Processing of DNA damage clusters in human cells: current status of knowledge
Alexandros G.
Georgakilas
Department of Biology, Thomas Harriot College of Arts and Sciences, East Carolina University, Greenville NC 27858, USA. E-mail: georgakilasa@ecu.edu; Fax: +1 252-328-4178; Tel: +1 252-328-5446
First published on 7th November 2007
Abstract
Eukaryotic cells exposed to DNA damaging agents activate important defensive pathways by inducing multiple proteins involved in DNA repair, cell cycle checkpoint control and potentially apoptosis. After the acceptance of the hypothesis that oxidatively generated clustered DNA lesions (OCDL: closely spaced DNA lesions) can be induced even by low doses of ionizing radiation or even endogenously, and significant advances have been made in the understanding of the biochemistry underlying the repair of closely spaced DNA lesions, many questions still remain unanswered. The major questions that have to be answered in the near future are: 1) how human cells process these types of DNA damage if they repair them at all, 2) under what conditions a double strand break (DSB) may be created during the processing of two closely spaced DNA lesions and 3) what type of repair protein interactions govern the processing of complex DNA damage? The data existing so far on human cells and tissues are very limited and in some cases contradicting. All of them though agree however on the major importance of gaining mechanistic insights on the pathways used by the cell to confront and process complex DNA damage located in a small DNA volume and the need of more in depth analytical studies. We selectively review recently-obtained data on the processing of non-DSB DNA damage clusters in human cells and tissues and discuss the current status of knowledge in the field.
 Alexandros Georgakilas | Dr Alexandros Georgakilas is an Assistant Professor in the Department of Biology at East Carolina University in Greenville, North Carolina. He received his PhD in Biology in 1998 from the University of Athens, Greece. His research has focused on the role of complex DNA damage in human cells and the overall effects of ionizing radiation on DNA which is considered the key target of radiation in the cell. He is particularly interested in the role of specific repair genes like BRCA1, DNA-PKcs in the processing of clustered DNA lesions especially in the case of breast cancer. |
Types of oxidatively generated DNA damage
One of the main etiological hypotheses in the promotion of mutagenesis and carcinogenesis is the lower fidelity or deficiencies of cellular repair mechanisms which are responsible for removing or bypassing the DNA damage sites and restoring the original sequence after exposure to mutagens such as estrogen metabolites, ionizing radiation, cigarette smoke and environmental chemicals.1 Numerous studies suggest the important role of oxidatively generated DNA damage and its repair in cancer and aging.2,3 This pool of oxidatively generated DNA damage can contain a wide variety of single or clustered DNA lesions, including single strand breaks (SSBs), oxidized bases and/or apurinic–apyrimidinic (abasic, AP) sites.4 Even at doses as low as ∼1 Gy (100 rad) ionizing radiation is capable of inducing all of the above types of DNA damage in the form of isolated lesions as well as in the form of clustered ones (1–10 bp apart).5,6The idea of ‘clustered DNA lesions’
Although similar types of DNA lesions are produced by endogenous processes and various physical and chemical exogenous agents like ionizing radiation,7 the relative yield and spatial arrangement of damaged nucleotides can be quite different.8,9 The idea of clustered DNA damage was first introduced by Ward as locally multiple damaged sites (LMDS), i.e., several closely spaced damages within a short DNA segment that could be produced by ionizing radiation.10,11 Ward introduced the idea of clustered DNA lesions to account for the increased lethality induced by ionizing radiation, which cannot be fully explained by the amount of DNA double stand breaks formed, although the specific lesions, if unrepaired or misrepaired, can lead to a lethal event. Fig. 1 shows an idealized representation of various types of clustered damage, including complex single strand breaks (SSBs) and double strand breaks (DSBs) that can be formed by ionizing radiation. The very short DNA fragments within complex SSB (1–2 nt) and within complex DSB (2–6 nt) will be probably lost during DNA extraction and likely in the cell within the chromatin too. A complex DSB, as proposed, may result in a DSB with loss of material. The complex SSB may be seen as a 4 nt gap. In any case, such complex lesions have no chance to be repaired in an error-free manner and are expected to result in mutations. Experiments have shown that clustered lesions are estimated to be 50–80% of the total complex DNA damage (DSB and non-DSB clustered DNA lesions) produced by ionizing radiation.12,13 Based on theoretical calculations, it is predicted that in addition to isolated lesions, low-LET