Systematic screens for human disease genes, from yeast to human and back
Fabiana
Perocchi†
,
Eugenio
Mancera†
and
Lars M.
Steinmetz
*
European Molecular Biology Laboratory, Meyerhofstrasse 1, 69117 Heidelberg, Germany. E-mail: larsms@embl.de
First published on 25th September 2007
Abstract
Systematic screens for human disease genes have emerged in recent years, due to the wealth of information provided by genome sequences and large scale datasets. Here we review how integration of genomic data in yeast and human is helping to elucidate the genetic basis of mitochondrial diseases. The identification of nearly all yeast mitochondrial proteins and many of their functional interactions provides insight into the role of mitochondria in cellular processes. This information enables prioritization of the candidate genes underlying mitochondrial disorders. In an iterative fashion, the link between predicted human candidate genes and their disease phenotypes can be experimentally tested back in yeast.
 Fabiana Perocchi | Fabiana Perocchi grew up in Rome, Italy. She earned a BS in Biology in 2003 from the University of Tor Vergata in Rome and is currently completing her PhD in Functional Genomics at EMBL, Heidelberg. Her thesis work, under the guidance of Dr Lars M. Steinmetz, focused on the identification of mitochondrial components and the analysis of their transcriptional regulation and functional interactions through the integration of genomic technologies in yeast. |
 Eugenio Mancera | Eugenio Mancera grew up in Guadalajara, Mexico, and received his BS degree in biology from UNAM in Mexico City in 2002. He earned his MS in molecular and cellular biology from the University of Heidelberg, Germany, in 2005. Thereafter he moved to the group of Lars Steinmetz at EMBL where he is currently doing his PhD. |
 Lars M. Steinmetz | Lars Steinmetz grew up in Germany, Switzerland and the USA. He earned a BS in molecular biophysics and biochemistry from Yale University and a PhD in genetics from Stanford University. Since 2003 he has been a group leader in gene expression and developmental biology at EMBL, Heidelberg. His group focuses on using and developing functional genomic approaches and high-throughput methods in yeast to study complex traits and the mitochondrial organelle at a systems level. |
Introduction
Genetic factors contribute to nearly all diseases, either directly—through, for example, malfunctioning genes—or by influencing our susceptibility to disease. Identifying the genetic variants associated with a disorder is often the first step towards an understanding of the molecular basis of the disease, and frequently leads to the development of diagnostic methods and effective treatments. Since the development of positional cloning about 20 years ago, more than 2400 genes with mutations causing mainly Mendelian disorders have been discovered (http://www.ncbi.nlm.nih.gov/omim/).1 However, the task ahead is not straightforward and the disease genes identified so far may be the “easy ones”.2 The Online Mendelian Inheritance in Man (OMIM) database reports close to 3700 Mendelian or suspected-Mendelian diseases with as yet unidentified genetic underpinnings. Even more demanding will be the identification of the responsible genes for common complex-trait diseases, such as type 1 diabetes, obesity and multiple sclerosis.2 Complexities arise from the lack of simple correspondence between phenotype and genotype caused by multifactorial inheritance, epistasis, pleiotropy, genetic heterogeneity and gene-environment interactions, among other factors.3Genomics offers new opportunities to accelerate the identification of genes and the molecular mechanisms underlying human diseases. At its fundamental level, identifying a disease gene requires establishing a link between phenotype and genotype. In candidate gene approaches, knowledge about gene function is employed to prioritize genes in the process of associating them with a disease. New approaches, which investigate properties of more than one gene at a time and systematically transfer this information across species, permit a more comprehensive prediction of gene function and informed prioritization of disease candidate genes. These approaches integrate information from sequence conservation, gene expression, protein interactions and gene knockout effects, among other properties.
The yeast Saccharomyces cerevisiae is a powerful model organism because of its comparatively simple genome, its genetic tractability and a range of unsurpassed genetic tools that can be applied. These advantages enabled the development of numerous genomic technologies in yeast, which have contributed to identifying human disease genes.4 One example is the identification of SURF–1, a gene involved in Leigh’s disease, a condition associated with cytochrome c oxidase deficiency (LD[COX–]). SURF–1 was singled out among genes within the linkage interval for Leigh disease because its yeast homologue encodes a product that is targeted to the mitochondria and, when mutated, impairs mitochondrial respiration.5 This kind of information transfer is possible for several processes that are conserved between yeast and human, including DNA replication, recombination and repair, RNA transcription and translation, intracellular trafficking, general metabolism, and mitochondrial function and biogenesis.6
The aim of this review is to highlight how advances in yeast genomics have been applied to the systematic analysis of human disease genes. We place a particular focus on the mitochondrial organelle, as it plays a central role in disease and cellular metabolism, and its study serves as a stepping-stone for the analysis of the entire cell. Many of the genetically uncharacterized diseases are likely linked to mitochondrial dysfunction. However, an impediment to finding the causative genes is that only one-third of the human mitochondrial proteome has been characterized in detail.7 This review will detail how large-scale data integration has catalyzed identification of nearly all yeast mitochondrial proteins and many of their functional interactions, as well as how this knowledge has aided the search for new disease genes (Fig. 1). In turn, the review also explores how the human candidate genes proposed can be tested back in yeast, where cell-based assays can be performed in a high-throughput manner.
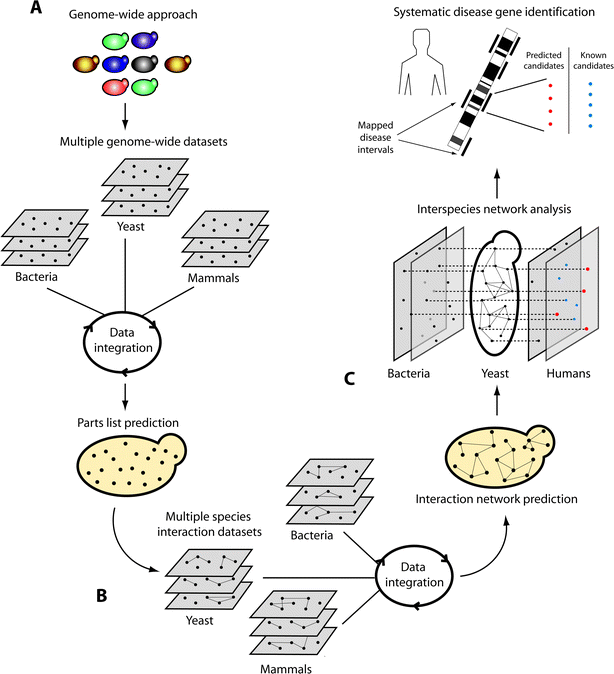 | ||
| Fig. 1 Systematic approach to identify disease genes: from yeast to human. (A) For any biological process, the prediction of parts (nodes) and (B) their interactions (lines) can be achieved by the integration of multiple datasets across several organisms. (C) The ensuing network enables extracting properties of its proteins, for example their conservation (doted lines) and interactions, and can be used for prioritizing disease candidate genes (red nodes). By searching mapped disease intervals in the human genome for genes previously known to be implicated in the process under study (blue nodes) as well as components newly predicted to be part of the process from the integrative analysis in yeast (red nodes), a list of genes for further investigation can be identified. These genes are then examined for mutations responsible for disease. | ||
Mitochondrial biology and disease
Mitochondria are best known as the “powerhouses” of eukaryotic cells. Besides production of ATP, the organelle is a center for the metabolism of nitrogen, heme and steroid hormones, as well as for the storage of calcium, iron and copper. While the biochemistry of mitochondria has been subject to decades of research, more recent insights into a mitochondrial role in signal transduction, oxygen sensing, antiviral responses and programmed cell death are expanding our view of mitochondria beyond their existence as isolated compartments of cells.8 The organelle is intimately involved in cell sensing and signaling and it undergoes dynamic morphological changes which are critical in disease, aging and development.9Mitochondrial biogenesis requires the coordinated action of two genomes: the mitochondrial powerhouses and the nuclear.10,11 Since its origin as an aerobic prokaryote that was engulfed in a eukaryotic cell about 2 billion years ago,12 the mitochondrion has lost most of the ancestral bacterial genome.13 Notably, only 13 proteins in human and 8 proteins in yeast are encoded by the mitochondrial DNA (mtDNA). Nevertheless, roughly 1500 proteins are expected to localize within human mitochondria14 and about half this number are needed for mitochondria in the simpler eukaryotic organism yeast15 (Table 1). As the majority of mitochondrial proteins are encoded by nuclear genes, a comprehensive knowledge of this organelle demands the integration of information about its resident proteins and proteins localized outside the organelle (e.g.transcriptional regulators, metabolic branches and signaling pathways involved in mitochondrial function).
| Database | Description | Website |
|---|---|---|
| MitoDat | Database of nuclear-encoded proteins involved in mitochondrial biogenesis and function | http://www-lecb.ncifcrf.gov/mitoDat/ |
| MITOMAP | Database of human mitochondrial genomic sequences, including polymorphisms and mutations | http://www.MITOMAP.org |
| Human Mitochondrial Genome Database | Database of human mitochondrial genomes | http://www.genpat.uu.se/mtDB |
| HMPDb (Human Mitochondrial Protein Database) | Database of human mitochondrial and nuclear encoded proteins involved in the biogenesis and function of the organelle | http://bioinfo.nist.gov/hmpd/ |
| MitoProteome | Database of human mitochondrial protein sequences, mtDNA- and nuclear-encoded | http://www.mitoproteome.org/ |
| MitoP2 | Database of mitochondria-related genes, proteins, and diseases. MitoP2 contains numerous datasets relevant to the study of mitochondrial proteins in yeast, mouse, Arabidopsis, Neurospora and human | http://www.mitop.de |
| YMPD (Yeast Mitochondrial Protein Database) | Database containing a curated list of mitochondrial proteins in budding yeast. The list includes mtDNA-encoded proteins and nuclear DNA-encoded proteins that localize in mitochondria as well as outside it (nuclear or cytoplasmic) with a role in mitochondrial biogenesis or function | http://bmerc-www.bu.edu/projects/mito/ |
| YDPM (Yeast Deletion Project and Mitochondria) | Database supporting the yeast deletion project and mitochondrial proteomics and expression projects to study mitochondrial protein composition and transcriptional regulation | http://www-deletion.stanford.edu/YDPM/ |
| OGRe (Organellar Genome Retrieval) | Database containing the complete mitochondrial genome sequences for over 250 Metazoan species | http://www.bioinf.man.ac.uk/ogre |
| MitoDrome | Database of Drosophila melanogasternuclear genes encoding mitochondrial | http://www2.ba.itb.cnr.it/MitoDrome/ |
| MiGenes | Database of mitochondrial proteins from several organisms curated using gene ontology | http://www.pharm.stonybrook.edu/migenes/ |
Due to the genetic dichotomy of the mitochondrial proteome, mitochondrial diseases can be caused by mutations in both mtDNA- and nuclear DNA-encoded genes, and follow maternal, nuclear or a combined mode of inheritance.16 While hundreds of point mutations and large-scale rearrangements in mtDNA have been successfully associated with several maternally inherited diseases, mutations in mtDNA are found in less than 15% of patients with symptoms consistent with a mitochondrial dysfunction.17 Culprits of mitochondrial disorders, therefore, likely predominate in nuclear genes.18 So far, mutations in over 150 nuclear genes that encode proteins localized to the organelle have been implicated in mitochondrial diseases7 (Table 1).
Due to the wide range of functions carried out by mitochondria, the spectrum of mitochondrial disorders is broad. Epidemiological studies suggest that overall mitochondrial disorders are among the most common inherited diseases, with a minimum prevalence of 1 in 5000 individuals.19 Dysfunctions of the mitochondrial oxidative phosphorylation pathway (OXPHOS) can drastically affect organs with high energy demands, such as brain and muscles, but the ubiquitous tissue requirement for ATP can result in almost any organ being affected.20 To name a few examples: defects in the electron transport chain are common in mitochondrial encephalomyopathies;21 increased generation of free oxygen radicals (ROS) is associated with neuronal cell death, Parkinson's disease, Alzheimer's disease, carcinogenesis, and aging;11,22 and dysregulation of glucose and mitochondrial fatty acid metabolism are implicated in type II diabetes and obesity.23
Integrative genomics to define the mitochondrial parts-list
Genome-scale approaches in cell biology, biochemistry, genetics and computational biology have catalyzed the identification of mitochondrial proteins in several organisms.24 Specific examples of these technologies include analysis of subcellular localization,25,26 deletion phenotypes,27,28 gene expression,29–31 mass spectrometry-based proteomics30–34 and computational prediction of mitochondrial signal peptides.35 Using fluorescence microscopy of proteins genetically tagged with either GFP or an immunologically detectable epitope, over 10% of the yeast proteome could be localized to mitochondria in yeast.25 Organellar proteomics, which relies on the isolation, separation and identification of proteins in organelles,36,37 has assigned over 800 proteins to yeast mitochondria30,32 and roughly 700 to human heart mitochondria.33,38 Analysis of yeast deletion mutants defective in respiration and measurement of gene transcript levels during respiration expanded the catalog of mitochondrial proteins to include proteins from other compartments that are essential for the regulation, processing and turnover of mitochondria.27–30,39To complete the inventory of mitochondrial proteins, genome-wide approaches need to be combined so as to complement their strengths and compensate for their shortcomings24 (Fig. 1A). This has been shown in yeast, mouse and human.7,30,31,40,41 Each individual approach is biased towards different functional subsets of proteins, as illustrated by a comparison of proteomic and deletion datasets in yeast.30 Mass-spectrometry based proteomics exhibits bias towards proteins that are soluble and abundant, and therefore it does not encompass the wide range of hydrophobicity, copy number, molecular weight, isoelectric point and membrane association found in mitochondrial proteins.42 Furthermore, this method has difficulty detecting proteins that execute important mitochondrial functions but do not localize stably to the organelle, such as transcription factors and cytoplasmic regulators. Instead, these proteins can be captured by single gene deletion phenotype screening, which identifies genes that function in mitochondrial processes irrespective of their localization.28 However, deletion phenotype analysis overlooks genes that do not have a specific knockout phenotype because they are redundant or essential, which is the case for components of mitochondrial outer membrane transport.28,41 In addition high-throughput datasets are prone to false discoveries. For example, mitochondrial proteomic datasets often include highly abundant proteins from other cellular compartments, such as proteins of the cytosolic ribosome and the cytoskeleton, which are possible contaminants in mitochondrial preparations.36,43
Due to the diversity of genome-wide data available for yeast, it is possible to systematically assess both the coverage and accuracy of each data type. This was addressed for 22 genome-wide datasets, mostly centered on mitochondria, which showed great heterogeneity in the number and type of mitochondrial proteins identified.30 While measurements of mRNA levels during growth on non-fermentable media captured only 10% of previously-known mitochondrial localized proteins, deletion phenotype screening, microscopy-based localization studies and mass-spectrometry-based proteomics covered from 50 to 80%.30 Remarkably, although some datasets show relatively high coverage, they have modest overlap due to biases in the methods. Mass-spectrometry-based proteomics and deletion phenotype screening, for instance, shared only 30% of their protein hits. Therefore, a combined analysis of several datasets can increase coverage by considering proteins that have been identified by multiple, but not necessarily all methods.
One means of systematic data integration employs algorithms which are trained to discriminate between mitochondrial and non-mitochondrial proteins using a benchmark dataset. A set of proteins unambiguously localized to mitochondria by single-gene studies can provide a high confidence reference set for benchmarking7,44 (Table 1). The most comprehensive integration of genome-wide data for the prediction of mitochondrial components has been performed in yeast.7,30,41 Machine-learning approaches have been applied to integrate over 22 genome-wide datasets ranging from expression, protein localization and deletion phenotype to computational predictions based on signal peptides. This has resulted in a score representing the probability of mitochondrial localization for every gene in the genome.7,30,41 So far, integrative analysis has covered over 90% of the previously known mitochondrial proteome and has yielded high-confidence predictions for hundreds of previously uncharacterized components.41
Since the size, shape, number and metabolism of mitochondria exhibit tissue specificity, completing the annotation of the mammalian mitochondrial proteome will require tissue-based identification of mitochondrial proteins.31,45 Less than half of the mitochondrial proteins from brain, liver, heart and kidney were found to be expressed across all cell types by proteomic analysis in mice.31 Characterization was considerably improved by integrating genome-wide datasets from different organisms and mammalian cell types.40 Predictions of mitochondrial targeting signals were combined with protein domain enrichments, yeast homology, ancestry, proteomics, co-expression and transcriptional induction during mitochondrial biogenesis to expand the catalog of 654 known human mitochondrial proteins by 490 novel predictions with an estimated false discovery rate of 10%.40
Despite the diversity of integrated datasets in yeast and human, a few known mitochondrial proteins have remained undetected by integrative approaches; these are mainly proteins with dual localization, low abundance and low solubility. This indicates a need for new genome-wide datasets that identify both transient and resident proteins and that reflect the changes in the mitochondrial proteome in response to a wider range of environmental conditions. Finally, through an iterative process, predicted mitochondrial proteins, once experimentally confirmed, can be then added to the reference set, improving future data integration.
Integrative genomics to define the mitochondrial interactome
Understanding the complexity of a whole organelle requires knowledge about how its individual parts interact to build up the hierarchical organization of the organelle. The most basic level of this organization is represented by its functional modules, which are groups of proteins that work together, for example protein complexes, transcription factors and gene targets, or enzymes in a pathway.46 Identifying these modules requires establishing the type, timing and mode of interaction among all the parts of the system. The reconstruction of a global interaction network allows (1) placement of both known and uncharacterized components into functional contexts from which hypotheses can be generated about gene function (Fig. 1B); and (2) prediction of phenotypic changes in response to perturbations of cellular state, such as gene mutations or environmental stresses.Yeast has been the model organism of choice for global mapping of gene, protein and metabolic networks, largely because of the abundance and quality of diverse high-throughput interaction data. A large number of gene-expression profiles have been generated using both genetic (e.g. deletion) and environmental (e.g. media type) perturbations. These allow the grouping of genes into co-expression modules that may share a common role and similar transcriptional regulation.47,48 Key regulators of gene expression and their target genes (protein–DNA interactions) have been identified genome-wide in yeast by combining chromatin immunoprecipitation (ChIP) with DNA microarray analysis (ChIP-on-chip).49 Screening double deletion mutants for cell death or reduced fitness has identified genetic interactions among genes whose products buffer one another and are involved in the same biological process.50 Functional proteomic technologies developed or implemented in yeast, such as yeast two-hybrid screens, large-scale protein-tagged affinity purifications and protein chips, resulted in an en masse detection of transient binary protein–protein interactions,51 stable physical protein complexes52,53 and substrate specificity of two-thirds of yeast kinases, as well as interactions among protein and lipids, and proteins and small molecules.54
Interactions can also be computationally predicted. The wealth of genomic information available for several organisms has driven the development of computational gene context analyses that infer functional associations based on the comparison of multiple genomes.55 The assumption is that proteins are most likely to interact if: (1) their genes are either present or absent together across genomes (gene co-occurrences or phylogenetic profiles);56 (2) a gene fusion event occurred in other species;57 or (3) the genes are conserved in physical proximity in phylogenetically distant genomes (gene neighborhood).58 Moreover, automatic methods for extracting different types of associations from scientific literature (text mining) have been used to infer biological interactions.59
Defining the mitochondrial parts list cannot rely on a sole interaction data source, and neither can a comprehensive characterization of interaction networks (Fig. 1B). Using manually-curated catalogues of known binary interactions (e.g.protein complexes)60 and pathways,61 systematic comparisons of interaction data types show that the interactions predicted with the highest accuracy are those supported by more than one dataset.62,63 To increase sensitivity and to improve confidence in predicted protein interactions, computational tools have been developed which integrate many approaches.64,65 Integrative approaches can be enhanced further by collecting and transferring interaction information across multiple species, based on the assumption that conserved proteins tend to have conserved interactions.65
Integrative analysis of heterogeneous yet complementary data has been a key strategy for reconstructing the mitochondrial interactome, given that mitochondrial metabolism is highly condition-dependent in yeast and tissue-specific in human.66 In particular, many mitochondrial pathways, like respiration, are repressed when yeast is grown in the presence of glucose (rich media). Since most of the protein interaction data have been generated in rich media, individual datasets are especially scarce for interactions between mitochondrial proteins. As an example, two genome-wide screens for protein complexes provided valuable insights into cellular machineries for over 70% of the yeast proteome, but well-known mitochondrial protein complexes such as the respiratory chain complexes were not detected52. Similar constraints arise with in silico predictions, the majority of which are based on comparisons of bacterial genomes. Only 13% of the yeast mitochondrial proteome appears to be ancient and evolutionarily conserved in proteobacteria, limiting the number and type of functional associations that can be identified through inference from bacterial interactions.41 Text mining, however, can assist in completing annotation of the mitochondrial interactome, given that most results from over 50 years of biochemical and genetic studies on mitochondrial proteins and their interactions are stored in the primary literature.41
An initial version of the yeast mitochondrial network has recently been reconstructed from over 15 data sources, including physical protein interactions, mRNA co-expression associations, functional associations from literature mining, and genome context analyses.41 The coverage of annotated mitochondrial protein complexes and metabolic pathways was improved by the integration of functional networks from about 200 organisms through inter-species data transfer.41 A total of 876 proteins, both known and predicted as mitochondrial, were placed into 164 functional modules, providing the first clue about the function of over 150 uncharacterized mitochondrial components. Moreover, by reconstructing the mitochondrial network in the cellular context, the network could be used to formulate hypotheses on the proteins that mediate cross-talk between mitochondria and other cellular compartments.41,66
Although high-throughput protein–protein interaction assays can be applied to mammals, most of these assays are not yet scalable to the whole human interactome.67 Therefore, the application of integrative strategies in human remains limited.62,68 Nevertheless, the integration of data from the mitochondrial genome, the heart mitochondrial proteome, the annotated human genome, and literature on mitochondrial metabolism enabled reconstruction of a human mitochondrial metabolic network comprised of 189 biochemical reactions and 230 metabolites.69 Furthermore, genome-scale atlases of RNA expression across diverse tissues have been used to measure transcriptional coexpression of uncharacterized components with known mitochondrial genes.31 This co-expression network grouped 386 genes into functional modules that share functional and regulatory mechanisms.31
As interactions often remodel in response to genetic or environmental stresses, it will be an informative next step to investigate the way in which mitochondrial modules change in composition, mode of action and regulation over time. A key idea is that diseases are caused by the operation of perturbed networks. Through a comparison of normal and diseased networks, critical nodes (proteins) can be identified which, if modulated, are likely to reconfigure the perturbed network structure toward its normal state or specifically kill the diseased cells.70 Moreover, from the integration of multiple types of networks (e.g. physical-interaction, regulatory, metabolic networks), cellular responses to genetic or environmental perturbations can be predicted, as has been reported for bacteria and yeast.71
Systematic analysis of disease genes
Positional cloning is commonly invoked for genetic mapping of human diseases. However, due to limited recombination resolution, the linkage of disease phenotypes to genetic loci often results in genomic intervals that contain many candidate genes. Patient samples and screening costs often constitute limiting factors to pinpointing disease genes. Any knowledge about the biological process in which a gene product is implicated can be used to guide the selection of candidate genes (Fig. 1C). As an example, a genome-wide association study had successfully mapped a form of autosomal recessive cytochrome c oxidase (COX) deficiency, known as Leigh syndrome French-Canadian type (LSFC), to a chromosomal region containing 30 known and predicted genes.72 The clinical manifestations and biochemical features of LSFC had suggested that mitochondrial dysfunction underlies this disorder, but none of the genes encoding known mitochondrial proteins were found within the disease interval. Since only a third of the mitochondrial proteome was known at that time, mitochondrial gene products were overlooked. A few years later, the integration of functional data of mRNA expression and subcellular localization identified new human mitochondrial components73, and named LRPPRC as the top-ranking candidate gene within the LSFC linkage region, based on its observed mitochondrial localization and the remarkably high co-regulation with known mitochondrial genes. Subsequent sequencing and genotyping analysis provided the genetic evidence that LRPPRC is responsible for LSFC. Such an example illustrates that completing the catalog of mitochondrial proteins holds great potential to accelerate the identification of mitochondrial disease genes.Given the successes in the identification and functional characterization of mitochondrial proteins in yeast, it is beneficial to integrate orthology to known yeast mitochondrial proteins into the analysis of mitochondrial diseases (Fig. 1C). Indeed, a dataset based on homology to yeast mitochondrial proteins performed second-best in predicting human mitochondrial proteins, outperforming proteomics and co-expression datasets in mammals40. Moreover, by exploiting orthology to yeast genes, mitochondrial protein predictions in yeast have been systematically combined with genetic linkage mapping data of mitochondrial disorders.28 This approach prioritized 14 genes among a few hundred candidates annotated in a linkage interval associated with seven putative human mitochondrial disorders. Clearly the transfer of information from yeast to human is limited to conserved mitochondrial proteins. Human mitochondrion is a more complex system with a predicted proteome two times larger than yeast.15 Furthermore, mechanisms such as alternative splicing that are common in human can increase polypeptide diversity. Nevertheless, using yeast orthology has been extremely powerful.
Recently, an integrative approach that directly predicted mitochondrial proteins in human, prioritized eight candidates among 151 genes annotated in a genomic locus associated with hepatic mtDNA depletion syndrome (MDDS).40,74 Among the eight candidates, the gene MPV17 was shown to have a causative mutation, which most likely causes MDDS74. Both MPV17 and LRPPRC have orthologs in yeast that are characterized as mitochondrial proteins. In fact, further elucidation of the pathogenetic mechanism underlying MDDS was achieved through experimental analysis in yeast.74 Similarly, early clues on the role and localization of LRPPRC in human came from its orthology to a well-known yeast mitochondrial translational activator of COX1mRNA, PET309.75
Apart from mitochondrial localization, additional properties of yeast orthologs can be used to prioritize candidate genes for mitochondrial diseases (Fig. 1C). For a significant fraction of the known mitochondrial disease genes, the yeast orthologs have α-proteobacterial ancestry and a mild deletion phenotype change during respiration.41 In particular, among the yeast mitochondrial proteins with proteobacterial ancestry and a non-severe growth defect of the mutant strain, 25 have orthologs in human of which 8 have already been identified as genes involved in mitochondrial disorders. Given that lethal or ‘petite’ phenotypes (small colony size) are under-represented, it is likely that essential genes for yeast mitochondrial function correspond to loss-of-function mutants incompatible with human development.41
Further correlations can be extracted from comparison of disease genes and yeast protein interaction partners. The interconnectivity between disease-causing genes can identify additional candidates based on the assumption that diseases which share a common biochemical mechanism could be caused by mutations in one of several genes that interact in a pathway or protein complex.76 Recently, through the reconstruction of a yeast mitochondrial network, human mitochondrial disease genes have been analyzed for their functional relatedness in yeast.41 The orthologs of human disease genes were found to cluster together. Moreover, human disease genes that had yeast orthologs enriched in certain functional modules showed similarities in clinical symptoms. These findings enable ranking of candidate genes for specific mitochondrial disorders even in the absence of genetic linkage data. In principle new patients could be associated with known mitochondrial disease genes based on phenotype similarity. In addition to the known disease genes, the candidates to be screened in the patients could be extended to interactors of these disease genes. In human, the reconstruction of a network has the potential to explain even complex mechanisms of pathogenesis. As shown for human spinocerebellar ataxias,77 highly interconnected proteins reveal key players of pathogenesis that could be targeted for therapy. Finally, the candidate genes proposed can be tested back in yeast, where high-throughput cell-based assays are feasible.
Testing of human mitochondrial disease genes in yeast
Experimental evaluation of the role of candidate genes in human disease can be achieved by heterologous expression of human proteins in yeast (Fig. 2). Human cDNA was expressed in yeast for the first time two decades ago.78 The objective of these initial studies was to identify human homologs of known yeast genes, exploiting the ability of many human gene products to restore growth of their respective yeast null mutant. This method, known as functional complementation (Fig 2A), has also rapidly become effective for revealing the role of mutations in the performance of human enzymes.79 So far, several studies have applied yeast functional complementation assays to testing candidate disease genes for human mitochondrial disorders.80–82 Two illustrative examples involve the nuclear-encoded genes COX10 and BCS1L. A mutation in COX10 was associated with tubulopathy and leukodystrophy,81 and several polymorphisms in BCS1L were related to heterogeneous clinical presentations including GRACILE syndrome.82 In both cases, yeast complementation assays were central in defining the role of the genes and their polymorphisms in mitochondrial disorders when the mutant alleles, unlike the wild-type, were unable to restore growth of their respective yeast null mutants in non-fermentable medium.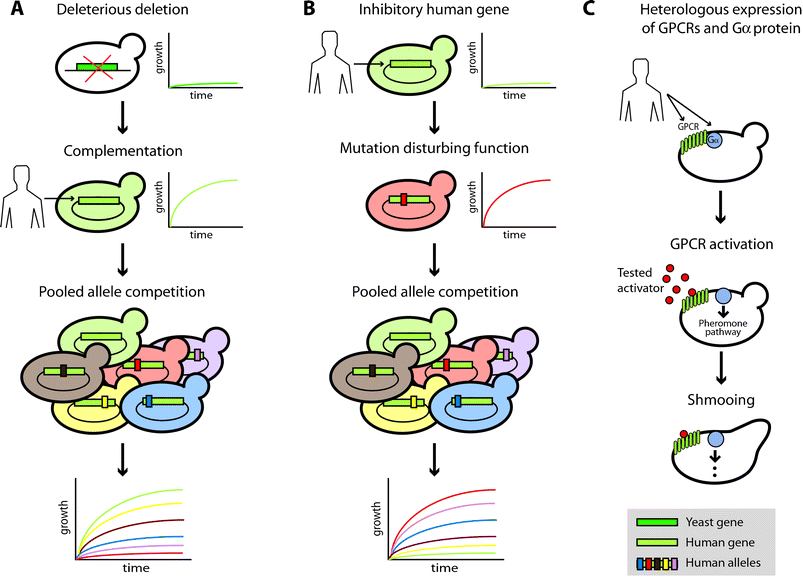 | ||
| Fig. 2 Evaluating disease candidate genes by heterologous expression in yeast. (A) Scheme of functional complementation to test the functional effect of different polymorphisms in a human gene. (B, C) Examples of alternative yeast cell-based assays to study human genes that may not have a yeast ortholog. (B) Scheme of a yeast growth inhibition assay to test the effect of polymorphisms in a human gene. (C) Scheme of an assay that measures human GPCR activity by its coupling to the pheromone yeast pathway through human Gα proteins. | ||
Recent methodological advances have suggested that yeast complementation assays can be performed in a high-throughput manner. With the use of two regulatable promoters (tetO and pMET3), a versatile complementation system to clone human cDNAs was developed.83 The system employs the promoters to independently tune the expression level of the yeast and human genes, maximizing complementation. As a proof of concept, 25 yeast strains in which an essential gene is controlled by the tetO promoter were used to screen two cDNA human libraries expressed from the pMET3 promoter. The screens succeeded in the isolation of six complementing human cDNAs. Further efforts have resulted in the construction of more than 1000 strains in which essential genes are under the control of such a regulatable promoter.48,84 These strains could be the first part of a collection of yeast lines in which to study human genes. Another interesting approach combined the complementation of the yeast deletion strain, zwf1, by human glucose-6-phosphate dehydrogenase (hG6PD) with yeast competition assays, in order to functionally test several alleles of the human enzyme85 (Fig. 2A). The functionality of each allele was calculated from the growth of the mutant strain transformed with the human allele when grown in a competition pool under conditions selective for active hG6PD. This strategy revealed the sensitivity to missense mutations of the hG6PD active site positions and demonstrated that yeast competition assays can be used to test in parallel human enzymes that differ by a single point mutation.
An important limitation when scaling up yeast functional complementation assays to test disease candidate genes is that not all human genes are able to complement their respective yeast deletion strains. The Princeton Protein Orthology Database (P-POD; http://ortholog.princeton.edu/) reports over 200 human genes that complement a yeast function. These cases were collected from literature documenting cross-species expression experiments and good complementing examples like COX10 and BCS1L are still not in the database. Another drawback of functional complementation assays in yeast is the difference in complexity between yeast and human mitochondria (larger protein number, alternative splicing, etc.). Higher human protein numbers would result in unique pathways and in more players in the common pathways, partially restricting the conclusions that can be drawn from the results of the complementation assays.
Although a complementation experiment is the “classic” yeast cell-based assay, a variety of other yeast cell-based methods have been shown to be effective in the study of human genes and their associated diseases. Taking advantage of the fact that yeast is an experimentally amenable eukaryotic cell, these methods couple the function of the human protein to a measurable yeast readout such as growth, shmooing or the expression of a reporter gene, irrespective of whether the human protein has a yeast counterpart (Fig. 2B and C).86–89 For example, the human protein p53, which has no ortholog in S. cerevisiae, causes a growth defect when overexpressed in yeast.90 This phenotype was employed to find hyperactive variants of the tumor suppressor among alleles generated by random mutagenesis.91 Similar strategies have also been efficient in screening for inhibitors of apoptotic factors92 and poly(ADP-ribose) polymerases (Fig. 2B).93 In order to increase the scale of this approach, a rapid method to introduce human cDNAs into yeast was developed for the purpose of identifying human genes causing yeast growth inhibition.94 Of the 29 human proteins tested, which belonged to a variety of functional categories, around 30% caused a growth inhibition of 55% or higher. In all such cases, mutations or small molecule inhibitors were used to show that the growth defect is caused by the protein function itself and is not related to the nonspecific toxicity caused by overexpression of heterologous proteins.
Several methods that couple more sophisticated readouts have been developed even though such assays require further engineering of the yeast system and a better understanding of the biology of the tested human protein (Fig. 2C). A notable example is the replacement of proteins at the top end of the mating pheromone signal transduction pathway of yeast by human G-protein-coupled receptors (GPCR) and accessory Gα subunits95 (reviewed in ref. 96). Due to the resemblance between the pheromone yeast pathway and the human G-protein initiated MAP kinase cascade, activation of the human GPCRs can trigger the pheromone signaling pathway, which itself will result in the activation of mating-specific genes or reporter genes with mating-specific promoters. Several studies have used this system to screen for agonists, antagonists and native ligands of human GPCRs97 as well as mammalian nonreceptor modulators of G-protein signaling pathways.98 As an extension, the system could also be used to test human GPCRalleles for functionality. Other successful examples include assays to screen ligands of the estrogen receptor,99 to find anti-prion disease drugs,100 to test the activity of the human beta-secretase BACE in the study of Alzheimer's disease101 and to detect p53 mutations102 (for more extensive reviews of yeast cell-based assays please see ref. 86–88). Interestingly, the method to assess p53 function permits screening for mutations from patient samples, including blood, tumors or cell lines.103 The fact that at least one yeast-cell based assay exists for each of the four major classes of human drug targets (ion channels, nuclear receptors, GPCRs and enzymes)87 demonstrates the versatility of this approach.
The success with yeast cell-based assays illustrated here and the considerable availability of high-throughput approaches in yeast (deletion strain collections, sets of strains with genes under a regulatable promoter, competition assays, growth-inhibition assays, etc.) indicate that S. cerevisiae is a promising system in which to experimentally assess disease candidate genes. For example, one can foresee the generation of a collection of yeast strains in which each mitochondrial disease candidate gene is replaced by its complementary human ortholog. Identification of disease mutations could then ensue by comparing patient and wildtype gene complemention ability. This could be done by systematically competing strains carrying patient and control genes and may be a useful tool to ascertain genetic determinants of disease.
Conclusion and outlook
The use of yeast as a model system has made significant contributions in at least three ways to our systemic understanding of mitochondria and disease. First, most genomic technologies and approaches for data integration have been piloted in yeast. These efforts have shown that (a) integration of genomic data accelerates the identification of mitochondrial parts and their function; (b) only the integration of complementary datasets that capture different functional categories of proteins provides the most thorough characterization; and (c) comprehensive knowledge of the parts and interactions of a biological system facilitates the identification of disease-causing genes. Second, due to the significant conservation between yeast and human mitochondria, the information on mitochondrial components, interactions and their properties gained from yeast integrative genomics have helped characterize human orthologs, in particular human disease candidate genes. Third, employment of simple model organisms has been beneficial in experimentally evaluating the effect of mutations in disease genes.The task ahead includes the development of new technologies and approaches to address the limitations of available datasets and the complexity of mitochondria in both yeast and human. For example, studies on changes undergone by the organelle in response to stresses will be instrumental in modeling the dynamics that human mitochondria experience in disease states. In addition, tissue-specific measures will reveal the variation in mitochondrial components and their interactions throughout the human body. Imminent advances in technology will burst the speed and quality of genomic, proteomic and metabolic data acquisition, and promises to catalyze mitochondrial research over the next couple years. Sequencing by synthesis (such as 454 Life Sciences' and Solexa's techniques), one of several dozen ideas for high-throughput sequencing currently under development, is already about 100 times faster than the fastest Sanger sequencing techniques.104 The new ease of sequence acquisition will not only allow sequence variation to be measured across more species and individuals but also yield an increase in biological information at other levels, including gene expression and protein–DNA interactions. Likewise, new mass-spectrometry-based proteomics methods offer opportunities to obtain quantitative measures on large numbers of mitochondrial proteins. Maturation of both stable isotope labeling methods and label-free strategies, as well as the development of statistical methods for interpretation of quantitative protein data, promise to enable application to whole mitochondrial proteomes.105 At the metabolite level, mass spectrometry combined with chromatographic technologies already provide a powerful means to systematically identify and quantify complex mixtures of hundreds of metabolites, even from nanomolar sample concentrations. Since physiological status is directly reflected in the metabolome, metabolic profiles may be used to monitor the biochemistry of mitochondria, as well as disease states, drug efficacy and side effects. So-called targeted approaches that focus on known metabolites are already being applied to analyze pathway-related enzyme substrates and products which are involved for example in amyotrophic lateral sclerosis (ALS), schizophrenia and aging.106 The challenge remains of scaling up current technologies to uncover novel biomarkers to obtain a comprehensive and time-related biochemical snapshot from hundreds of patients.
It is clear that no single technology will answer why diseases and their symptoms arise, or how we can treat them. Instead, the integration of all measurements—at the genetic, transcriptional, proteomic, metabolic and phentoypic level—is needed to move towards a personalized medicine that assesses genetic predisposition, monitors health biomarkers and develops individualized treatments for patients with mitochondrial disorders. To achieve these goals the transfer of information and technical know-how between model systems and human will continue to play a pivotal role.
Acknowledgements
We thank Lior David, Raeka Aiyar, Himanshu Sinha, Julien Gagneur and Anna-Lynn Wegener for helpful comments on the manuscript. Research in the authors' laboratory is supported by grants from the National Institutes of Health and the Deutsche Forschungsgemeinschaft (LMS).References
- A. Hamosh, A. F. Scott, J. S. Amberger, C. A. Bocchini and V. A. McKusick, Nucleic Acids Res., 2005, 33, D514–517 CAS.
- D. Botstein and N. Risch, Nat. Genet., 2003, 33, 228–237 CrossRef CAS.
- E. S. Lander and N. J. Schork, Science, 1994, 265, 2037–2048 CrossRef CAS.
- D. E. Bassett, Jr., M. S. Boguski and P. Hieter, Nature, 1996, 379, 589–590 CrossRef CAS.
- V. Tiranti, K. Hoertnagel, R. Carrozzo, C. Galimberti, M. Munaro, M. Granatiero, L. Zelante, P. Gasparini, R. Marzella, M. Rocchi, M. P. Bayona-Bafaluy, J. A. Enriquez, G. Uziel, E. Bertini, C. Dionisi-Vici, B. Franco, T. Meitinger and M. Zeviani, Am. J. Hum. Genet., 1998, 63, 1609–1621 CrossRef CAS.
- A. Barrientos, IUBMB Life, 2003, 55, 83–95 Search PubMed.
- H. Prokisch, C. Andreoli, U. Ahting, K. Heiss, A. Ruepp, C. Scharfe and T. Meitinger, Nucleic Acids Res., 2006, 34, D705–711 CrossRef CAS.
- H. M. McBride, M. Neuspiel and S. Wasiak, Curr. Biol., 2006, 16, R551–560 CrossRef CAS.
- D. C. Chan, Cell, 2006, 125, 1241–1252 CrossRef CAS; H. Chen and D. C. Chan, Curr. Opin. Cell Biol., 2006, 18, 453–459 CrossRef CAS.
- G. Burger, M. W. Gray and B. F. Lang, Trends Genet., 2003, 19, 709–716 CrossRef CAS; O. Karlberg, B. Canback, C. G. Kurland and S. G. Andersson, Yeast, 2000, 17, 170–187 CrossRef CAS.
- D. C. Wallace, Annu. Rev. Genet., 2005, 39, 359–407 CrossRef CAS.
- M. W. Gray, G. Burger and B. F. Lang, Science, 1999, 283, 1476–1481 CrossRef CAS.
- S. G. Andersson, A. Zomorodipour, J. O. Andersson, T. Sicheritz-Ponten, U. C. Alsmark, R. M. Podowski, A. K. Naslund, A. S. Eriksson, H. H. Winkler and C. G. Kurland, Nature, 1998, 396, 133–140 CrossRef CAS; M. W. Gray, G. Burger and B. F. Lang, GenomeBiology, 2001, 2, 1018 Search PubMed; S. G. Andersson, O. Karlberg, B. Canback and C. G. Kurland, Philos. Trans. R. Soc. London, Ser. B, 2003, 358, 165–177 CrossRef CAS discussion 177–169.
- M. F. Lopez, B. S. Kristal, E. Chernokalskaya, A. Lazarev, A. I. Shestopalov, A. Bogdanova and M. Robinson, Electrophoresis, 2000, 21, 3427–3440 CrossRef CAS.
- B. Westermann and W. Neupert, Nat. Biotechnol., 2003, 21, 239–240 CrossRef CAS.
- D. C. Wallace, Science, 1999, 283, 1482–1488 CrossRef CAS.
- S. Dimauro and G. Davidzon, Ann. Med., 2005, 37, 222–232 Search PubMed.
- M. Zeviani, A. Spinazzola and V. Carelli, Curr. Opin. Genet. Dev., 2003, 13, 262–270 CrossRef CAS.
- P. F. Chinnery and D. M. Turnbull, Am. J. Med. Genet., 2001, 106, 94–101 CrossRef CAS; A. M. Schaefer, R. W. Taylor, D. M. Turnbull and P. F. Chinnery, Biochim. Biophys. Acta, 2004, 1659, 115–120 CAS.
- R. McFarland, R. W. Taylor and D. M. Turnbull, Curr. Top. Dev. Biol., 2007, 77, 113–155 Search PubMed.
- S. Dimauro, S. Tay and M. Mancuso, Ann. N. Y. Acad. Sci., 2004, 1011, 217–231 CrossRef CAS; J. Smeitink, L. van den Heuvel and S. DiMauro, Nat. Rev. Genet., 2001, 2, 342–352 CrossRef CAS.
- J. W. Lustbader, M. Cirilli, C. Lin, H. W. Xu, K. Takuma, N. Wang, C. Caspersen, X. Chen, S. Pollak, M. Chaney, F. Trinchese, S. Liu, F. Gunn-Moore, L. F. Lue, D. G. Walker, P. Kuppusamy, Z. L. Zewier, O. Arancio, D. Stern, S. S. Yan and H. Wu, Science, 2004, 304, 448–452 CrossRef CAS; J. St-Pierre, S. Drori, M. Uldry, J. M. Silvaggi, J. Rhee, S. Jager, C. Handschin, K. Zheng, J. Lin, W. Yang, D. K. Simon, R. Bachoo and B. M. Spiegelman, Cell, 2006, 127, 397–408 CrossRef CAS; R. S. Balaban, S. Nemoto and T. Finkel, Cell, 2005, 120, 483–495 CrossRef CAS; T. M. Dawson and V. L. Dawson, Science, 2003, 302, 819–822 CrossRef CAS; E. M. Valente, P. M. Abou-Sleiman, V. Caputo, M. M. Muqit, K. Harvey, S. Gispert, Z. Ali, D. Del Turco, A. R. Bentivoglio, D. G. Healy, A. Albanese, R. Nussbaum, R. Gonzalez-Maldonado, T. Deller, S. Salvi, P. Cortelli, W. P. Gilks, D. S. Latchman, R. J. Harvey, B. Dallapiccola, G. Auburger and N. W. Wood, Science, 2004, 304, 1158–1160 CrossRef CAS.
- V. K. Mootha, C. M. Lindgren, K. F. Eriksson, A. Subramanian, S. Sihag, J. Lehar, P. Puigserver, E. Carlsson, M. Ridderstrale, E. Laurila, N. Houstis, M. J. Daly, N. Patterson, J. P. Mesirov, T. R. Golub, P. Tamayo, B. Spiegelman, E. S. Lander, J. N. Hirschhorn, D. Altshuler and L. C. Groop, Nat. Genet., 2003, 34, 267–273 CrossRef CAS; P. Maechler and C. B. Wollheim, Nature, 2001, 414, 807–812 CrossRef CAS.
- A. S. Reichert and W. Neupert, Trends Genet., 2004, 20, 555–562 CrossRef CAS.
- A. Kumar, S. Agarwal, J. A. Heyman, S. Matson, M. Heidtman, S. Piccirillo, L. Umansky, A. Drawid, R. Jansen, Y. Liu, K. H. Cheung, P. Miller, M. Gerstein, G. S. Roeder and M. Snyder, Genes Dev., 2002, 16, 707–719 CrossRef CAS; W. K. Huh, J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S. Weissman and E. K. O'Shea, Nature, 2003, 425, 686–691 CrossRef CAS.
- T. Ozawa, Y. Sako, M. Sato, T. Kitamura and Y. Umezawa, Nat. Biotechnol., 2003, 21, 287–293 CrossRef CAS.
- K. S. Dimmer, S. Fritz, F. Fuchs, M. Messerschmitt, N. Weinbach, W. Neupert and B. Westermann, Mol. Biol. Cell, 2002, 13, 847–853 CrossRef CAS.
- L. M. Steinmetz, C. Scharfe, A. M. Deutschbauer, D. Mokranjac, Z. S. Herman, T. Jones, A. M. Chu, G. Giaever, H. Prokisch, P. J. Oefner and R. W. Davis, Nat. Genet., 2002, 31, 400–404 CAS.
- J. L. DeRisi, V. R. Iyer and P. O. Brown, Science, 1997, 278, 680–686 CrossRef CAS; R. Lascaris, H. J. Bussemaker, A. Boorsma, M. Piper, H. van der Spek, L. Grivell and J. Blom, GenomeBiology, 2003, 4, R3 Search PubMed.
- H. Prokisch, C. Scharfe and D. G. Camp, PLoS Biol., 2004, 2, e160 Search PubMed.
- V. K. Mootha, J. Bunkenborg, J. V. Olsen, M. Hjerrild, J. R. Wisniewski, E. Stahl, M. S. Bolouri, H. N. Ray, S. Sihag, M. Kamal, N. Patterson, E. S. Lander and M. Mann, Cell, 2003, 115, 629–640 CrossRef CAS.
- J. Reinders, R. P. Zahedi, N. Pfanner, C. Meisinger and A. Sickmann, J. Proteome Res., 2006, 5, 1543–1554 CrossRef CAS; A. Sickmann, J. Reinders, Y. Wagner, C. Joppich, R. Zahedi, H. E. Meyer, B. Schonfisch, I. Perschil, A. Chacinska, B. Guiard, P. Rehling, N. Pfanner and C. Meisinger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13207–13212 CrossRef CAS.
- S. W. Taylor, E. Fahy, B. Zhang, G. M. Glenn, D. E. Warnock, S. Wiley, A. N. Murphy, S. P. Gaucher, R. A. Capaldi, B. W. Gibson and S. S. Ghosh, Nat. Biotechnol., 2003, 21, 281–286 CrossRef CAS.
- J. L. Heazlewood, J. S. Tonti-Filippini, A. M. Gout, D. A. Day, J. Whelan and A. H. Millar, Plant Cell, 2004, 16, 241–256 CrossRef CAS.
- C. Guda, E. Fahy and S. Subramaniam, Bioinformatics, 2004, 20, 1785–1794 CrossRef CAS; M. G. Claros and P. Vincens, Eur. J. Biochem., 1996, 241, 779–786 CrossRef CAS; O. Emanuelsson, H. Nielsen, S. Brunak and G. von Heijne, J. Mol. Biol., 2000, 300, 1005–1016 CrossRef CAS.
- S. W. Taylor, E. Fahy and S. S. Ghosh, Trends Biotechnol., 2003, 21, 82–88 CrossRef CAS.
- J. R. Yates, Nat. Rev. Mol. Cell Biol., 2005, 6, 702–714 CAS; J. S. Andersen and M. Mann, EMBO Rep., 2006, 7, 874–879 Search PubMed.
- S. P. Gaucher, S. W. Taylor, E. Fahy, B. Zhang, D. E. Warnock, S. S. Ghosh and B. W. Gibson, J. Proteome Res., 2004, 3, 495–505 CrossRef CAS.
- P. Marc, A. Margeot, F. Devaux, C. Blugeon, M. Corral-Debrinski and C. Jacq, EMBO Rep., 2002, 3, 159–164 Search PubMed.
- S. Calvo, M. Jain, X. Xie, S. A. Sheth, B. Chang, O. A. Goldberger, A. Spinazzola, M. Zeviani, S. A. Carr and V. K. Mootha, Nat. Genet., 2006, 38, 576–582 CrossRef CAS.
- F. Perocchi, L. J. Jensen, J. Gagneur, U. Ahting, C. von Mering, P. Bork, H. Prokisch and L. M. Steinmetz, PLoS Genet., 2006, 2, e170 Search PubMed.
- S. Da Cruz, P. A. Parone and J. C. Martinou, Expert Rev. Proteomics, 2005, 2, 541–551 Search PubMed.
- S. Da Cruz, I. Xenarios, J. Langridge, F. Vilbois, P. A. Parone and J. C. Martinou, J. Biol. Chem., 2003, 278, 41566–41571 CrossRef.
- S. Basu, E. Bremer, C. Zhou and D. F. Bogenhagen, Bioinformatics, 2006, 22, 485–492 CAS; P. Guda, S. Subramaniam and C. Guda, Methods Mol. Biol., 2007, 357, 375–383 Search PubMed.
- T. Kislinger, B. Cox, A. Kannan, C. Chung, P. Hu, A. Ignatchenko, M. S. Scott, A. O. Gramolini, Q. Morris, M. T. Hallett, J. Rossant, T. R. Hughes, B. Frey and A. Emili, Cell, 2006, 125, 173–186 CrossRef CAS.
- A. L. Barabasi and Z. N. Oltvai, Nat. Rev. Genet., 2004, 5, 101–113 CrossRef CAS.
- J. Ihmels, S. Bergmann and N. Barkai, Bioinformatics, 2004, 20, 1993–2003 CrossRef CAS; M. B. Eisen, P. T. Spellman, P. O. Brown and D. Botstein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14863–14868 CrossRef CAS; S. Tavazoie, J. D. Hughes, M. J. Campbell, R. J. Cho and G. M. Church, Nat. Genet., 1999, 22, 281–285 CrossRef CAS; L. F. Wu, T. R. Hughes, A. P. Davierwala, M. D. Robinson, R. Stoughton and S. J. Altschuler, Nat. Genet., 2002, 31, 255–265 CrossRef CAS; E. Segal, M. Shapira, A. Regev, D. Pe'er, D. Botstein, D. Koller and N. Friedman, Nat. Genet., 2003, 34, 166–176 CrossRef CAS.
- T. R. Hughes, M. J. Marton, A. R. Jones, C. J. Roberts, R. Stoughton, C. D. Armour, H. A. Bennett, E. Coffey, H. Dai, Y. D. He, M. J. Kidd, A. M. King, M. R. Meyer, D. Slade, P. Y. Lum, S. B. Stepaniants, D. D. Shoemaker, D. Gachotte, K. Chakraburtty, J. Simon, M. Bard and S. H. Friend, Cell, 2000, 102, 109–126 CrossRef CAS.
- B. Ren, F. Robert, J. J. Wyrick, O. Aparicio, E. G. Jennings, I. Simon, J. Zeitlinger, J. Schreiber, N. Hannett, E. Kanin, T. L. Volkert, C. J. Wilson, S. P. Bell and R. A. Young, Science, 2000, 290, 2306–2309 CrossRef CAS; T. I. Lee, N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J. B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford and R. A. Young, Science, 2002, 298, 799–804 CrossRef CAS.
- A. H. Tong, M. Evangelista, A. B. Parsons, H. Xu, G. D. Bader, N. Page, M. Robinson, S. Raghibizadeh, C. W. Hogue, H. Bussey, B. Andrews, M. Tyers and C. Boone, Science, 2001, 294, 2364–2368 CrossRef CAS; A. H. Tong, G. Lesage, G. D. Bader, H. Ding, H. Xu, X. Xin, J. Young, G. F. Berriz, R. L. Brost, M. Chang, Y. Chen, X. Cheng, G. Chua, H. Friesen, D. S. Goldberg, J. Haynes, C. Humphries, G. He, S. Hussein, L. Ke, N. Krogan, Z. Li, J. N. Levinson, H. Lu, P. Menard, C. Munyana, A. B. Parsons, O. Ryan, R. Tonikian, T. Roberts, A. M. Sdicu, J. Shapiro, B. Sheikh, B. Suter, S. L. Wong, L. V. Zhang, H. Zhu, C. G. Burd, S. Munro, C. Sander, J. Rine, J. Greenblatt, M. Peter, A. Bretscher, G. Bell, F. P. Roth, G. W. Brown, B. Andrews, H. Bussey and C. Boone, Science, 2004, 303, 808–813 CrossRef CAS; X. Pan, D. S. Yuan, D. Xiang, X. Wang, S. Sookhai-Mahadeo, J. S. Bader, P. Hieter, F. Spencer and J. D. Boeke, Mol. Cell, 2004, 16, 487–496 CrossRef CAS.
- T. Ito, T. Chiba, R. Ozawa, M. Yoshida, M. Hattori and Y. Sakaki, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4569–4574 CrossRef CAS; P. Uetz, L. Giot, G. Cagney, T. A. Mansfield, R. S. Judson, J. R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, A. Qureshi-Emili, Y. Li, B. Godwin, D. Conover, T. Kalbfleisch, G. Vijayadamodar, M. Yang, M. Johnston, S. Fields and J. M. Rothberg, Nature, 2000, 403, 623–627 CrossRef CAS.
- N. J. Krogan, G. Cagney, H. Yu, G. Zhong, X. Guo, A. Ignatchenko, J. Li, S. Pu, N. Datta, A. P. Tikuisis, T. Punna, J. M. Peregrin-Alvarez, M. Shales, X. Zhang, M. Davey, M. D. Robinson, A. Paccanaro, J. E. Bray, A. Sheung, B. Beattie, D. P. Richards, V. Canadien, A. Lalev, F. Mena, P. Wong, A. Starostine, M. M. Canete, J. Vlasblom, S. Wu, C. Orsi, S. R. Collins, S. Chandran, R. Haw, J. J. Rilstone, K. Gandi, N. J. Thompson, G. Musso, P. St Onge, S. Ghanny, M. H. Lam, G. Butland, A. M. Altaf-Ul, S. Kanaya, A. Shilatifard, E. O'Shea, J. S. Weissman, C. J. Ingles, T. R. Hughes, J. Parkinson, M. Gerstein, S. J. Wodak, A. Emili and J. F. Greenblatt, Nature, 2006, 440, 637–643 CrossRef CAS; A. C. Gavin, P. Aloy, P. Grandi, R. Krause, M. Boesche, M. Marzioch, C. Rau, L. J. Jensen, S. Bastuck, B. Dumpelfeld, A. Edelmann, M. A. Heurtier, V. Hoffman, C. Hoefert, K. Klein, M. Hudak, A. M. Michon, M. Schelder, M. Schirle, M. Remor, T. Rudi, S. Hooper, A. Bauer, T. Bouwmeester, G. Casari, G. Drewes, G. Neubauer, J. M. Rick, B. Kuster, P. Bork, R. B. Russell and G. Superti-Furga, Nature, 2006, 440, 631–636 CrossRef CAS.
- A. C. Gavin, M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J. M. Rick, A. M. Michon, C. M. Cruciat, M. Remor, C. Hofert, M. Schelder, M. Brajenovic, H. Ruffner, A. Merino, K. Klein, M. Hudak, D. Dickson, T. Rudi, V. Gnau, A. Bauch, S. Bastuck, B. Huhse, C. Leutwein, M. A. Heurtier, R. R. Copley, A. Edelmann, E. Querfurth, V. Rybin, G. Drewes, M. Raida, T. Bouwmeester, P. Bork, B. Seraphin, B. Kuster, G. Neubauer and G. Superti-Furga, Nature, 2002, 415, 141–147 CrossRef CAS.
- J. Ptacek, G. Devgan, G. Michaud, H. Zhu, X. Zhu, J. Fasolo, H. Guo, G. Jona, A. Breitkreutz, R. Sopko, R. R. McCartney, M. C. Schmidt, N. Rachidi, S. J. Lee, A. S. Mah, L. Meng, M. J. Stark, D. F. Stern, C. De Virgilio, M. Tyers, B. Andrews, M. Gerstein, B. Schweitzer, P. F. Predki and M. Snyder, Nature, 2005, 438, 679–684 CrossRef CAS; H. Zhu, J. F. Klemic, S. Chang, P. Bertone, A. Casamayor, K. G. Klemic, D. Smith, M. Gerstein, M. A. Reed and M. Snyder, Nat. Genet., 2000, 26, 283–289 CrossRef CAS.
- M. Huynen, B. Snel and W. Lathe, Genome Res., 2000, 10, 1204–1210 CrossRef CAS.
- M. Pellegrini, E. M. Marcotte, M. J. Thompson, D. Eisenberg and T. O. Yeates, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4285–4288 CrossRef CAS; M. A. Huynen and P. Bork, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5849–5856 CrossRef CAS.
- E. M. Marcotte, M. Pellegrini, H. L. Ng, D. W. Rice, T. O. Yeates and D. Eisenberg, Science, 1999, 285, 751–753 CrossRef CAS; A. J. Enright, I. Iliopoulos, N. C. Kyrpides and C. A. Ouzounis, Nature, 1999, 402, 86–90 CrossRef CAS.
- T. Dandekar, B. Snel, M. Huynen and P. Bork, Trends Biochem. Sci., 1998, 23, 324–328 CrossRef CAS; R. Overbeek, M. Fonstein, M. D'Souza, G. D. Pusch and N. Maltsev, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2896–2901 CrossRef CAS.
- L. J. Jensen, J. Saric and P. Bork, Nat. Rev. Genet., 2006, 7, 119–129 CrossRef CAS.
- U. Guldener, M. Munsterkotter, M. Oesterheld, P. Pagel, A. Ruepp, H. W. Mewes and V. Stumpflen, Nucleic Acids Res., 2006, 34, D436–441 CrossRef.
- M. Kanehisa, S. Goto, M. Hattori, K. F. Aoki-Kinoshita, M. Itoh, S. Kawashima, T. Katayama, M. Araki and M. Hirakawa, Nucleic Acids Res., 2006, 34, D354–357 CrossRef CAS.
- G. T. Hart, A. K. Ramani and E. M. Marcotte, GenomeBiology, 2006, 7, 120 Search PubMed.
- C. von Mering, R. Krause, B. Snel, M. Cornell, S. G. Oliver, S. Fields and P. Bork, Nature, 2002, 417, 399–403 CrossRef CAS.
- C. L. Myers, D. Robson, A. Wible, M. A. Hibbs, C. Chiriac, C. L. Theesfeld, K. Dolinski and O. G. Troyanskaya, GenomeBiology, 2005, 6, R114 Search PubMed; I. Lee, S. V. Date, A. T. Adai and E. M. Marcotte, Science, 2004, 306, 1555–1558 CrossRef CAS.
- C. von Mering, L. J. Jensen, M. Kuhn, S. Chaffron, T. Doerks, B. Kruger, B. Snel and P. Bork, Nucleic Acids Res., 2007, 35, D358–362 CrossRef.
- T. E. Shutt and G. S. Shadel, GenomeBiology, 2007, 8, 203 Search PubMed.
- U. Stelzl, U. Worm, M. Lalowski, C. Haenig, F. H. Brembeck, H. Goehler, M. Stroedicke, M. Zenkner, A. Schoenherr, S. Koeppen, J. Timm, S. Mintzlaff, C. Abraham, N. Bock, S. Kietzmann, A. Goedde, E. Toksoz, A. Droege, S. Krobitsch, B. Korn, W. Birchmeier, H. Lehrach and E. E. Wanker, Cell, 2005, 122, 957–968 CrossRef CAS; J. F. Rual, K. Venkatesan, T. Hao, T. Hirozane-Kishikawa, A. Dricot, N. Li, G. F. Berriz, F. D. Gibbons, M. Dreze, N. Ayivi-Guedehoussou, N. Klitgord, C. Simon, M. Boxem, S. Milstein, J. Rosenberg, D. S. Goldberg, L. V. Zhang, S. L. Wong, G. Franklin, S. Li, J. S. Albala, J. Lim, C. Fraughton, E. Llamosas, S. Cevik, C. Bex, P. Lamesch, R. S. Sikorski, J. Vandenhaute, H. Y. Zoghbi, A. Smolyar, S. Bosak, R. Sequerra, L. Doucette-Stamm, M. E. Cusick, D. E. Hill, F. P. Roth and M. Vidal, Nature, 2005, 437, 1173–1178 CrossRef CAS.
- D. R. Rhodes, S. A. Tomlins, S. Varambally, V. Mahavisno, T. Barrette, S. Kalyana-Sundaram, D. Ghosh, A. Pandey and A. M. Chinnaiyan, Nat. Biotechnol., 2005, 23, 951–959 CrossRef CAS.
- T. D. Vo, H. J. Greenberg and B. O. Palsson, J. Biol. Chem., 2004, 279, 39532–39540 CrossRef CAS.
- L. Hood, J. R. Heath, M. E. Phelps and B. Lin, Science, 2004, 306, 640–643 CrossRef CAS.
- M. W. Covert, E. M. Knight, J. L. Reed, M. J. Herrgard and B. O. Palsson, Nature, 2004, 429, 92–96 CrossRef CAS; T. Ideker, V. Thorsson, J. A. Ranish, R. Christmas, J. Buhler, J. K. Eng, R. Bumgarner, D. R. Goodlett, R. Aebersold and L. Hood, Science, 2001, 292, 929–934 CrossRef CAS.
- N. Lee, M. J. Daly, T. Delmonte, E. S. Lander, F. Xu, T. J. Hudson, G. A. Mitchell, C. C. Morin, B. H. Robinson and J. D. Rioux, Am. J. Hum. Genet., 2001, 68, 397–409 CrossRef CAS.
- V. K. Mootha, P. Lepage, K. Miller, J. Bunkenborg, M. Reich, M. Hjerrild, T. Delmonte, A. Villeneuve, R. Sladek, F. Xu, G. A. Mitchell, C. Morin, M. Mann, T. J. Hudson, B. Robinson, J. D. Rioux and E. S. Lander, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 605–610 CrossRef CAS.
- A. Spinazzola, C. Viscomi, E. Fernandez-Vizarra, F. Carrara, P. D'Adamo, S. Calvo, R. M. Marsano, C. Donnini, H. Weiher, P. Strisciuglio, R. Parini, E. Sarzi, A. Chan, S. DiMauro, A. Rotig, P. Gasparini, I. Ferrero, V. K. Mootha, V. Tiranti and M. Zeviani, Nat. Genet., 2006, 38, 570–575 CrossRef CAS.
- J. Ostrowski, L. Wyrwicz, L. Rychlewski and K. Bomsztyk, J. Biol. Chem., 2002, 277, 6303–6310 CrossRef CAS; G. M. Manthey, B. D. Przybyla-Zawislak and J. E. McEwen, Eur. J. Biochem., 1998, 255, 156–161 CrossRef CAS.
- H. G. Brunner and M. A. van Driel, Nat. Rev. Genet., 2004, 5, 545–551 CrossRef CAS; N. Jamshidi and B. O. Palsson, Mol. Syst. Biol., 2006, 2, 38; K. I. Goh, M. E. Cusick, D. Valle, B. Childs, M. Vidal and A. L. Barabasi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8685–8690 CrossRef CAS; L. Franke, H. Bakel, L. Fokkens, E. D. de Jong, M. Egmont-Petersen and C. Wijmenga, Am. J. Hum. Genet., 2006, 78, 1011–1025 CrossRef CAS.
- J. Lim, T. Hao, C. Shaw, A. J. Patel, G. Szabo, J. F. Rual, C. J. Fisk, N. Li, A. Smolyar, D. E. Hill, A. L. Barabasi, M. Vidal and H. Y. Zoghbi, Cell, 2006, 125, 801–814 CrossRef CAS.
- M. G. Lee and P. Nurse, Nature, 1987, 327, 31–35 CrossRef CAS; G. Draetta, L. Brizuela, J. Potashkin and D. Beach, Cell, 1987, 50, 319–325 CrossRef CAS; Y. Tanaka, T. Ashikari, Y. Shibano, T. Amachi, H. Yoshizumi and H. Matsubara, J. Biochem., 1988, 103, 954–961 CAS; M. J. Castanon, W. Spevak, G. R. Adolf, E. Chlebowicz-Sledziewska and A. Sledziewski, Gene, 1988, 66, 223–234 CrossRef CAS.
- Y. Tanaka, T. Ashikari, Y. Shibano, T. Amachi, H. Yoshizumi and H. Matsubara, J. Biochem., 1988, 104, 477–480 CAS.
- R. Allikmets, W. H. Raskind, A. Hutchinson, N. D. Schueck, M. Dean and D. M. Koeller, Hum. Mol. Genet., 1999, 8, 743–749 CrossRef CAS; P. Cavadini, C. Gellera, P. I. Patel and G. Isaya, Hum. Mol. Genet., 2000, 9, 2523–2530 CrossRef CAS.
- I. Valnot, J. C. von Kleist-Retzow, A. Barrientos, M. Gorbatyuk, J. W. Taanman, B. Mehaye, P. Rustin, A. Tzagoloff, A. Munnich and A. Rotig, Hum. Mol. Genet., 2000, 9, 1245–1249 CrossRef CAS.
- P. de Lonlay, I. Valnot, A. Barrientos, M. Gorbatyuk, A. Tzagoloff, J. W. Taanman, E. Benayoun, D. Chretien, N. Kadhom, A. Lombes, H. O. de Baulny, P. Niaudet, A. Munnich, P. Rustin and A. Rotig, Nat. Genet., 2001, 29, 57–60 CrossRef; I. Visapaa, V. Fellman, J. Vesa, A. Dasvarma, J. L. Hutton, V. Kumar, G. S. Payne, M. Makarow, R. Van Coster, R. W. Taylor, D. M. Turnbull, A. Suomalainen and L. Peltonen, Am. J. Hum. Genet., 2002, 71, 863–876 CrossRef.
- N. Zhang, M. Osborn, P. Gitsham, K. Yen, J. R. Miller and S. G. Oliver, Gene, 2003, 303, 121–129 CrossRef CAS.
- E. Gari, L. Piedrafita, M. Aldea and E. Herrero, Yeast, 1997, 13, 837–848 CrossRef CAS; S. Mnaimneh, A. P. Davierwala, J. Haynes, J. Moffat, W. T. Peng, W. Zhang, X. Yang, J. Pootoolal, G. Chua, A. Lopez, M. Trochesset, D. Morse, N. J. Krogan, S. L. Hiley, Z. Li, Q. Morris, J. Grigull, N. Mitsakakis, C. J. Roberts, J. F. Greenblatt, C. Boone, C. A. Kaiser, B. J. Andrews and T. R. Hughes, Cell, 2004, 118, 31–44 CrossRef CAS; J. A. Wishart, M. Osborn, M. E. Gent, K. Yen, Z. Vujovic, P. Gitsham, N. Zhang, J. Ross Miller and S. G. Oliver, Yeast, 2006, 23, 325–331 CrossRef CAS.
- J. Merritt, J. A. Butz, B. A. Ogunnaike and J. S. Edwards, Biotechnol. Bioeng., 2005, 92, 519–531 CrossRef CAS.
- C. L. Tucker, Drug Discovery Today, 2002, 7, S125–130 CrossRef CAS.
- T. R. Hughes, Funct. Integr. Genomics, 2002, 2, 199–211 Search PubMed.
- T. Melese and P. Hieter, Trends Pharmacol. Sci., 2002, 23, 544–547 CrossRef CAS.
- W. H. Mager and J. Winderickx, Trends Pharmacol. Sci., 2005, 26, 265–273 CrossRef CAS; S. G. Oliver, Curr. Opin. Genet. Dev., 1997, 7, 405–409 CrossRef CAS; S. G. Oliver, Philos. Trans. R. Soc. London, Ser., 2002, 357, 17–23 Search PubMed.
- J. M. Nigro, R. Sikorski, S. I. Reed and B. Vogelstein, Mol. Cell. Biol., 1992, 12, 1357–1365 CAS.
- A. Inga and M. A. Resnick, Oncogene, 2001, 20, 3409–3419 CrossRef CAS.
- Q. Xu, N. Ke, S. Matsuyama and J. C. Reed, Methods Enzymol., 2000, 322, 283–296 CAS; Q. Xu, J. M. Jurgensmeier and J. C. Reed, Methods, 1999, 17, 292–304 CrossRef CAS; Q. Xu and J. C. Reed, Mol. Cell, 1998, 1, 337–346 CrossRef CAS; H. Zhang, S. W. Cowan-Jacob, M. Simonen, W. Greenhalf, J. Heim and B. Meyhack, J. Biol. Chem., 2000, 275, 11092–11099 CrossRef CAS; M. Sawada, W. Sun, P. Hayes, K. Leskov, D. A. Boothman and S. Matsuyama, Nat. Cell Biol., 2003, 5, 320–329 CrossRef CAS; M. L. Brezniceanu, K. Volp, S. Bosser, C. Solbach, P. Lichter, S. Joos and M. Zornig, FASEB J., 2003, 17, 1295–1297 CAS.
- E. Perkins, D. Sun, A. Nguyen, S. Tulac, M. Francesco, H. Tavana, H. Nguyen, S. Tugendreich, P. Barthmaier, J. Couto, E. Yeh, S. Thode, K. Jarnagin, A. Jain, D. Morgans and T. Melese, Cancer Res., 2001, 61, 4175–4183 CAS.
- S. Tugendreich, E. Perkins, J. Couto, P. Barthmaier, D. Sun, S. Tang, S. Tulac, A. Nguyen, E. Yeh, A. Mays, E. Wallace, T. Lila, D. Shivak, M. Prichard, L. Andrejka, R. Kim and T. Melese, Genome Res., 2001, 11, 1899–1912 CAS.
- K. King, H. G. Dohlman, J. Thorner, M. G. Caron and R. J. Lefkowitz, Science, 1990, 250, 121–123 CrossRef CAS.
- I. I. Klein, Drug Discovery Today, 2000, 5, 37–38 CrossRef; M. H. Pausch, Trends Biotechnol., 1997, 15, 487–494 CrossRef CAS; S. J. Dowell and A. J. Brown, Recept. Channels, 2002, 8, 343–352 Search PubMed.
- J. R. Erickson, J. J. Wu, J. G. Goddard, G. Tigyi, K. Kawanishi, L. D. Tomei and M. C. Kiefer, J. Biol. Chem., 1998, 273, 1506–1510 CrossRef CAS; R. M. Campbell, C. Cartwright, W. Chen, Y. Chen, E. Duzic, J. M. Fu, M. Loveland, R. Manning, B. McKibben, C. M. Pleiman, L. Silverman, J. Trueheart, D. R. Webb, V. Wilkinson, D. J. Witter, X. Xie and A. L. Castelhano, Bioorg. Med. Chem. Lett., 1999, 9, 2413–2418 CrossRef CAS; P. G. Szekeres, Recept. Channels, 2002, 8, 297–308 Search PubMed.
- M. J. Cismowski, A. Takesono, C. Ma, J. S. Lizano, X. Xie, H. Fuernkranz, S. M. Lanier and E. Duzic, Nat. Biotechnol., 1999, 17, 878–883 CrossRef CAS.
- N. G. Coldham, M. Dave, S. Sivapathasundaram, D. P. McDonnell, C. Connor and M. J. Sauer, Environ. Health Perspect., 1997, 105, 734–742 CAS; T. F. Bovee, R. J. Helsdingen, P. D. Koks, H. A. Kuiper, R. L. Hoogenboom and J. Keijer, Gene, 2004, 325, 187–200 CrossRef CAS; K. O. Klein, J. Baron, M. J. Colli, D. P. McDonnell and G. B. Cutler, Jr., J. Clin. Invest., 1994, 94, 2475–2480 CrossRef CAS; S. F. Arnold, M. K. Robinson, A. C. Notides, L. J. Guillette, Jr. and J. A. McLachlan, Environ. Health Perspect., 1996, 104, 544–548 CAS.
- S. Bach, N. Talarek, T. Andrieu, J. M. Vierfond, Y. Mettey, H. Galons, D. Dormont, L. Meijer, C. Cullin and M. Blondel, Nat. Biotechnol., 2003, 21, 1075–1081 CrossRef CAS.
- U. Luthi, C. Schaerer-Brodbeck, S. Tanner, O. Middendorp, K. Edler and A. Barberis, Biochim. Biophys. Acta, 2003, 1620, 167–178 CrossRef CAS; D. Edbauer, E. Winkler, J. T. Regula, B. Pesold, H. Steiner and C. Haass, Nat. Cell Biol., 2003, 5, 486–488 CrossRef CAS; O. Middendorp, C. Ortler, U. Neumann, P. Paganetti, U. Luthi and A. Barberis, Biochim. Biophys. Acta, 2004, 1674, 29–39 CrossRef CAS.
- J. Watanabe, H. Nishiyama, K. Okubo, T. Takahashi, Y. Toda, T. Habuchi, Y. Kakehi, M. Tada and O. Ogawa, Urology, 2004, 63, 989–993 CrossRef; T. Shibata, D. Nakata, I. Chiba, T. Yamashita, Y. Abiko, M. Tada and T. Moriuchi, J. Oral Pathol. Med., 2002, 31, 534–538 CrossRef CAS; Y. Mitsumoto, T. Nakajima, M. Marutani, H. Kashiwazaki, M. Moriguchi, H. Kimura, T. Okanoue, K. Kagawa and M. Tada, Hum. Pathol., 2004, 35, 350–356 CrossRef CAS.
- J. M. Flaman, T. Frebourg, V. Moreau, F. Charbonnier, C. Martin, P. Chappuis, A. P. Sappino, I. M. Limacher, L. Bron and J. Benhattar, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3963–3967 CrossRef CAS.
- R. Dalton, Nature, 2006, 443, 258–259 CrossRef CAS.
- M. Bantscheff, M. Schirle, G. Sweetman, J. Rick and B. Kuster, Anal. Bioanal. Chem., 2007 Search PubMed; S. E. Ong and M. Mann, Nat. Chem. Biol., 2005, 1, 252–262 CrossRef CAS.
- R. Bowser, M. Cudkowicz and R. Kaddurah-Daouk, Expert Rev. Mol. Diagn., 2006, 6, 387–398 Search PubMed; R. Kaddurah-Daouk, PLoS Med., 2006, 3, e363 Search PubMed; B. S. Kristal, Y. I. Shurubor, R. Kaddurah-Daouk and W. R. Matson, Methods Mol. Biol., 2007, 371, 393–409 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2008 |