Metabolic flux analysis and metabolic engineering of microorganisms
Hyun Uk
Kim
ab,
Tae Yong
Kim
ab and
Sang Yup
Lee
*abc
aDepartment of Chemical and Biomolecular Engineering (BK21 Program), Metabolic and Biomolecular Engineering National Research Laboratory, Korea Advanced Institute of Science and Technology, Daejeon 305-701, Republic of Korea. E-mail: leesy@kaist.ac.kr; Fax: +82 42 869 3910; Tel: +82 42 869 3930
bCenter for Systems and Synthetic Biotechnology, Institute for the BioCentury, Korea Advanced Institute of Science and Technology, Daejeon 305-701, Republic of Korea
cDepartment of Bio and Brain Engineering, BioProcess Engineering Research Center and Bioinformatics Research Center, Korea Advanced Institute of Science and Technology, Daejeon 305-701, Republic of Korea
First published on 16th October 2007
Abstract
Recent advances in metabolic flux analysis including genome-scale constraints-based flux analysis and its applications in metabolic engineering are reviewed. Various computational aspects of constraints-based flux analysis including genome-scale stoichiometric models, additional constraints used for the improved accuracy, and several algorithms for identifying the target genes to be manipulated are described. Also, some of the successful applications of metabolic flux analysis in metabolic engineering are reviewed. Finally, we discuss the limitations that need to be overcome to make the results of genome-scale flux analysis more realistically represent the real cell metabolism.
 Hyun Uk Kim | Hyun Uk Kim received his BS in biotechnology from Yonsei University, and is a graduate student at the Metabolic and Biomolecular Engineering Laboratory at Korea Advanced Institute of Science and Technology (KAIST). His current research is concerned with metabolic flux analysis, development of genome-scale stoichiometric models and integration of biological information for rational design of metabolic engineering experiments and identification of drug targets in silico. |
 Tae Yong Kim | Tae Yong Kim obtained his BS in chemical engineering from Korea University. He received MS in chemical and biomolecular engineering from KAIST in 2005, during which he conducted research on in silico metabolic engineering of microorganisms. He is currently a PhD candidate working on modeling genome-scale metabolic networks and their applications at KAIST. |
 Sang Yup Lee | Sang Yup Lee is a Distinguished Professor and LG Chem Chair Professor at the Department of Chemical and Biomolecular Engineering at KAIST. He is a Director of Center for Systems and Synthetic Biotechnology, Director of BioProcess Engineering Research Center, Director of Bioinformatics Research Center, and Co-Director of the Institute for the BioCentury. He is also serving as senior editor, associate editor, and editorial board member of 12 journals including Biotechnology and Bioengineering, Applied Microbiology and Biotechnology, and Biotechnology Journal. His research interests include metabolic engineering, systems biotechnology, synthetic biology, white biotechnology, and nanobiotechnology. |
Metabolic engineering and metabolic fluxes
Cells including microorganisms have been rigorously studied to unravel mysteries of life for more than a century. Cells have also been employed as miniaturized chemical plants that produce various chemicals useful for mankind.1–4 The sharp increase of oil price together with our increasing concerns over the environmental impacts of crude oil-based industries are prompting a shift towards bio-based processes.5–7 However, the bio-based processes are generally inefficient due to the limited metabolic capacity of the cell towards the production of a desired product. This is because the objective of microbial metabolism (e.g., survival and replication) is different from that of ours (e.g., enhanced production of our desired product).Metabolic engineering has emerged to fulfill this purpose, which can be defined as purposeful modification of metabolic and cellular networks by employing various experimental techniques to achieve desired goals.8–10 What distinguishes metabolic engineering from genetic engineering and old-fashioned strain improvement before the emergence of recombinant DNA technique is that it considers metabolic and other cellular network as a whole to identify targets to be engineered. In this sense, metabolic flux is an essential concept in the practice of metabolic engineering. Although gene expression levels and the concentrations of proteins and metabolites in the cell can provide clues to the status of metabolic network, they have inherent limitations in fully describing the cellular phenotype due to the lack of information on the correlations among these cellular components. Metabolic fluxes represent the reaction rates in metabolic pathways, and serve to integrate these factors through a mathematical framework.9,11,12 Thus, metabolic fluxes can be considered as one way of representing the phenotype of the cell as a result of interplays among various cell components; the observed metabolic flux profiles reflect the consequences of interconnected transcription, translation, and enzyme reactions incorporating complex regulations.
Metabolic flux analysis (MFA) is an analytical technique that quantifies intracellular metabolic fluxes and dissects the functional aspects of metabolic network into greater details. MFA is based on mass balances around intracellular metabolites under the pseudo-steady state assumption, and two methods have generally been used: 13C-based flux analysis and constraints-based flux analysis. The former utilizes an isotope labeled carbon source and analyzes 13C enrichment patterns of metabolites with nuclear magnetic resonance (NMR) or gas chromatography–mass spectrometry (GC–MS). The outcome is used as input data for mathematical calculations that estimate the in vivo fluxes.11–15 Even though this allows relatively accurate estimation of intracellular fluxes, difficulties in experiments and subsequent calculations using a large-sized metabolic network are limiting the wide-spread use.
Constraints-based flux analysis is a general term for optimization-based simulation techniques, and various algorithms are available for this (Table 1). First, a stoichiometric model is reconstructed based on genomic information and literature. It is then simulated by linear optimization technique using an appropriate objective function (e.g. maximization of cell growth rate) and constraints that restrict the solution space within cell's capacity16 (Fig. 1). Importantly, it enables systematically predicting and evaluating the effects of genetic and/or environmental perturbations on the cell on a global scale, and suggests what parts of the system (e.g.genes) need to be modified for the directed improvement of the system. This cannot be intuitively gained by handling individual reactions, and thus this method enables more rational and efficient design of real metabolic engineering experiments. In addition to genome-scale stoichiometric modeling and its use in constraints-based flux analysis, there are two approaches that allow pathway analysis: elementary (flux) mode analysis17,18 and extreme pathway analysis.19 They allow identification of minimal set of systemic pathways and all possible steady-state flux distributions that the network can inherently achieve.20,21
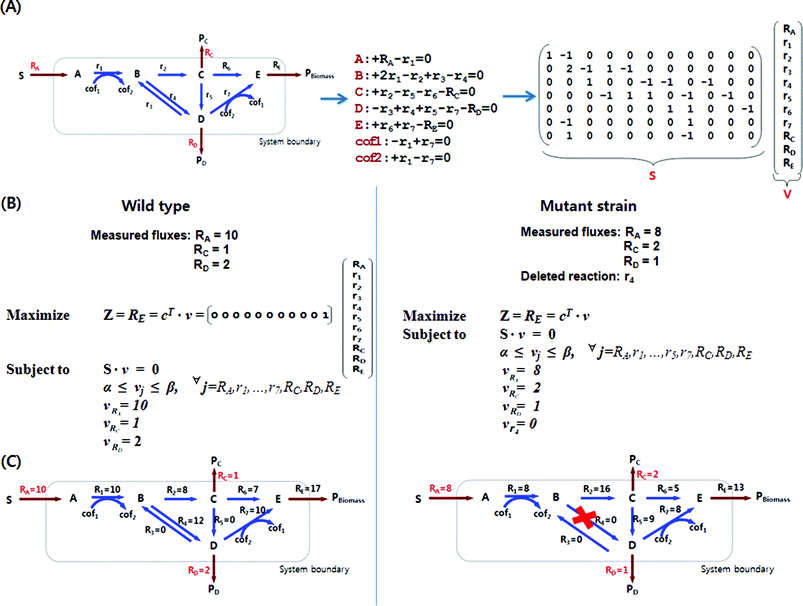 | ||
| Fig. 1 Construction of stoichiometric matrix for a model metabolic network and constraints-based flux analysis. (A) A model metabolic network consists of 7 internal and 4 external metabolites with 11 reactions. From this network, mass balances for each metabolite can be set up as linear equations, and they can be converted into the form of matrix S on the right. Reactants and products in the reaction have negative and positive stoichiometric coefficients, respectively. In this example, internal reactions are denoted by r, and reactions that span the system boundary are denoted by R. The stoichiometric coefficient for metabolite B is 2 as the reaction r1 generates two molecules of B from one molecule of A. (B) Optimization by linear programming is formulated with an objective function (Z) of maximizing the biomass formation (growth) rate (RE) subject to mass balances and additional constraints. Each reaction flux, vi, is subject to lower and upper bound constraints, represented as αi and βi, respectively. Also, measured fluxes, RA, Rc and RD in this example, can be used as additional constraints. c is a vector that specifies which flux to optimize. Other objective functions such as maximization of product formation and minimization of byproduct formation can be used. Knock-out mutant strains can be similarly simulated by setting the reaction flux of the knocked-out gene to zero. Mutant strain shown in this example has a deleted reaction r4 to increase the production rate of the metabolite Pc. Be noted that the measured fluxes change due to the deleted reaction, which is often observed in real examples. (C) Intracellular metabolic flux distributions calculated by constraints-based flux analysis are shown. Fluxes are represented in mmol (g dry cell weight h)−1. | ||
| Algorithma | Input | Output | Objective | Ref. |
|---|---|---|---|---|
| a The inputs shown for FBA are common for all algorithms, and thus are not shown. Abbreviations of algorithms are: FBA, flux balance analysis; SR-FBA, steady-state regulatory flux balance analysis; ROOM, regulatory on/off minimization; TMFA, thermodynamic–metabolic flux analysis; OMNI, optimal metabolic network identification; MOMA, minimization of metabolic adjustment. Abbreviations of solvers are: LP, linear programming; QP, quadratic programming, MILP, mixed integer linear programming. | ||||
| FBA | Various constraints such as substrate uptake rate, metabolite excretion rate, maintenance energy and capacity limits | Metabolic fluxes | Predicts intracellular flux distribution with maximization of an objective function (e.g. biomass, metabolite production, etc.) using LP | 16 |
| SR-FBA | Constraints for regulations, genes to reactions mapping, reaction enzyme state, and reaction predicates | Metabolic fluxes, gene expression status | Predicts gene expression and metabolic fluxes in a genome-scale integrated metabolic-regulatory model using MILP | 35 |
| ROOM | Thresholds determining significance of the flux change and their relative and absolute ranges of tolerance | Metabolic fluxes of the knock-out mutant | Minimizes the number of significant flux changes in the knock-out mutant compared to the wild-type using MILP | 36 |
| TMFA | Gibbs free energy change of a reaction | Metabolic fluxes free of thermodynamic infeasibilities | Predicts intracellular flux distribution with additional thermodynamic constraints using MILP | 42 |
| OMNI | Experimentally determined flux distribution, number of reactions to be knocked-out | Bottleneck reactions to be removed in the model | Identifies a set of reactions in the model whose removal improves the agreement between the model predictions and experimental data using MILP | 45 |
| MOMA | Metabolic flux profile of the wild-type | Metabolic fluxes of the knock-out mutant | Minimizes the Euclidian distance from a wild type flux distribution under knock-out condition using QP | 49 |
| OptKnock | Minimal growth rate, number of genes to be knocked-out | Gene knock-out targets for biochemical production | Predicts gene knock-out targets through bilevel optimization framework using MILP | 51 |
| OptGene | Number of individuals forming a population, | Gene knock-out targets for biochemical production | Predicts gene knock-out targets using genetic algorithm and constraints-based flux analysis (e.g.FBA, MOMA, ROOM, etc.) | 48 |
| OptReg | Minimal growth rate, regulation strength parameter, number of reactions to be modulated or knocked-out | Gene knock-out or up/down-regulation targets for biochemical production | Determines the activation/inhibition and elimination reaction set for biochemical production using MILP | 52 |
This Highlight focuses on the recent developments in constraints-based flux analysis of stoichiometric models and its applications in metabolic engineering of microorganisms. We describe the current status of genome-scale stoichiometric models, the strategies of incorporating additional constraints for more accurate flux simulation, gene targeting algorithms for metabolic pathway engineering, and their applications in actual metabolic engineering. Readers can refer to other excellent reviews9,11,12,14,16 for more general information on MFA, which is not repeated in this Highlight.
Reconstruction of the genome-scale stoichiometric metabolic models
A starting point of MFA is the development of a stoichiometric model that can describe the cellular metabolism. This can be achieved by setting mass balance equations for the cellular metabolites and accounting for their stoichiometry in the form of matrices and vectors22 (Fig. 1(A)). It should be noted that the stoichiometric model we are discussing here is not just a metabolic network, but is the validated network which can successfully describe the metabolic phenotypes by MFA. The scale of such genome-scale stoichiometric models has been greatly expanding in order to incorporate newly discovered biological knowledge and information into the model, thereby improving its accuracy (Fig. 2). The stoichiometric models of several microorganisms including Escherichia coli, by far the best characterized organism, have been reconstructed, but were limited to mainly central metabolic pathways23 until the mid-1990s. The situation drastically changed with the breakthrough of genome sequencing technique and bioinformatic analyses, which accelerated generation of systems-wide data. In case of E. coli, its stoichiometric model has gradually been expanded towards a genome-scale by incorporating more genetic and biochemical information24–26 (Fig. 2). Recently, Feist et al.26 reported the development of so far the most comprehensive metabolic model of E. coli. This model composed of 2077 reactions and 1039 metabolites describes the metabolism of E. coli in a much more expanded manner than the previous models in terms of the number of genes (1260 genes) incorporated in the model. In particular, thermodynamic analyses were performed for most reactions to confirm their directionality. It was shown that this new model allowed better prediction results compared with the previous model developed by Reed et al.25 The stoichiometric models for H. influenza,27,28H. pylori,29,30M. succiniciproducens31 and S. cerevisiae32,33 were similarly expanded by incorporating more genes and their corresponding reactions, and more accurately describing biochemical reactions (Fig. 2). In addition, the expanded versions of H. pylori30 and S. cerevisiae32,33genome-scale models have element- and charge-balanced reactions, while that of M. succiniciproducens31 was rigorously updated using the reannotated genomic information and validation experiments.25Genome-scale stoichiometric models for other microorganisms and even mammalian cells have also been developed, and are shown in Fig. 2.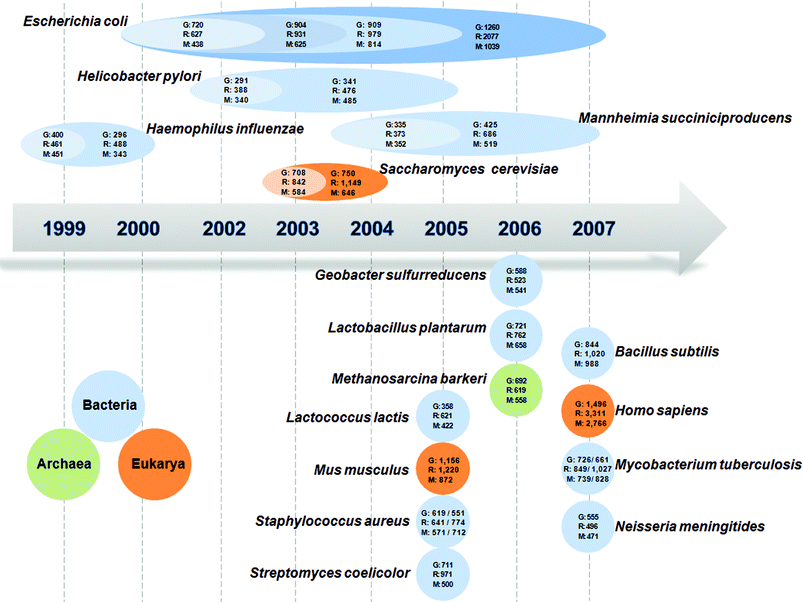 | ||
| Fig. 2 Development of genome-scale stoichiometric models for various organisms, which are continually expanding. The models are available for bacteria including E. coli,24–26H. influenza,27,28H. pylori,29,30M. succiniciproducens,31,61B. subtilis,62G. sulfurreducens,63L. plantarum,64L. lactis,65M. tuberculosis,66,67N. meningitides,68S. aureus,69,70 and S. coelicolor,71 for archae including M. barkeri,72 and for eukaryotes including S. cerevisiae,32,33H. sapiens73 and M. musculus.74 ‘G’ stands for the number of genes incorporated in the model, ‘R’ for the number of reactions, and ‘M’ for the number of metabolites. For S. aureus, the stoichiometric model on the left side of the slash was developed by Becker and Palsson,69 while that on the right side was by Heinemann et al.70 For M. tuberculosis, left model information before the slash is from Beste et al.67 and the right one is from Jamshidi and Palsson.66 | ||
Additional constraints to the stoichiometric model
As comprehensive genome-scale stoichiometric models have been expected to be a promising tool for systems-level understanding of metabolism, they are constantly being elaborated. Particularly, the solution space of metabolic fluxes, which represents all physiologically plausible states, is further reduced by employing additional constraints so as to make the simulation results become more realistic (Fig. 3). There has been much effort to achieve this in several different ways (Table 1). The first method is the addition of regulatory constraints (Fig. 3(D)). The metabolic network can be combined with a transcriptional regulatory network, in which biological correlations among gene, proteins and reactions are described by Boolean logics AND, OR and NOT34,35 (Fig. 3). Expression of a gene, and thus the presence of enzyme or protein is represented by the value 1, while the opposite state is described by the value 0. Once the genes in the transcriptional regulatory network are appropriately assigned with binary values under specific conditions, other remaining genes can also be automatically assigned with binary values according to the regulatory rules. The flux can be set to zero if its corresponding gene is inactive (= 0), thereby reducing the solution space. A method called steady-state regulatory flux balance analysis (SR-FBA) implements this Boolean logic by integrating it with a conventional genome-scale metabolic model.35 Regulatory on/off minimization (ROOM) is another method that accounts for regulatory information to predict flux distributions in gene knock-out mutant, but does not use Boolean logic. Instead, it resorts to the observation that the mutant minimizes the number of significant flux changes compared to the wild-type.36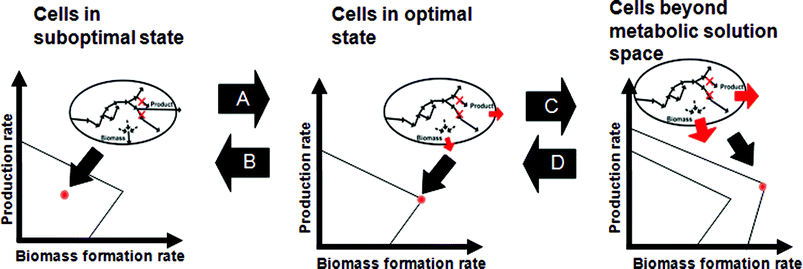 | ||
| Fig. 3 Changing states of cellular physiology through environmental and genetic perturbations within biologically feasible solution space. (A) Under a given condition, cells are at their suboptimal state, and can reach their optimal state through adaptive evolution. (B) MOMA allows identification of the state with the minimal metabolic adjustment as a result of gene knock-outs leading to suboptimal state of the cell. (C) In the rightmost graph, the outermost boundary refers to the biological solution space beyond the current metabolic capacity, which is possible by altered regulation/metabolic engineering. (D) In the opposite direction, superior cells beyond metabolic solution space can approach the real cell by returning the regulatory circuits back to that of wild-type. Or, they can be interpreted as reducing the solution space by altered regulation/metabolic engineering and/or imposing thermodynamic constraints. | ||
The second method is to reduce the solution space of metabolic fluxes by the addition of thermodynamic constraints37–42 (Fig. 3(D)). Conventional MFA relies only on mass balance of metabolites, but does not account for thermodynamics of reactions; this makes several reactions to take place even though they are not thermodynamically feasible. Based on the first and second laws of thermodynamics, the sum of Gibbs free energy changes around flux loops should be zero, and the Gibbs free energy change should be negative for a reaction to proceed. Consequently, mass balance constraints can be augmented with thermodynamic constraints such that the metabolic network no longer contains thermodynamically infeasible flux loops and reactions. In particular, thermodynamics-based metabolic flux analysis (TMFA) recently developed by Henry et al.42 allows genome-scale calculation of intracellular fluxes by employing additional linear constraints that segregate fluxes that violate the above two thermodynamic criteria.
The third method is to constrain the solution space by employing experiments-based metabolic flux data, which are typically obtained by 13C-based flux analysis.43–45 Although constraints-based flux analysis can predict the intracellular fluxes on a genome-scale, the fluxes calculated may not represent real metabolic fluxes because the model cannot perfectly describe the cellular metabolism. Furthermore, the optimal results obtained by using the objective function during the constraints-based flux analysis may be different from real cellular metabolism (i.e., the cellular metabolism may operate in a suboptimal mode).46,47 The 13C-based flux analysis calculates in vivo fluxes using isotope-labeled substrate, such as 13C-labeled glucose, and its resulting fluxes are believed to be more reliable.11,12 Thus, the 13C-based metabolic fluxes can serve as realistic constraints to reduce the solution space of constraints-based genome-scale flux analysis. Herrgard et al.45 devised a method called optimal metabolic network identification (OMNI) that identifies a set of reaction changes to be made to the model based on the principle described above, so that predictions from the modified model and experimental data become coherent. Diagnosis of E. coli strains engineered for lactate overproduction with OMNI suggested several factors that limit their performance and the strategies to overcome these limits. Thus, this approach may be useful for designing metabolic engineering strategies.
Predicting gene targets based on metabolic flux analysis
Constraints-based flux analysis can be a powerful tool for predicting the target genes to be engineered by analyzing the re-distributed metabolic fluxes upon specific gene modifications. In particular, the effects of knocking-out genes on metabolic flux distribution can be easily examined by setting the fluxes of the knocked-out reactions to zero (Fig. 1). Several algorithms have been developed for this purpose.48The minimization of metabolic adjustment (MOMA)49 algorithm allows prediction of the suboptimal distribution of metabolic fluxes in knock-out mutants by minimizing the changes in the flux distribution of the mutant with respect to the wild type instead of maximizing the biomass formation in the mutant. This algorithm has successfully been applied to construct intensively engineered microorganisms for improved production of lycopene50 and L-valine.4
OptKnock51 and OptGene48 allow identification of target genes to be knocked out, while more recently developed OptReg52 allows prediction of the genes to be down- and up-regulated in addition to simple knock-outs. OptKnock method is an approach that identifies knock-out targets by formulating a bi-level linear optimization problem with mixed integer linear programming (MILP).51 OptKnock usually finds a set of gene deletions that maximizes the flux towards a desired product, while the internal flux distribution is still operated such that growth is optimized. Thus, the identified gene deletions will force the microorganism to produce the desired bioproduct in order to achieve maximum growth. Using this method, the gene knock-out targets for enhancing the production of various metabolites could be predicted; for example, a quadruple gene deletion mutant E. coli (pta–adhE–pfk–glk mutant) capable of high level production of lactate could be identified.53 OptGene48 is an improved algorithm of OptKnock developed by employing genetic algorithm to reduce computational time.
Pharkya and Maranas introduced a simulation method called OptReg52 that allows examination of the effects of homologous gene amplification (up-regulation of genes) in addition to those of down-regulation and knock-out of genes. This can be considered as an improved version of OptKnock by employing additional constraints that describe the magnitude of fluxes to be regulated. The proposed strategy was used to identify the genes to be engineered for enhancing ethanol production in E. coli. Pharkya and co-workers also developed a computational framework termed OptStrain for the overproduction of a wide range of biochemicals, which may be novel to the host microorganism by adding heterologous metabolic pathways and/or deleting pathways that hamper the production of targeted compounds.54 It was used to identify three heterologous reactions that need to be introduced into E. coli for the production of vanillin, which is followed by systematic gene knock-out studies to enhance the yield. The characteristics of these algorithms are comparatively summarized in Table 1.
Metabolic engineering based on metabolic flux analysis
As described above, MFA allows systems-level understanding of cellular metabolism, and thus allows identification of the target genes to be engineered at the systems level; it considers extensive stoichiometric linking of local and distant reactions through cofactors and energetics, so that it predicts gene targets on a global scale, which cannot be intuitively determined. Recently, several successful examples of metabolic engineering based on the results of metabolic flux analysis have appeared for the production of succinic acid,2,55lactic acid,53lycopene,3,50 and L-valine4 by E. coli, and ethanol56 by S. cerevisiae.Fong et al.53 reported an interesting observation that a strain engineered based on MFA predictions is at suboptimal state, which can undergo adaptive evolution to reach the state of the metabolically optimal in silicocell (Fig. 3(A)). Strong evidences were found that cells maximize their biomass formation as they undergo adaptive evolution.57,58 In other words, incorrect predictions of MFA may be partly due to incomplete adaptive evolution of the cell under the condition examined. Based on this concept, Fong et al.53 first constructed mutants overproducing lactic acid as predicted by OptKnock, and then conducted the adaptive evolution experiments. It was confirmed that mutants did actually evolve towards the maximization of growth rate and lactic acid secretion rate.
Additional gene manipulations associated with various types of regulations can be combined with the above in silico prediction methods to result in more drastic metabolic engineering results beyond the normal limit (Fig. 3(C)).3,4 In one of the recent examples, genome engineering to knock out negative regulations, engineering of new target genes identified by transcriptome profiling, and knocking-out the metabolic genes based on MOMA were all combined to develop an E. coli strain capable of enhanced L-valine production.4 First, an L-valine producing E. coli base strain was constructed by knocking out all known feedback inhibitions, removing attenuation controls, and amplifying activities of the direct L-valine biosynthetic enzymes. This base strain was further improved in a stepwise manner using new information deciphered from transcriptome profiling; a global regulator leucine responsive protein (Lrp) and an L-valine exporter protein were identified. Then, MOMA was employed to identify the genes to be further knocked out. The resulting triple knock-out mutant possessing upgraded regulatory circuits and exporter system produced a remarkable yield of valine (0.378 g L-valine per g glucose). It should be noted that engineering targets for regulatory circuits, global regulator and exporter were selected based on the previous literature information accumulated by many years of research and/or new data generated by omics studies. Understanding the regulatory circuits and subsequent engineering based on the simulation will be one of the topics to which future research should be directed.
Future prospects
Recent advances in genomics, transcriptomics, proteomics and metabolomics are providing us with unprecedentedly large amounts of biological data, which serve as driving forces of systems biology.59,60 An important role of systems biology in metabolic engineering is to comprehensively collect such global cellular information, and to combine them in quantitative and system-wide manner in order to generate new knowledge of the biological system suitable for developing metabolic engineering strategies. Constraints-based flux analysis serves this role of systems biology in metabolic engineering. The key issue here would be how to accurately describe and predict metabolic flux distributions inside a cell. Therefore, current limitations of simulating genome-scale stoichiometric models with constraints-based flux analysis suggest future research direction. First, the current in silicocell lacks in the ability of simulating sophisticated gene regulatory and signaling pathways, which makes it difficult to accurately predict the cell phenotype in response to changing environment. This could be largely resolved by employing more equations and conditions as constraints mentioned throughout this article that reflect these regulations and signaling during the pseudo-steady state simulation. However, much effort needs to be devoted to more accurately incorporate these regulations and signaling, which are inherently heterogeneous information. Second, there is a need to develop a method to simulate the cell growth states other than those in exponential phase or chemostat culture because metabolic engineering is sometimes applied to the cells from a stationary phase, for instance to produce secondary metabolites. Third, a new validation method is needed to monitor the accuracy of the MFA results. As the constraints-based flux analysis relies on linear programming-based optimization of an underdetermined system, the absolute values of fluxes calculated may not be the real ones. A simple solution is the use of flux values determined by isotopomer analysis as additional constraints for improving the accuracy of constraints-based flux analysis.44 A better, yet simple way, of improving the accuracy of flux calculation is needed. Despite of these problems to be solved, the MFA of the genome-scale model will play more and more important roles in designing metabolic engineering strategies to achieve improved metabolic performance.Acknowledgements
This work was supported by the Korean Systems Biology Program from the Ministry of Science and Technology through the Korea Science and Engineering Foundation (No. M10309020000-03B5002-00000). Further support by LG Chem Chair Professorship is appreciated.References
- S. H. Hong, S. J. Park, S. Y. Moon, J. P. Park and S. Y. Lee, Biotechnol. Bioeng., 2003, 83, 854–863 CrossRef CAS.
- S. J. Lee, D. Y. Lee, T. Y. Kim, B. H. Kim, J. Lee and S. Y. Lee, Appl. Environ. Microbiol., 2005, 71, 7880–7887 CrossRef CAS.
- H. Alper, K. Miyaoku and G. Stephanopoulos, Nat. Biotechnol., 2005, 23, 612–616 CrossRef CAS.
- J. H. Park, K. H. Lee, T. Y. Kim and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7797–7802 CrossRef CAS.
- D. R. Lovley, Nat. Rev. Microbiol., 2003, 1, 35–44 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, Jr., J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef.
- G. Stephanopoulos, Science, 2007, 315, 801–804 CrossRef CAS.
- J. E. Bailey, Science, 1991, 252, 1668–1675 CrossRef CAS.
- G. Stephanopoulos, Metab. Eng., 1999, 1, 1–11 CrossRef CAS.
- S. Y. Lee, D. Y. Lee and T. Y. Kim, Trends Biotechnol., 2005, 23, 349–358 CrossRef CAS.
- J. Nielsen, J. Bacteriol., 2003, 185, 7031–7035 CrossRef CAS.
- U. Sauer, Mol. Syst. Biol., 2006, 2, 62.
- K. Schmidt, A. Marx, A. A. de Graaf, W. Wiechert, H. Sahm, J. Nielsen and J. Villadsen, Biotechnol. Bioeng., 1998, 58, 254–257 CrossRef CAS.
- W. Wiechert, Metab. Eng., 2001, 3, 195–206 CrossRef CAS.
- E. Fischer, N. Zamboni and U. Sauer, Anal. Biochem., 2004, 325, 308–316 CrossRef CAS.
- N. D. Price, J. L. Reed and B. O. Palsson, Nat. Rev. Microbiol., 2004, 2, 886–897 Search PubMed.
- S. Schuster, T. Dandekar and D. A. Fell, Trends Biotechnol., 1999, 17, 53–60 CrossRef CAS.
- S. Schuster, D. A. Fell and T. Dandekar, Nat. Biotechnol., 2000, 18, 326–332 CrossRef CAS.
- C. H. Schilling, J. S. Edwards, D. Letscher and B. O. Palsson, Biotechnol. Bioeng., 2000, 71, 286–306 CrossRef CAS.
- S. Klamt and J. Stelling, Trends Biotechnol., 2003, 21, 64–69 CrossRef CAS.
- J. A. Papin, J. Stelling, N. D. Price, S. Klamt, S. Schuster and B. O. Palsson, Trends Biotechnol., 2004, 22, 400–405 CrossRef CAS.
- G. N. Stephanopoulos, A. A. Aristidou and J. Nielsen, Metabolic Engineering – Principles and Methodologies, Academic Press, San Diego, CA, 1998 Search PubMed.
- A. Varma and B. O. Palsson, Appl. Environ. Microbiol., 1994, 60, 3724–3731 CAS.
- J. S. Edwards and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5528–5533 CrossRef CAS.
- J. L. Reed, T. D. Vo, C. H. Schilling and B. O. Palsson, Genome Biol., 2003, 4, R54 Search PubMed.
- A. M. Feist, C. S. Henry, J. L. Reed, M. Krummenacker, A. R. Joyce, P. D. Karp, L. J. Broadbelt, V. Hatzimanikatis and B. O. Palsson, Mol. Syst. Biol., 2007, 3, 121.
- J. S. Edwards and B. O. Palsson, J. Biol. Chem., 1999, 274, 17410–17416 CrossRef CAS.
- C. H. Schilling and B. O. Palsson, J. Theor. Biol., 2000, 203, 249–283 CrossRef CAS.
- C. H. Schilling, M. W. Covert, I. Famili, G. M. Church, J. S. Edwards and B. O. Palsson, J. Bacteriol., 2002, 184, 4582–4593 CrossRef CAS.
- I. Thiele, T. D. Vo, N. D. Price and B. O. Palsson, J. Bacteriol., 2005, 187, 5818–5830 CrossRef CAS.
- T. Y. Kim, H. U. Kim, J. M. Park, H. Song, J. S. Kim and S. Y. Lee, Biotechnol. Bioeng., 2007, 97, 657–671 CrossRef CAS.
- J. Forster, I. Famili, P. Fu, B. O. Palsson and J. Nielsen, Genome Res., 2003, 13, 244–253 CrossRef CAS.
- N. C. Duarte, M. J. Herrgard and B. O. Palsson, Genome Res., 2004, 14, 1298–1309 CrossRef CAS.
- M. W. Covert, E. M. Knight, J. L. Reed, M. J. Herrgard and B. O. Palsson, Nature, 2004, 429, 92–96 CrossRef CAS.
- T. Shlomi, Y. Eisenberg, R. Sharan and E. Ruppin, Mol. Syst. Biol., 2007, 3, 101.
- T. Shlomi, O. Berkman and E. Ruppin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7695–7700 CrossRef CAS.
- D. A. Beard, S. D. Liang and H. Qian, Biophys. J., 2002, 83, 79–86 CrossRef CAS.
- H. Qian, D. A. Beard and S. D. Liang, Eur. J. Biochem., 2003, 270, 415–421 CrossRef CAS.
- D. A. Beard, E. Babson, E. Curtis and H. Qian, J. Theor. Biol., 2004, 228, 327–333 CrossRef CAS.
- D. A. Beard and H. Qian, Am. J. Physiol. Endocrinol. Metab., 2005, 288, E633–644 CAS.
- C. S. Henry, M. D. Jankowski, L. J. Broadbelt and V. Hatzimanikatis, Biophys. J., 2006, 90, 1453–1461 CAS.
- C. S. Henry, L. J. Broadbelt and V. Hatzimanikatis, Biophys. J., 2007, 92, 1792–1805 CAS.
- L. M. Blank, L. Kuepfer and U. Sauer, Genome Biol., 2005, 6, R49 Search PubMed.
- T. Y. Kim and S. Y. Lee, J. Microbiol. Biotechnol., 2006, 16, 1139–1143 CAS.
- M. J. Herrgard, S. S. Fong and B. O. Palsson, PLoS Comput. Biol., 2006, 2, e72 Search PubMed.
- B. O. Palsson, N. D. Price and J. A. Papin, Trends Biotechnol., 2003, 21, 195–198 CrossRef CAS.
- E. Fischer and U. Sauer, Nat. Genet., 2005, 37, 636–640 CrossRef CAS.
- K. R. Patil, I. Rocha, J. Forster and J. Nielsen, BMC Bioinformatics, 2005, 6, 308 CrossRef.
- D. Segre, D. Vitkup and G. M. Church, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15112–15117 CrossRef CAS.
- H. Alper, Y. S. Jin, J. F. Moxley and G. Stephanopoulos, Metab. Eng., 2005, 7, 155–164 CrossRef CAS.
- A. P. Burgard, P. Pharkya and C. D. Maranas, Biotechnol. Bioeng., 2003, 84, 647–657 CrossRef CAS.
- P. Pharkya and C. D. Maranas, Metab. Eng., 2006, 8, 1–13 CrossRef CAS.
- S. S. Fong, A. P. Burgard, C. D. Herring, E. M. Knight, F. R. Blattner, C. D. Maranas and B. O. Palsson, Biotechnol. Bioeng., 2005, 91, 643–648 CrossRef CAS.
- P. Pharkya, A. P. Burgard and C. D. Maranas, Genome Res., 2004, 14, 2367–2376 CrossRef CAS.
- Q. Wang, X. Chen, Y. Yang and X. Zhao, Appl. Microbiol. Biotechnol., 2006, 73, 887–894 CrossRef CAS.
- C. Bro, B. Regenberg, J. Forster and J. Nielsen, Metab. Eng., 2006, 8, 102–111 CrossRef CAS.
- R. U. Ibarra, J. S. Edwards and B. O. Palsson, Nature, 2002, 420, 186–189 CrossRef CAS.
- S. S. Fong and B. O. Palsson, Nat. Genet., 2004, 36, 1056–1058 CrossRef CAS.
- H. Kitano, Science, 2002, 295, 1662–1664 CrossRef CAS.
- G. Stephanopoulos, H. Alper and J. Moxley, Nat. Biotechnol., 2004, 22, 1261–1267 CrossRef CAS.
- S. H. Hong, J. S. Kim, S. Y. Lee, Y. H. In, S. S. Choi, J. K. Rih, C. H. Kim, H. Jeong, C. G. Hur and J. J. Kim, Nat. Biotechnol., 2004, 22, 1275–1281 CrossRef CAS.
- Y. K. Oh, B. O. Palsson, S. M. Park, C. H. Schilling and R. Mahadevan, J. Biol. Chem., 2007, 282, 28791–28799 CrossRef CAS.
- R. Mahadevan, D. R. Bond, J. E. Butler, A. Esteve-Nunez, M. V. Coppi, B. O. Palsson, C. H. Schilling and D. R. Lovley, Appl. Environ. Microbiol., 2006, 72, 1558–1568 CrossRef CAS.
- B. Teusink, A. Wiersma, D. Molenaar, C. Francke, W. M. de Vos, R. J. Siezen and E. J. Smid, J. Biol. Chem., 2006, 281, 40041–40048 CrossRef CAS.
- A. P. Oliveira, J. Nielsen and J. Forster, BMC Microbiol., 2005, 5, 39 Search PubMed.
- N. Jamshidi and B. O. Palsson, BMC Syst. Biol., 2007, 1, 26 Search PubMed.
- D. J. Beste, T. Hooper, G. Stewart, B. Bonde, C. Avignone-Rossa, M. E. Bushell, P. Wheeler, S. Klamt, A. M. Kierzek and J. McFadden, Genome Biol., 2007, 8, R89 Search PubMed.
- G. J. Baart, B. Zomer, A. de Haan, L. A. van der Pol, E. C. Beuvery, J. Tramper and D. E. Martens, Genome Biol., 2007, 8, R136 Search PubMed.
- S. A. Becker and B. O. Palsson, BMC Microbiol., 2005, 5, 8 Search PubMed.
- M. Heinemann, A. Kummel, R. Ruinatscha and S. Panke, Biotechnol. Bioeng., 2005, 92, 850–864 CrossRef CAS.
- I. Borodina, P. Krabben and J. Nielsen, Genome Res., 2005, 15, 820–829 CrossRef CAS.
- A. M. Feist, J. C. Scholten, B. O. Palsson, F. J. Brockman and T. Ideker, Mol. Syst. Biol., 2006, 2, 0004.
- N. C. Duarte, S. A. Becker, N. Jamshidi, I. Thiele, M. L. Mo, T. D. Vo, R. Srivas and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1777–1782 CrossRef CAS.
- K. Sheikh, J. Forster and L. K. Nielsen, Biotechnol. Prog., 2005, 21, 112–121 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |