16s.rt.F: 5′- CGTGTTGTGAAATGTTGGGTTAA-3′, the reverse primer 16s.rt.R, and the Taqman probe 16s.rt.p: 5′- TCCCGCAACGAGCGCAACC-3′. ROX (Invitrogen) was used as a passive reference dye for all samples. Average ΔC(T) values for each sample were calculated from 3 independent cultures grown in parallel. Fold induction in gfpm2mRNA levels from uninduced (aTc minus) to induced (aTc plus) samples was calculated as: fold induction = mRNAinduced/mRNAuninducedTranslation competition assays
Coupled transcription/translation reactions were performed with the Rapid Translation System RTS 100 E. coli HY Kit (Roche, Indianapolis IN) according to manufacturer instructions. 75 fmol GFPm2 double stranded DNA template bearing the T7 promoter and ribosome binding site sequence from pACYCDuet-1 was added with increasing molar ratios of rcRNA DNA sequence bearing the same promoter and RBS elements. GFPm2 fluorescence was read on a Tecan SAFIRE fluorescence microplate reader using a clear flat bottomed 96-well plate and an excitation wavelength of 488 nm and an emission wavelength of 509 nm. The autofluorescence of a reaction without GFPm2 DNA template was measured and subtracted from all samples. All reactions were performed and measured in triplicate.In-line probing
In-line RNA structure assays were performed on rcRNA transcripts as described in ref. 22 unless otherwise noted. rcRNAs were amplified using primers flanking the T7 promoter and terminator from pACYCDuet-1, transcribed in vitro by a T7 RNA polymerase transcription kit (Epicentre, Madison WI), purified on a 12% denaturing polyacrylamide gel, phosphatased with Antarctic Phosphatase (NEB), radiolabeled on the 5′-most nucleotide , and gel purified as before. In-line degradation was performed at 25 °C for 41 h, and equivalent counts of 32P containing RNA were added to each lane of a 10% denaturing polyacrylamide gel. The gel was dried under vacuum at 75 °C for 1 h and imaged using a Phosphorimager (Amersham, Piscataway NJ).Acknowledgements
We thank K. E. Griswold for assistance with two-channel flow cytometry, E. A. Davidson for the GFP expression plasmid, J. J. Bull, J. J. Collins, E. M. Marcotte and C. A. Voigt for critical reading of an earlier version of the manuscript. We also thank several anonymous reviewers for insightful commentary and suggestions. This work was supported by NIH grant #9R01GM077040-05, the Welch Foundation and the Texas Higher Education Coordinating Board TDT-003658-0611-2003.References
- H. H. McAdams and A. Arkin, Stochastic mechanisms in gene expression, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 814–9 CrossRef CAS.
- A. Becskei, B. B. Kaufmann and A. van Oudenaarden, Contributions of low molecule number and chromosomal positioning to stochastic gene expression, Nat. Genet., 2005, 37, 937–44 CrossRef CAS.
- W. J. Blake, K. A. M, C. R. Cantor and J. J. Collins, Noise in eukaryotic gene expression, Nature, 2003, 422, 633–7 CrossRef CAS.
- I. Golding, J. Paulsson, S. M. Zawilski and E. C. Cox, Real-time kinetics of gene activity in individual bacteria, Cell, 2005, 123, 1025–36 CrossRef CAS.
- J. M. Raser and E. K. O'Shea, Control of stochasticity in eukaryotic gene expression, Science, 2004, 304, 1811–4 CrossRef CAS.
- W. J. Blake, G. Balazsi, M. A. Kohanski, F. J. Isaacs, K. F. Murphy, Y. Kuang, C. R. Cantor, D. R. Walt and J. J. Collins, Phenotypic consequences of promoter-mediated transcriptional noise, Mol. Cell, 2006, 24, 853–65 CrossRef CAS.
- J. M. Pedraza and J. Paulsson, Effects of molecular memory and bursting on fluctuations in gene expression, Science, 2008, 319, 339–43 CrossRef CAS.
- A. Bar-Even, J. Paulsson, N. Maheshri, M. Carmi, E. O'Shea, Y. Pilpel and N. Barkai, Noise in protein expression scales with natural protein abundance, Nat. Genet., 2006, 38, 636–43 CrossRef CAS.
- J. R. Newman, S. Ghaemmaghami, J. Ihmels, D. K. Breslow, M. Noble, J. L. Derisi and J. S. Weissman, Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise, Nature, 2006 Search PubMed.
- J. Paulsson, Summing up the noise in gene networks, Nature, 2004, 427, 415–8 CrossRef CAS.
- P. S. Swain, Efficient attenuation of stochasticity in gene expression through post-transcriptional control, J. Mol. Biol., 2004, 344, 965–76 CrossRef CAS.
- L. Cai, N. Friedman and X. S. Xie, Stochastic protein expression in individual cells at the single molecule level, Nature, 2006, 440, 358–62 CrossRef CAS.
- J. Yu, J. Xiao, X. Ren, K. Lao and X. S. Xie, Probing gene expression in live cells, one protein molecule at a time, Science, 2006, 311, 1600–3 CrossRef CAS.
- E. M. Ozbudak, M. Thattai, I. Kurtser, A. D. Grossman and A. van Oudenaarden, Regulation of noise in the expression of a single gene, Nat. Genet., 2002, 31, 69–73 CrossRef CAS.
- M. Thattai and A. van Oudenaarden, Intrinsic noise in gene regulatory networks, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8614–9 CrossRef CAS.
- U. N. Singh, Polyribosomes and unstable messenger RNA: a stochastic model of protein synthesis, J. Theor. Biol., 1969, 25, 444–60 CrossRef CAS.
- M. B. Elowitz, A. J. Levine, E. D. Siggia and P. S. Swain, Stochastic gene expression in a single cell, Science, 2002, 297, 1183–6 CrossRef CAS.
- J. M. Pedraza and A. van Oudenaarden, Noise propagation in gene networks, Science, 2005, 307, 1965–9 CrossRef CAS.
- D. Volfson, J. Marciniak, W. J. Blake, N. Ostroff, L. S. Tsimring and J. Hasty, Origins of extrinsic variability in eukaryotic gene expression, Nature, 2005 Search PubMed.
- N. J. Guido, P. Lee, X. Wang, T. C. Elston and J. J. Collins, A pathway and genetic factors contributing to elevated gene expression noise in stationary phase, Biophys. J., 2007 Search PubMed.
- A. Colman-Lerner, A. Gordon, E. Serra, T. Chin, O. Resnekov, D. Endy, C. G. Pesce and R. Brent, Regulated cell-to-cell variation in a cell-fate decision system, Nature, 2005, 437, 699–706 CrossRef CAS.
- N. Rosenfeld, J. W. Young, U. Alon, P. S. Swain and M. B. Elowitz, Gene regulation at the single-cell level, Science, 2005, 307, 1962–5 CrossRef CAS.
- J. R. Chabot, J. M. Pedraza, P. Luitel and A. van Oudenaarden, Stochastic gene expression out-of-steady-state in the cyanobacterial circadian clock, Nature, 2007, 450, 1249–52 CrossRef CAS.
- D. A. Drubin, J. C. Way and P. A. Silver, Designing biological systems, Genes Dev., 2007, 21, 242–54 CrossRef CAS.
- D. Sprinzak and M. B. Elowitz, Reconstruction of genetic circuits, Nature, 2005, 438, 443–8 CrossRef CAS.
- B. J. Yeh and W. A. Lim, Synthetic biology: lessons from the history of synthetic organic chemistry, Nat. Chem. Biol., 2007, 3, 521–5 CrossRef CAS.
- N. J. Guido, X. Wang, D. Adalsteinsson, D. McMillen, J. Hasty, C. R. Cantor, T. C. Elston and J. J. Collins, A bottom-up approach to gene regulation, Nature, 2006, 439, 856–60 CrossRef CAS.
- T. S. Gardner, C. R. Cantor and J. J. Collins, Construction of a genetic toggle switch in Escherichia coli, Nature, 2000, 403, 339–42 CrossRef CAS.
- M. B. Elowitz and S. Leibler, A synthetic oscillatory network of transcriptional regulators, Nature, 2000, 403, 335–8 CrossRef CAS.
- G. M. Suel, R. P. Kulkarni, J. Dworkin, J. Garcia-Ojalvo and M. B. Elowitz, Tunability and noise dependence in differentiation dynamics, Science, 2007, 315, 1716–9 CrossRef.
- G. M. Suel, J. Garcia-Ojalvo, L. M. Liberman and M. B. Elowitz, An excitable gene regulatory circuit induces transient cellular differentiation, Nature, 2006, 440, 545–50 CrossRef.
- L. S. Weinberger, J. C. Burnett, J. E. Toettcher, A. P. Arkin and D. V. Schaffer, Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity, Cell, 2005, 122, 169–82 CrossRef CAS.
- F. J. Isaacs, D. J. Dwyer, C. Ding, D. D. Pervouchine, C. R. Cantor and J. J. Collins, Engineered riboregulators enable post-transcriptional control of gene expression, Nat. Biotechnol., 2004, 22, 841–7 CrossRef CAS.
- E. Masse and S. Gottesman, A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4620–5 CrossRef CAS.
- T. Moller, T. Franch, C. Udesen, K. Gerdes and P. Valentin-Hansen, Spot 42 RNA mediates discoordinate expression of the E. coli galactose operon, Genes Dev., 2002, 16, 1696–706 CrossRef CAS.
- W. Winkler, A. Nahvi and R. R. Breaker, Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression, Nature, 2002, 419, 952–6 CrossRef CAS.
- G. A. Soukup and R. R. Breaker, Relationship between internucleotide linkage geometry and the stability of RNA, RNA, 1999, 5, 1308–25 CrossRef CAS.
- S. T. Liang, Y. C. Xu, P. Dennis and H. Bremer, mRNA composition and control of bacterial gene expression, J. Bacteriol., 2000, 182, 3037–44 CrossRef CAS.
- B. Xia, J. P. Etchegaray and M. Inouye, Nonsense mutations in cspA cause ribosome trapping leading to complete growth inhibition and cell death at low temperature in Escherichia coli, J. Biol. Chem., 2001, 276, 35581–8 CrossRef CAS.
- J. R. Newman, S. Ghaemmaghami, J. Ihmels, D. K. Breslow, M. Noble, J. L. DeRisi and J. S. Weissman, Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise, Nature, 2006, 441, 840–6 CrossRef CAS.
- P. Regnier and C. M. Arraiano, Degradation of mRNA in bacteria: emergence of ubiquitous features, Bioessays, 2000, 22, 235–44 CrossRef CAS.
- A. Arkin, J. Ross and H. H. McAdams, Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells, Genetics, 1998, 149, 1633–48 CAS.
- A. D. Grossman, Genetic networks controlling the initiation of sporulation and the development of genetic competence in Bacillus subtilis, Annu. Rev. Genet., 1995, 29, 477–508 CrossRef CAS.
- R. Kemkemer, S. Schrank, W. Vogel, H. Gruler and D. Kaufmann, Increased noise as an effect of haploinsufficiency of the tumor-suppressor gene neurofibromatosis type 1 in vitro, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 13783–8 CrossRef CAS.
- H. Maughan and W. L. Nicholson, Stochastic processes influence stationary-phase decisions in Bacillus subtilis, J. Bacteriol., 2004, 186, 2212–4 CrossRef CAS.
- D. M. Wolf, V. V. Vazirani and A. P. Arkin, Diversity in times of adversity: probabilistic strategies in microbial survival games, J. Theor. Biol., 2005, 234, 227–53 CrossRef.
- A. M. Giuliodori, A. Brandi, C. O. Gualerzi and C. L. Pon, Preferential translation of cold-shock mRNAs during cold adaptation, RNA, 2004, 10, 265–276 CrossRef CAS.
- F. Fiedler, G. V. Mallo, H. Bodeker, V. Keim, J. C. Dagorn and J. L. Iovanna, Overexpression of the PC3/TIS21/BTG2 mRNA is part of the stress response induced by acute pancreatitis in rats, Biochem. Biophys. Res. Commun., 1998, 249, 562–5 CrossRef CAS.
- A. Brandi, R. Spurio, C. O. Gualerzi and C. L. Pon, Massive presence of the Escherichia coli ‘major cold-shock protein’ CspA under non-stress conditions, EMBO J., 1999, 18, 1653–9 CrossRef CAS.
- K. Yamanaka and M. Inouye, Induction of CspA, an E. coli major cold-shock protein, upon nutritional upshift at 37 degrees C, Genes Cells, 2001, 6, 279–90 CrossRef CAS.
- W. J. Welch, H. S. Kang, R. P. Beckmann and L. A. Mizzen, Response of mammalian cells to metabolic stress; changes in cell physiology and structure/function of stress proteins, Curr. Top. Microbiol. Immunol., 1991, 167, 31–55 Search PubMed.
- L. Y. Wei and P. D. Roepe, Low external pH and osmotic shock increase the expression of human MDR protein, Biochemistry, 1994, 33, 7229–38 CrossRef CAS.
- P. G. Jones, R. A. VanBogelen and F. C. Neidhardt, Induction of proteins in response to low temperature in Escherichia coli, J. Bacteriol., 1987, 169, 2092–5 CAS.
- D. A. Colon-Ramos, C. L. Shenvi, D. H. Weitzel, E. C. Gan, R. Matts, J. Cate and S. Kornbluth, Direct ribosomal binding by a cellular inhibitor of translation, Nat. Struct. Mol. Biol., 2006, 13, 103–111 CrossRef CAS.
- J. G. Lawrence, Shared strategies in gene organization among prokaryotes and eukaryotes, Cell, 2002, 110, 407–13 CrossRef CAS.
- T. Dandekar, B. Snel, M. Huynen and P. Bork, Conservation of gene order: a fingerprint of proteins that physically interact, Trends Biochem. Sci., 1998, 23, 324–8 CrossRef CAS.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Production of the antimalarial drug precursor artemisinic acid in engineered yeast, Nature, 2006, 440, 940–3 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary Table 1 and details on reporter independence. See DOI: 10.1039/b801245h |
| ‡ Current address: Department of Pharmaceutical Chemistry, University of California – San Francisco, San Francisco, California 94158 |
| This journal is © The Royal Society of Chemistry 2008 |