radiation can create clusters with as many as 10 lesions while high-LET radiation is capable of producing damage of even greater complexity.14 The maximal 10 or more lesions per cluster are predicted to occur though with much lower frequency compared to isolated lesions. In addition to the DSB formed directly by ionizing radiation (“prompt DSB”), the attempted excision repair of clusters containing sugar and base residues can sometimes produce additional (enzymatic) DSB,15 also reviewed in ref. 16. Clustered DNA damages are hypothesized to be poorly repaired lesions which produce cytotoxic and mutagenic effects,6,17,18 as well as chromosomal instability.19 Although multiple pathways for the induction and perpetuation of genomic instability are possible,20 studies conducted with repair-deficient cell lines suggest that DSB and other forms of clustered damage are involved in genomic instability.21,22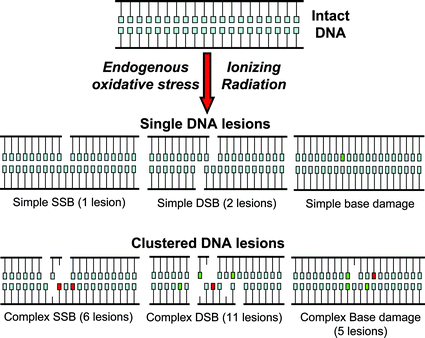 | ||
| Fig. 1 Examples of the oxidatively generated DNA lesions single and complex induced by ionizing radiation or endogenous oxidative stress. Light squares denote undamaged bases. Damage to individual nucleotides comprise missing or damaged (red squares) bases and strand breaks (accompanied by base loss). As shown and based also on theoretical studies many neighboring lesions can accompany one single strand break (SSB) or double strand break (DSB) (complex DNA lesions).14,72 | ||
DNA repair refers to a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. The DNA repair ability of a cell is vital to the integrity of its genome and thus to its normal functioning and that of the organism. Many genes that were initially shown to influence lifespan have turned out to be involved in DNA damage repair and protection. Failure to correct molecular lesions in cells that form gametes can introduce mutations into the genomes of the offspring and thus influence the rate of evolution. Experimental animals with genetic deficiencies in DNA repair often show decreased lifespan and increased cancer incidence.23
Processing of oxidatively generated clustered DNA lesions (OCDL)
Lethal and mutagenic effects of ionizing radiation are thought to result principally from incompletely or incorrectly repaired DNA lesions.24 While isolated damages are generally repaired efficiently, clustered DNA lesions have been suggested to be more difficult to repair, and in general are considered as DNA damages that are highly repair-resistant or non-repairable and therefore considered as highly significant biological lesions.5,10,25 Bistranded or tandem (in the same strand) DNA clusters could be resistant to processing by glycosylases or endonucleases, as shown for synthetic oligonucleotides containing clusters of specific composition.26–32 Such repair-resistant clusters could persist for a substantial time after irradiation (up to several days).17,33,34The pool of oxidatively generated base lesions and AP sites induced by endogenous sources or ionizing radiation are predominantly repaired by base excision repair (BER), which is a multi-step procedure involving the sequential action of several proteins . The BER pathways are initiated by a DNA glycosylase (in human cells: mainly hOGG1 or hNTH1) that recognizes and hydrolytically cleaves and removes the altered base, giving rise to an AP site. The AP site is then processed by AP endonuclease (APE1), which incises the DNA strand 5′ to the baseless sugar . Then, DNA polymerase β catalyzes the β-elimination of the 5′-sugar phosphate residue and at the same time fills the one-nucleotide gap (single-nucleotide patch BER pathway). Finally, the nick is sealed by the DNA ligase III/XRCC1 complex.35 This repair process results in the removal and replacement of a single damaged nucleotide with a normal one.24 In addition to the above pathway (A) an additional pathway (B) may occur. DNA glycosylases that process oxidatively generated DNA lesions have an intrinsic AP lyase activity that incises AP sites 3′ to the baseless sugar leaving a 3′ (2,3-didehydro-2,3-dideoxyribose) terminus that is then removed by AP endonuclease. As in pathway (A), the gap is filled by DNA polymerase and the nick is sealed by DNA ligase. However, all major human glycosylases possess strong glycosylase activity relative to their lyase activity but in the presence of an AP endonuclease, pathway (A) is favored.36–39 Newly discovered human proteins like the glycosylases NEIL1 and NEIL2 have been hypothesized to be implicated in the repair of oxidatively generated DNA damage,40 and the nuclease Artemis and the variant form of histone H2A, the so-called H2AX, in DSB repair.41
There are many fundamental questions about the repair of clustered DNA lesions that remain unanswered. The most important are as follows: i) to what extent does the presence of one lesion affect the efficiency of recognition and simultaneous processing of the opposite neighboring lesion(s), ii) is there a hierarchy in repair of closely spaced DNA lesions,25 iii) are there any cellular strategies to avoid DSB formation that can trigger a pathway to cell death or genomic instability, and iv) which repair proteins participate in the processing of these lesions and what mechanisms are used by the cell to eliminate the presence of these lesions? To address these questions, several in vitro studies (using oligonucleotides carrying well-defined clustered lesions and human or E. coli repair enzymes or cell extracts) have considered the effect of neighboring lesions on the excision or repair of base damage or AP sites by the corresponding enzyme or cell extract. Focusing here only on the studies using human enzymes or human cell extracts, some major inferences can be made at this time:
a) Excision of 8-oxo-7,8-dihydroguanine (8-oxoGua) by human glycosylase hOGG1 or whole cell extracts is significantly inhibited or retarded by the presence of an AP site or SSB on the opposite strand up to five bases in the 3′ or 5′ direction, whereas a second base lesion has little or no influence on the rate of excision of 8-oxoGua thus minimizing DSB formation.31,42,43
b) hAPE1 which is expected to be the major AP endonuclease dealing with abasic clusters in human cells was found to be significantly retarded in processing of an AP site in the presence of an opposing AP site or SSB one to three bases in the 3′ direction whereas no inhibition was observed in the presence of base damage.26,29,44
All the above in vitro or in vivo studies are in agreement to some degree. However, to what extent DSBs, resulting from repair intermediates (mainly opposing SSBs), are produced in human cells is still a major open question. The general suggestion of all the above studies is that clustered DNA lesions in human cells may have a significantly increased lifetime compared with isolated ones, if they are finally repaired at all. Previous data for the repair of abasic DNA clusters in human cells agree with this suggestion.17,33 Simultaneous repair of both lesions constituting an oxybase or abasic cluster can lead to a DSB. Considering that repair of DSBs frequently results in deletions and loss of genetic information, it is possible that the BER mechanism includes features that reduce the probability of DSB formation during repair of closely spaced DNA lesions. As mentioned above, a number of studies, including the study by Georgakilas et al.,17 indicate that there should be a certain hierarchy in processing of clustered DNA lesions in order to minimize the possibility of DSB creation.31,45 What is the biochemical basis for this mechanism of “DSB avoidance” if it exists in human cells? The key role in this suggested mechanism seems to be played by the creation of an AP site or SSB (nt gap). Once an AP site or SSB is created (as repair intermediates) in the processing of clustered DNA lesions then this event is expected to significantly inhibit the processing of the opposing oxybase lesion or AP site by DNA glycosylase or AP endonuclease.25,43
Therefore it is expected that the lesions will be successively repaired i.e., as soon as the completion of one lesion occurs, the initiation of the repair of the remaining lesion will happen (Fig. 2). This idea of successive (‘sequential’) repair was first introduced by Chaudhry and Weinfeld.26 However, there are studies on the repair of clustered DNA lesions suggesting that DSBs can occur during repair of closely spaced DNA lesions forming a cluster.13,46–49
 | ||
| Fig. 2 Model for the repair of bistranded clustered DNA lesions in human cells based on current knowledge. Radiation induces different types of clustered lesions (A and B) as well as prompt DSBs (C). Here for simplicity, one type of oxybase (A) and abasic (B) cluster is shown, i.e. the oxybase cluster containing only oxybases and the abasic cluster containing one oxybase and one AP site. The highlighted arrows indicate the preferred repair pathways based on present knowledge and accepting the hypothesis that the human cell will try to avoid the pathway of simultaneous repair of both lesions leading to DSB formation (II). Instead it is suggested that pathway (I) may be chosen. In this pathway, each lesion will be processed in a separate successive (sequential) way: in the case (A) as soon as one oxybase is excised by DNA glycosylase and an AP site is created (repair intermediate B), the repair will continue with the processing of this AP site, completion of its repair and then processing of the remaining oxybase or AP site. Even in the case that both oxybases are excised and two AP sites are created a similar sequential pathway of repair is expected. The reason is that the existence of the AP site or SSB (when the AP site is incised by AP endonuclease) on one strand is expected to be a strong inhibitory factor for both DNA glycosylase and AP endonuclease willing to process the opposite oxybase lesion or created AP site respectively. Finally the formation of DSB may lead to cell death or genomic instability if not repaired efficiently as shown. | ||
Finally, Georgakilas et al.17 have shown that two mechanisms, replication and cell death, contribute to the ‘disappearance’ of clusters. The first, by diluting the bistranded DNA lesions and the second excluding from the cell population cells with high and resistant damage load (Fig. 2). In accordance with that finding, Tsao et al. have recently reported that the contribution of cell death and especially cell division (dilution) has been found increased for 56Fe high LET-induced OCDL compared to low LET (γ rays), reflecting a high percentage of practically non-repairable clusters of high complexity as also suggested by many theoretical studies.50
Detection of endogenous clusters in human or mammalian tissues
Substantial evidence supports the accumulation and persistence of different oxidatively generated DNA lesions (e.g., abasic sites or oxidized bases) in human or animal cells and tissues at values ranging from 100–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lesions Gbp−1.51–54 Rodents or cancer patients exposed to radiotherapy have elevated levels of different types of oxidatively generated DNA damage (e.g., 8-oxodG or double strand breaks). These lesions have been detected in blood cells or tissues even after several weeks (1–24 weeks) post-irradiation, indicating an induced chronic oxidative stress.55–60 There are very limited data on the possible accumulation of OCDLs in human malignant cells or tissues and in mammalian/human tissues exposed to ionizing radiation. Gollapalle et al.60 showed for the first time that endogenous OCDLs can be detected in non-irradiated mice tissues at a steady state of ∼0.5–0.9 clusters Mbp−1 and that exposure of these tissues to ionizing radiation (X-rays) leads to an accumulation of DNA clusters detected 20 weeks after irradiation. Measurement of endogenous OCDLs in the human breast cancer line MCF-7 revealed elevated levels compared with the non-malignant MCF-10A. These cluster frequencies are higher than the ones detected for DNA isolated from human skin primary cell cultures (20–40 clusters Gbp−1 or 0.02–0.04 clusters Mbp−1).61 However, a completely different isolation and enzyme-treatment method was followed embedding DNA in agarose plugs and using transverse alternating field electrophoresis for DNA analysis while the different tissue origin may be another explanation for this discrepancy. Interestingly, Cadet et al.62 reported a number of 1–3 8-oxoGua Mbp−1 for DNA isolated from liver or kidney of young or old mice. Even higher background numbers (∼5 8-oxoGua Mbp−1) have been reported by an independent group for mouse lung.63 Previous data for levels of total endogenous oxidatively generated lesions suggest higher frequencies (abasic sites: 10–60 lesions Mbp−1 and 8-oxodG: 0.1–4 lesions Mbp−1) for tissue DNA of mice, rat or human origin (for reviews see ref. 54,64,65). Specifically, Nakamura and Swenberg, using the ASB (Aldehyde Reactive Probe-Slot-Blot) assay ,51 detected 10–60 AP sites Mbp−1 in DNA isolated from different rat tissues (not derived from skin). Similarly Atamna et al.,53 using the ARP (Aldehyde Reactive Probe) assay , detected ∼35 AP sites Mbp−1 in rat liver DNA, while Klungland et al.52 detected ∼3 8-oxoGua Mbp−1 using HPLC-ECD again in liver DNA. These high numbers of endogenous AP sites must be compared with the reported background values of 0.26 SSBs and alkali-labile sites Mbp−1 measured in human monocytes using the alkaline comet assay .66 Using 8-oxoGua electrochemical detection (ECD) of this lesion in DNA isolated from cultured human skin fibroblasts, Kaneko et al.67 reported much higher values of ∼2–10 lesions Mbp−1, depending on the ‘age’ of cells. Overall, there is still a discrepancy between the values provided by either HPLC-electrochemical detection or HPLC-tandem mass spectrometry (MS-MS) and that inferred from the assessment of formamidopyrimidine (Fpg)-sensitive sites by either the comet assay or the alkaline elution technique.65,68 It is likely, as proposed in the conclusions of the ESCODD research network, that HPLC measurements led to an overestimation by several-fold of the cellular background of 8-oxoGua due to the occurrence of artifactual oxidation of overwhelming normal DNA constituents. Therefore, the endogenous level of 8-oxoGua is more likely to be within the range of 1 to several lesions per 10 Mbp whereas the total number of oxidized bases is close to 2–4 modifications per Mbp. This number of total oxidatively generated lesions gives an expected minimum frequency of 0.4–1 oxybase clusters Mbp−1 based on the suggested ratio of 5 : 1 to 10 : 1 of total oxidative lesions to oxybase clusters.69 Certainly and as also stated by Boucher et al.,70 the role of artifactual DNA oxidation is very important but based on the above different group data, relatively high numbers of endogenous clusters in mammalian or human tissues cannot be excluded. This possibility is obviously very important for the biological significance of endogenous oxidative stress especially in the case of clustered DNA lesions with their proven high mutagenic potential.
000 lesions Gbp−1.51–54 Rodents or cancer patients exposed to radiotherapy have elevated levels of different types of oxidatively generated DNA damage (e.g., 8-oxodG or double strand breaks). These lesions have been detected in blood cells or tissues even after several weeks (1–24 weeks) post-irradiation, indicating an induced chronic oxidative stress.55–60 There are very limited data on the possible accumulation of OCDLs in human malignant cells or tissues and in mammalian/human tissues exposed to ionizing radiation. Gollapalle et al.60 showed for the first time that endogenous OCDLs can be detected in non-irradiated mice tissues at a steady state of ∼0.5–0.9 clusters Mbp−1 and that exposure of these tissues to ionizing radiation (X-rays) leads to an accumulation of DNA clusters detected 20 weeks after irradiation. Measurement of endogenous OCDLs in the human breast cancer line MCF-7 revealed elevated levels compared with the non-malignant MCF-10A. These cluster frequencies are higher than the ones detected for DNA isolated from human skin primary cell cultures (20–40 clusters Gbp−1 or 0.02–0.04 clusters Mbp−1).61 However, a completely different isolation and enzyme-treatment method was followed embedding DNA in agarose plugs and using transverse alternating field electrophoresis for DNA analysis while the different tissue origin may be another explanation for this discrepancy. Interestingly, Cadet et al.62 reported a number of 1–3 8-oxoGua Mbp−1 for DNA isolated from liver or kidney of young or old mice. Even higher background numbers (∼5 8-oxoGua Mbp−1) have been reported by an independent group for mouse lung.63 Previous data for levels of total endogenous oxidatively generated lesions suggest higher frequencies (abasic sites: 10–60 lesions Mbp−1 and 8-oxodG: 0.1–4 lesions Mbp−1) for tissue DNA of mice, rat or human origin (for reviews see ref. 54,64,65). Specifically, Nakamura and Swenberg, using the ASB (Aldehyde Reactive Probe-Slot-Blot) assay ,51 detected 10–60 AP sites Mbp−1 in DNA isolated from different rat tissues (not derived from skin). Similarly Atamna et al.,53 using the ARP (Aldehyde Reactive Probe) assay , detected ∼35 AP sites Mbp−1 in rat liver DNA, while Klungland et al.52 detected ∼3 8-oxoGua Mbp−1 using HPLC-ECD again in liver DNA. These high numbers of endogenous AP sites must be compared with the reported background values of 0.26 SSBs and alkali-labile sites Mbp−1 measured in human monocytes using the alkaline comet assay .66 Using 8-oxoGua electrochemical detection (ECD) of this lesion in DNA isolated from cultured human skin fibroblasts, Kaneko et al.67 reported much higher values of ∼2–10 lesions Mbp−1, depending on the ‘age’ of cells. Overall, there is still a discrepancy between the values provided by either HPLC-electrochemical detection or HPLC-tandem mass spectrometry (MS-MS) and that inferred from the assessment of formamidopyrimidine (Fpg)-sensitive sites by either the comet assay or the alkaline elution technique.65,68 It is likely, as proposed in the conclusions of the ESCODD research network, that HPLC measurements led to an overestimation by several-fold of the cellular background of 8-oxoGua due to the occurrence of artifactual oxidation of overwhelming normal DNA constituents. Therefore, the endogenous level of 8-oxoGua is more likely to be within the range of 1 to several lesions per 10 Mbp whereas the total number of oxidized bases is close to 2–4 modifications per Mbp. This number of total oxidatively generated lesions gives an expected minimum frequency of 0.4–1 oxybase clusters Mbp−1 based on the suggested ratio of 5 : 1 to 10 : 1 of total oxidative lesions to oxybase clusters.69 Certainly and as also stated by Boucher et al.,70 the role of artifactual DNA oxidation is very important but based on the above different group data, relatively high numbers of endogenous clusters in mammalian or human tissues cannot be excluded. This possibility is obviously very important for the biological significance of endogenous oxidative stress especially in the case of clustered DNA lesions with their proven high mutagenic potential.
The additional recent findings by Francisco et al.71 about the deficient processing of DSBs and OCDLs in MCF-7 (reduced BRCA1 expression) and HCC1937 (truncated BRCA1) signify the importance of the processing of complex DNA damage, especially in the case that at least one repair gene is mutated or one repair pathway is compromised. Accumulation of clusters can increase significantly the mutation frequency and overall genomic instability in the cells.
Acknowledgements
The author would like to thank Dr R. D. Stewart for helpful discussions and suggestions. This work has been supported by funds provided to Dr Georgakilas by the Biology Department of East Carolina University, a Research/Creative Activity Grant to A. Georgakilas and a College Research Award (East Carolina University).References
- D. L. Ellsworth, R. E. Ellsworth, M. N. Liebman, J. A. Hooke and C. D. Shriver, Lancet, 2004, 5, 753–758 CrossRef CAS.
- M. B. Kastan and J. Bartek, Nature, 2004, 432, 316–323 CrossRef CAS.
- S. P. Hussain, L. J. Hofseth and C. C. Harris, Nat. Rev. Cancer, 2003, 3, 276–285 CrossRef CAS.
- J. F. Ward, Int. J. Radiat. Biol., 1994, 66, 427–432 CrossRef CAS.
- D. T. Goodhead, Int. J. Radiat. Biol., 1994, 65, 7–17 CAS.
- B. Sutherland, P. V. Bennett, O. Sidorkina and J. Laval, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 103–108 CrossRef CAS.
- E. C. Friedberg, G. C. Walker and W. Siede, DNA Repair and Mutagenesis, ASM Press, Washington DC, 1995 Search PubMed.
- J. F. Ward, G. D. D. Jones and J. R. Milligan, Radiat. Prot. Dosim., 1994, 52, 271–276 CAS.
- J. F. Ward, Prog. Nucleic Acid Res. Mol. Biol, 1988, 35, 95–125 Search PubMed.
- J. F. Ward, Radiat. Res., 1981, 86, 185–195 CAS.
- J. F. Ward, Radiat. Res., 1985, 104, S103–S111 CrossRef CAS.
- B. M. Sutherland, P. V. Bennett, J. C. Sutherland and J. Laval, Radiat. Res., 2002, 157, 611–616 CrossRef CAS.
- M. Gulston, C. de Lara, T. Jenner, E. Davis and P. O'Neill, Nucleic Acids Res., 2004, 32, 1602–1609 CrossRef CAS.
- V. A. Semenenko and R. D. Stewart, Radiat. Res., 2004, 161, 451–457 CrossRef CAS.
- S. S. Wallace, Radiat. Res., 1998, 150(suppl.), S60–S79 CrossRef CAS.
- V. A. Semenenko and R. D. Stewart, Radiat. Res., 2005, 164, 194–201 CrossRef CAS.
- A. G. Georgakilas, P. V. Bennett, D. M. Wilson, III and B. M. Sutherland, Nucleic Acids Res., 2004, 32, 5609–5620 CrossRef CAS.
- S. Malyarchuk, K. L. Brame, R. Youngblood, R. Shi and L. Harrison, Nucleic Acids Res., 2004, 32, 5721–5731 CrossRef CAS.
- B. K. Singleton, C. S. Griffin and J. Thacker, Cancer Res., 2002, 62, 6263–6269 CAS.
- M. I. Kaplan, C. L. Limoli and W. F. Morgan, Radiat. Oncol. Invest., 1997, 5, 124–128 CrossRef CAS.
- L. D. Attardi, Mutat. Res., 2005, 569, 145–157 CrossRef CAS.
- Z. Somodi, N. A. Zyuzikov, G. Kashino, K. R. Trott and K. M. Prise, Int. J. Radiat. Biol., 2005, 81, 929–936 CrossRef CAS.
- J. Boer, J. Andressoo, J. de Wit, J. Huijmans, R. B. Beems, H. W. van Steeg, G. G. T. van der Horst, W. van Leeuwen, A. P. Themmen, M. Meradji and J. H. Hoeijmakers, Science, 2002, 296, 1276–1279 CrossRef CAS.
- T. Lindahl and R. D. Wood, Science, 1999, 286, 1897–1905 CrossRef CAS.
- G. L. Dianov, P. O'Neill and D. T. Goodhead, BioEssays, 2001, 23, 745–749 CrossRef CAS.
- M. A. Chaudhry and M. Weinfeld, J. Biol. Chem., 1997, 272, 15650–15655 CrossRef CAS.
- L. Harrison, Z. Hatahet and S. Wallace, J. Mol. Biol., 1999, 290, 667–684 CrossRef CAS.
- A. A. McKenzie and P. R. Strauss, Biochemistry, 2001, 40, 13254–13261 CrossRef.
- M. H. David-Cordonnier, S. M. T. Cunniffe, I. D. Hickson and P. O'Neill, Biochemistry, 2002, 41, 634–642 CrossRef CAS.
- A. G. Georgakilas, P. V. Bennett and B. M. Sutherland, Nucleic Acids Res., 2002, 30, 2800–2808 CrossRef CAS.
- G. Eot-Houllier, S. Eon-Marchais, D. Gasparutto and E. Sage, Nucleic Acids Res., 2005, 33, 260–271 CrossRef CAS.
- S. M. Cunniffe, M. E. Lomax and P. O'Neill, DNA Repair, 2007 DOI:10.1016/j.dnarep.2007.07.003.
- A. G. Georgakilas, P. V. Bennett and B. M. Sutherland, Conference proceedings of 12th International Congress of Radiation Research (ed. ICRR), Brisbane, Australia, 2003, pp. 35–36 Search PubMed.
- H. Budworth, G. Matthewman, P. O'Neill and G. L. Dianov, J. Mol. Biol., 2005, 351, 1020–1029 CrossRef CAS.
- G. Slupphaug, B. Kavli and H. E. Krokan, Mutat. Res., 2003, 531, 231–251 CrossRef CAS.
- A. Klungland, M. Hoss, D. Gunz, A. Constantinou, S. G. Clarkson, P. W. Doetsch, P. H. Bolton, R. D. Wood and T. Lindahl, Mol. Cell, 1999, 3, 33–42 CrossRef CAS.
- B. Demple and M. S. DeMott, Oncogene, 2002, 21, 8926–8934 CrossRef CAS.
- M. T. A. Ocampo, W. Chaung, D. R. Marenstein, M. K. Chan, A. Altamirano, A. K. Basu, R. J. Boorstein, R. P. Cunningham and G. W. Teebor, Mol. Cell. Biol., 2002, 22, 6111–6121 CrossRef CAS.
- D. R. Marenstein, M. K. Chan, A. Altaminaro, A. K. Basu, R. J. Boorstein, R. P. Cunningham and G. W. Teebor, J. Biol. Chem., 2003, 278, 9005–9012 CrossRef CAS.
- L. Wiederhold, J. B. Leppard, P. Kedar, F. Karimi-Busheri, A. Rasouli-Nia, M. Weinfeld, A. E. Tomkinson, T. Izumi, R. Prasad, S. H. Wilson, S. Mitra and T. K. Hazra, Mol. Cell, 2004, 15, 209–220 CrossRef CAS.
- E. Riballo, M. Kuhne, N. Rief, A. Doherty, G. C. M. Smith, M. J. Recio, C. Reis, K. Dahm, A. Fricke, A. Krempler, A. R. Parker, S. P. Jackson, A. Gennery, P. A. Jeggo and M. Lobrich, Mol. Cell, 2004, 16, 715–724 CrossRef CAS.
- M. H. David-Cordonnier, S. Boiteux and P. O'Neill, Biochemistry, 2001, 40, 11811–11818 CrossRef CAS.
- G. Eot-Houllier, M. Gonera, D. Gasparutto, C. Giustranti and E. Sage, Nucleic Acids Res., 2007, 35, 3355–3366 CrossRef CAS.
- H. Budworth and G. L. Dianov, J. Biol. Chem., 2003, 278, 9378–9381 CrossRef CAS.
- S. Malyarchuk and L. Harrison, J. Mol. Biol., 2005, 345, 731–743 CrossRef CAS.
- G. L. Dianov, T. V. Timchenko, O. I. Sinitsina, A. V. Kuzminov, O. A. Medvedev and R. I. Salganik, Mol. Gen. Genet., 1991, 225, 448–452 CAS.
- J. O. Blaisdell and S. Wallace, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7426–7430 CrossRef CAS.
- N. Yang, H. Galick and S. S. Wallace, DNA Repair, 2004, 3, 1323–1334 CrossRef CAS.
- L. Harrison, K. L. Brame, L. E. Geltz and A. M. Landry, DNA Repair, 2006, 5, 324–335 CrossRef CAS.
- D. Tsao, P. Kalogerinis, I. Tabrizi, M. Dingfelder, R. D. Stewart and A. G. Georgakilas, Radiat. Res., 2007, 168, 87–97 CrossRef CAS.
- J. Nakamura and A. J. Swenberg, Cancer Res., 1999, 59, 2522–2526 CAS.
- A. Klungland, I. Rosewell, S. Hollenbach, E. Larsen, G. Daly, B. Epe, E. Seeberg, T. Lindahl and D. E. Barnes, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 13300–13305 CrossRef CAS.
- H. Atamna, I. Cheung and B. N. Ames, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 686–691 CrossRef CAS.
- R. De Bont and N. van Larebeke, Mutagenesis, 2004, 19, 169–185 CrossRef CAS.
- I. H. Zwingmann, I. J. Welle, J. J. M. Engelen, P. A. E. L. Schilderman, J. M. A. de Jong and J. C. S. Kleinjans, Mutat. Res., 1999, 431, 361–369 CrossRef CAS.
- M. E. C. Robbins, W. Zhao, C. S. Davis, S. Toyokuni and S. M. Bonsib, Micron, 2002, 33, 133–141 CrossRef CAS.
- C. N. Coleman, W. F. Blakely, J. R. Fike, T. J. MacVittie, N. F. Metting, J. B. Mitchell, J. E. Moulder, R. J. Preston, T. M. Seed, H. B. Stone, P. J. Tofilon and R. S. L. Wong, Radiat. Res., 2003, 159, 812–834 CrossRef CAS.
- R. Neal, R. H. Matthews, P. Lutz and N. Ercal, Free Radical Biol. Med., 2003, 34, 689–695 CrossRef CAS.
- M. Lobrich, N. Rief, M. Kuhne, M. Heckmann, J. Fleckenstein, C. Rube and M. Uder, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8984–8989 CrossRef.
- E. Gollapalle, R. Wong, R. Adetolu, D. Tsao, D. Francisco, G. Sigounas and A. G. Georgakilas, Radiat. Res., 2007, 167, 207–216 CrossRef CAS.
- P. V. Bennett, N. L. Cuomo, S. Paul, S. T. Tafrov and B. M. Sutherland, Free Radical Biol. Med., 2005, 39, 832–839 CrossRef CAS.
- J. Cadet, T. Douki, S. Frelon, S. Sauvaigo, J. P. Pouget and J. L. Ravanat, Free Radical Biol. Med., 2002, 33, 441–449 CrossRef CAS.
- L. Risom, C. Lundby, J. J. Thomsen, L. Mikkelsen, S. Loft, G. Friis and P. Møller, Free Radical Res., 2003, 37, 957–966 CrossRef CAS.
- J. Cadet, T. Douki, D. Gasparutto and J. Ravanat, Mutat. Res., 2003, 531, 5–23 CrossRef CAS.
- ESCODD (European Standards Committee on Oxidative DNA Damage), C. M. Gedik and A. Collins, FASEB J., 2005, 19, 82–84 CAS.
- J.-P. Pouget, J.-L. Ravanat, T. Douki, M.-J. Richard and J. Cadet, Int. J. Radiat. Biol., 1999, 75, 51–58 CrossRef CAS.
- T. Kaneko, S. Tahara, T. Taguchi and H. Kondo, Mutat. Res., 2001, 487, 19–30 CAS.
- A. R. Collins, J. Cadet, L. Möller, H. E. Poulsen and J. Viña, Arch. Biochem. Biophys., 2004, 423, 57–65 CrossRef CAS.
- B. M. Sutherland, P. V. Bennett, O. Sidorkina and J. Laval, Biochemistry, 2000, 39, 8026–8031 CrossRef CAS.
- D. Boucher, I. Testard and D. Averbeck, Radiat. Environ. Biophys., 2006, 45, 267–276 CrossRef CAS.
- D. Francisco, P. Peddi, J. Hair, B. A. Flood, G. Sigounas and A. G. Georgakilas, Free Radical Biol. Med., 2007 DOI:10.1016/j.freeradbiomed.2007.10.045.
- H. Nikjoo, P. O'Neill, E. W. Wilson and D. Goodhead, Radiat. Res., 2001, 156, 577–583 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |