Synthesis, evaluation and application of novel bifunctional N,N-di-isopropylbenzylamineboronic acid catalysts for direct amide formation between carboxylic acids and amines†‡
Kenny
Arnold
a,
Andrei S.
Batsanov
a,
Bryan
Davies
b and
Andrew
Whiting
*a
aDepartment of Chemistry, Durham University, South Road, Durham, UK, DH1 3LE. E-mail: andy.whiting@durham.ac.uk
bGlaxoSmithKline Research & Development Ltd., Gunnels Wood Road, Stevenage, Hertfordshire, UK, SG1 2NY
First published on 22nd November 2007
Abstract
Three new derivatives of N,N-di-isopropylbenzylamine-2-boronic acid have been prepared by directed metallation-borylation methods, to derive the 3-fluoro, 3-methoxy and 5-trifluoromethyl systems. The addition of an electron withdrawing group does increase the reactivity of such systems to act as improved direct amide formation catalysts under the most ambient conditions employed to date. In contrast, an electron donating group does result in considerable lowering of catalyst reactivity. DoE studies have been used to identify the ideal reaction conditions under which these types of catalysts should be used, typified by the parent system N,N-di-isopropylbenzylamine-2-boronic acid. This shows best performance at a 5 mol% loading and under higher dilution conditions, which most likely reflect the drying capacity of the solvent.
Introduction
A generally applicable, efficient direct amide formation from amines and carboxylic acids, via the corresponding ammonium salt, that proceeds under relatively ambient conditions remains elusive. Since the first report of ammonium carboxylate pyrolyses,1 there have been various developments in the intervening years, including relatively small reductions in temperature,2 the use of various acid catalysts3 and microwave assistance.4 However, none of these approaches have provided a solution to facile direct amide formation, and the more usual stoichiometric activation methods remain by far the most utilised approach.5 Initial reports of the application of organoboron compounds to assist amide formation,6 has resulted in the development of several useful boronic acid catalysts,7 however, the report that boric acid alone is an efficient catalyst under azeotropic conditions8 shows the need to be wary about the mode of action of electron deficient arylboronic acids and there is still a need to develop systems with higher reactivity, and at lower temperatures. To this end, we demonstrated the use of bifunctional amino-boronic acid derivatives9 for direct amide formation, and examined the previously essentially unaddressed issue of the competing degree of direct thermal amide formation. Our major aim was to develop catalytic systems which exhibit: (1) general reactivity for all types of amine and carboxylic acid substrates; (2) lower reaction temperatures to relatively ambient conditions, i.e. lower than refluxing toluene where possible, and certainly below refluxing xylene or mesitylene for example; and (3) zero propensity to proto-deboronation, and hence acting a source of boric acid. This resulted in the demonstration that ortho-N,N-di-isopropylbenzylaminoboronic acid system 1 has a bifunctional catalytic effect for direct amide formation reactions involving benzoic acid, and it could be used using refluxing fluorobenzene temperatures.10 Under these conditions, proto-deboronation does not compete and a comparison of the rates of amide formation between various boron-based catalysts under two different sets of reaction conditions clearly showed the enhanced activity of catalyst 1 for the more difficult substrates under the milder set of reaction conditions. In this paper, we report our further studies aimed at better understanding the factors that control catalyst activity, including the synthesis of further examples of these bifunctional aminoarylboronic acids and an examination of their comparative reactivity in the direct amide condensation versus existing systems. We also report associated optimisation of reaction conditions for different substrate combinations using statistical design of experiments (DoE) methods to gain a further insight into the key reaction parameters that affect reactivity.
Results and discussion
We decided to access novel derivatives of system 1, i.e. systems of type 2, in which the hindered N,N-di-isopropylaminobenzyl function was retained, since in previous work, clear benefit over the less hindered N,N-dimethyl system was demonstrated.10 In addition, the new systems 2 were chosen with the specific aim of testing the effect of changing the Lewis acidity of the boronic acid function by suitable choice of the functional group X, i.e. comparing electron withdrawing and electron donating substituents.Catalyst synthesis
The synthesis of new catalysts based on system 2, started with the fluoro- and methoxybenzylboronic acid, 6a and b, respectively (Scheme 1). Hence, N,N-di-isopropyl-3-fluorobenzylamine-2-boronic acid 6a was prepared starting with 3-fluorobenzoyl chloride 3a, resulting in the formation of amide 4a, upon reaction with di-isopropylamine, which was then subjected to directed metallation. Following several attempts to achieve directed ortho-metallation of 4a, attempts to directly isolate the corresponding boronic acid proved unsuccessful, and it was decided to access the pinacol ester 5a in order to simplify isolation and purification. Hence, reaction of amide 4a with sec-butyllithium followed by trimethylborate, followed by hydrolysis and esterification with pinacol derived the boronate ester 5a (Scheme 1).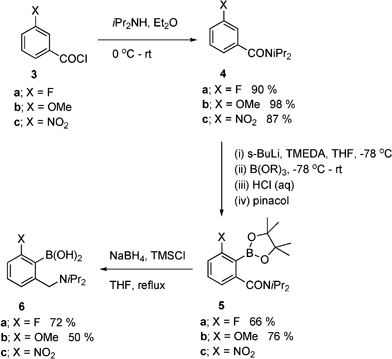 | ||
| Scheme 1 Synthesis of fluoro- and methoxy-substituted amino-boronic acids 6. | ||
The reduction method chosen to convert the pinacol ester 5a was selected in order to achieve both amide reduction and deprotection of the boronate ester in one step.9b Although the reduction proceeded well using sodium borohydride-trimethylsilyl chloride, isolation of the amino-boronic acid 6a proved troublesome, due to its high water solubility. However, use of an acidification-neutralisation sequence during the work up allowed direct extraction of the amino-boronic acid 6a, which could then be efficiently precipitated to give pure product in 72 % yield.
The same strategy was employed in order to synthesise N,N-di-isopropyl-3-methoxybenzylamine-2-boronic acid 6b. As before, the amidation of acid chloride 3b was facile, efficiently providing di-isopropylamide 4b (Scheme 1). However, the directed metallation of 4b proved problematic at first, and resulted in low conversion to the pinacol ester 5b, i.e.sec-butyllithium-TMEDA, –78 °C. This was solved by allowing much longer reaction times for the intermediate aryllithium to react with the borate electrophiles (ca. 12 h), resulting in the isolation of pinacol ester 5b in good yield. It is worth noting that metallation-borylation of amide 4b fails to proceed at all with n-butyllithium, whereas the alkyllithium source has no significant effect upon the metallation of 4a. In addition, attempted metallation of methoxy-substituted system 4b with lithium di-isopropylamide, followed by triisopropyl borate resulted in the formation of ester 5b, but in only 11 % yield.
Having obtained boronate ester 5b, reduction was attempted as for 5a, resulting in only low yields amino-boronic acid 6b (10 %). It appears that the methoxy system 6b has increased water solubility compared to 6a, and therefore, suitable changes to the work up procedure (THF evaporation and minimisation of aqueous solution and solvent volumes) resulted in a reasonably efficient isolation of 6b, which after subsequent recrystallisation gave a 50 % yield (Scheme 1).
In order to compare catalysts of type 2, systems with different electronic properties were required. Several attempts were made to access the system 6c, via formation of the corresponding amide 4c (Scheme 1). However, all attempts to achieve deprotonation of 4c to access boronate 5c led to the formation of intractable complex products, which is not in contradiction with other unsuccessful attempts to achieve lithiation of nitroaryl systems.11 As an alternative to the nitro-substituted system, the final catalyst prepared was the trifluoromethyl-substituted system, i.e.10, which proved reasonably straightforward to access, as outlined in Scheme 2. Thus, amidation of acid chloride 7 provided amide 8 in 98 % yield. Directed ortho-metallation under the same conditions as used for systems 5 (n- or s-BuLi-TMEDA) led to a mixture of ortho- and para-CF3 boronates, with low total conversion (ca. 10 %). A number of attempts at improvement (increasing reaction temperature, metallation time, etc.) failed to improve matters and alternative metallating agents were examined. A mixture of potassium tert-butoxide and BuLi12 gave no reaction, however, use of lithium di-isopropylamide afforded the para-CF3-substituted boronate 9 selectively and in high yield (95 %). Subsequent initial attempts at reduction of the amide 9 using borane-dimethyl sulfide in THF at reflux failed to give an observable reaction, however, using the TMSCl-borohydride conditions resulted in formation of boronic acid 10 after work up, albeit via a slow reaction, which resulted in only 40 % conversion in 40 h. The solution to this problem was to simply decrease the reaction concentration used for the reduction conditions, which resulted in complete conversion in 24 h (Scheme 2), and after modification of the work up conditions used for amino-boronic acids 6, the trifluoromethyl-boronic acid 10 was obtained in 60 % yield after recrystallisation.
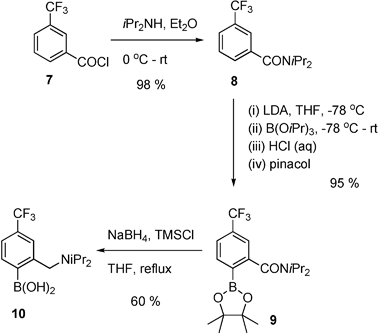 | ||
| Scheme 2 Synthesis of trifluoromethyl-substituted amine-boronic acid. | ||
Comparison of crystal structures
Crystals of both 6a and 10, which were suitable for single crystal X-ray analysis, were readily prepared, however, the methoxy derivative 6b evaded attempts to derive good quality crystals. However, fluoro and trifluoro derivatives 6a and 10, respectively, are essentially isostructural with one another and also with the analogue 1.9b Molecular conformations are also similar (Fig. 1). The C(1)BO(1)O(2) moiety is planar and inclined to the benzene ring plane by 25.8° (6a) or 21.8° (10). The larger twist in 6a obviously is caused by steric repulsion between the F and O(2) atoms. One hydroxyl group, O(1)H, forms an intramolecular hydrogen bond with the amino N atom, which is slightly pyramidalised, with the mean C–N–C angle of 113°. Molecules are linked into centrosymmetric dimers by pairs of hydrogen bonds O(2)–H⋯O(1)[1–x, 1–y, 1–z]. There are only marginal differences in bond distances between 1, 6a and 10 (Table 1). It is interesting to note that the trifluoromethyl system 10 exhibits a shorter C–B bond length compared with the fluoro system 6a, however, it is little different to the non-substituted system 1 (see Table 1). This probably suggests that there is not a major difference in Lewis acidity between systems 1 and 10, though 6a might be slightly less Lewis acidic. Unfortunately, we cannot compare these solid state structures with the methoxy derivative 6b, and 11B NMR also does not seem to suggest significant differences in the properties of the boronic acid function, which show resonances at δ 28.8,9b 28.4, 29.3 and 28.5 for 1, 6a, 6b and 10, respectively.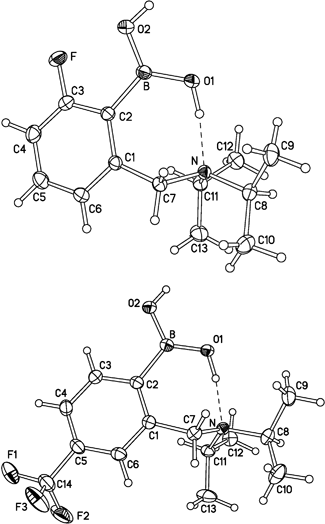 | ||
| Fig. 1 X-ray molecular structures of 6a (top) and 10. Thermal ellipsoids are drawn at 50 % probability level. | ||
| 1 | 6a | 10 | |
|---|---|---|---|
| C(2)–B | 1.588(2) | 1.5965(14) | 1.5876(17) |
| B–O(1) | 1.356(2) | 1.3547(13) | 1.3575(15) |
| B(1)–O(2) | 1.366(2) | 1.3581(13) | 1.3609(16) |
| O(1)⋯N | 2.637(1) | 2.607(1) | 2.623(1) |
| C(1)–C(2) | 1.412(2) | 1.4204(12) | 1.4169(16) |
| C(1)–C(7) | 1.522(2) | 1.5171(13) | 1.5187(16) |
| C(7)–N | 1.480(1) | 1.4837(12) | 1.4795(15) |
Catalyst optimisation using design of experiments (DoE)
To gain an insight into the key reaction parameters that influence the direct amide condensation, a DoE study was carried out on the formation of amide 13 catalysed by 1 (eqn (1)).13 Four factors were examined (Table 2) and to simplify subsequent analysis of the results, separate designs were carried out for fluorobenzene and toluene. Temperature could not be included as a factor in these individual studies, as it has been shown previously that azeotropic reflux is desired to enable the reaction to proceed. A 2-level fractional factorial design of 8 experiments was selected14 and 4 centre points were included to provide a measure of variation and indicate curvature.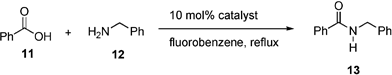 | (1) |
| Factor | Low | Centre | High |
|---|---|---|---|
| 1 (mol%) | 1 | 5.5 | 10 |
| Acid (equiv) | 0.8 | 1.1 | 1.4 |
| Conc/M | 0.1 | 0.3 | 0.5 |
| Time/h | 4 | 12 | 20 |
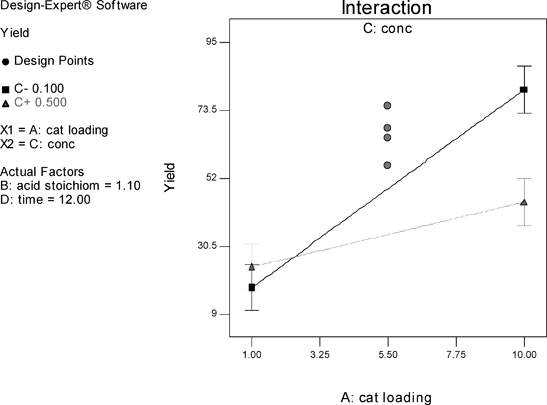
The results of the study of the reaction in fluorobenzene are summarised in Fig. 2 in the form of a half-normal probability plot in which the factors having the greatest influence appear towards the right-hand side of the graph. It can be seen that catalyst loading, time, and the two-factor interaction of catalyst loading and time are the most important factors, whilst acid stoichiometry and concentration have no significant effect.15 The centre points in the interaction graph (Fig. 3) show no significant deviation from the linear relationship plotted, i.e. there is no significant curvature, and the linear relationship plotted is a good representative model. Thus, the DoE suggests, in order to increase yield of amide 13, catalyst loading and time should be increased. Although increasing reaction time is a possibility, increasing the loading of 1 beyond 10 mol% would be undesirable.
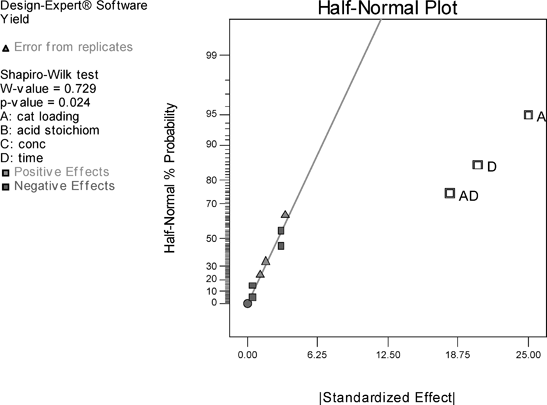 | ||
| Fig. 2 Half normal plot visualizing the most important factors determining yield in the direct amide condensation in fluorobenzene. | ||
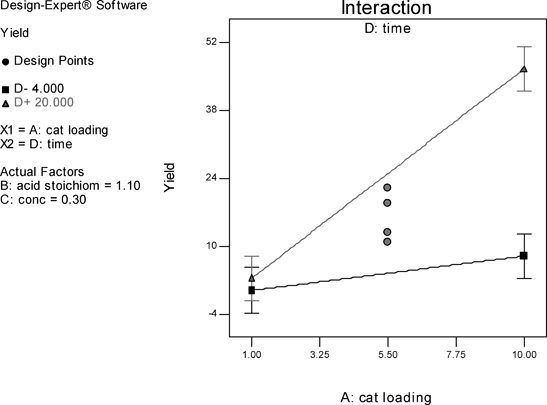 | ||
| Fig. 3 Interaction graph for the direct amide condensation in fluorobenzene. | ||
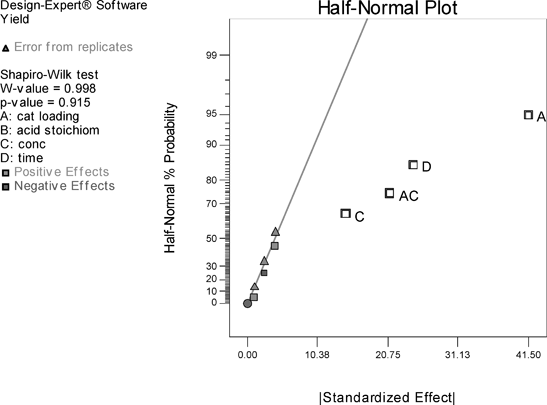
The same design run in toluene shows catalyst loading is again the most important effect, followed by time, and acid stoichiometry still has no significant effect (Fig. 4).15 However, concentration and catalyst loading are now involved in an interaction, with the model suggesting that in order to increase yields of amide 13, both low concentration and higher catalyst loading are required (Fig. 5). The interaction graph shows significant curvature indicating that the design space is over an optimum. Since catalyst loading is the most important effect, this appears to indicate that similar yields of amide 13 could be obtained if the loading of 1 is decreased below 10 mol%. In fact, the centre points indicate that a catalyst loading of 5.5 mol% gives a similar effect, whilst the concentration effect suggests that reaction rate is limited by azeotropic removal of water, and therefore, low concentration is beneficial (vide infra). In order to model the curvature, a central composite design (CCD) could be employed to augment the existing design. Although the DoE study was carried out on the formation of amide 13, the information obtained can clearly be used as a basis to start to improve the isolated yields of more problematic substrates (vide infra) and hence, these results have been taken into account in the catalyst evaluation.
Catalyst evaluation
We have previously shown10 that the boronic acid 1, and related systems, promote amidation reactions, which are highly substrate dependent and that the use of more difficult substrates under lower temperature conditions (refluxing fluorobenzene) exposes differences in catalyst activity. With this in mind, the new catalysts were evaluated and compared using the reaction between benzoic acid and benzylamine (eqn (1)), and it had already been shown that the direct (uncatalysed) thermal formation of amide 13 under these conditions is non-existent.10 The results of this comparison are shown in Fig. 6.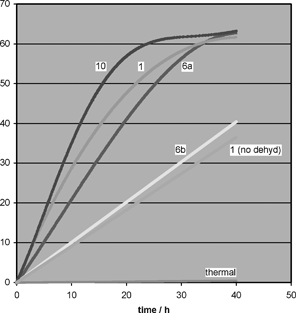 | ||
| Fig. 6 Yield verus time data for catalysed and thermal direct amide condensation between benzoic acid and benzylamine in refluxing fluorobenzene. | ||
The addition of a para-trifluoromethyl group (catalyst 10) is beneficial to rate of formation of N-benzylbenzamide 13, compared to the unsubstituted system 1, whereas the addition of the ortho-fluorine function, i.e.catalyst 6a, decreases reaction rate to a small extent (Fig. 6). While the differences between catalysts 1, 6a and 10 are not substantial, increasing the electron density of the phenyl ring by incorporating a methoxy group, i.e.catalyst 6b, leads to a significant decrease in reaction rate. This results in only 28 % conversion to amide 11 in 28 h using 6b compared to ca. 63 % conversion for catalysts 1, 10 and 6a, which have all reached their equilibrium conversions at around this time period. The efficiency of water removal determines the equilibrium position, therefore affecting both final conversion and reaction rate, as demonstrated by the reaction catalysed by 1 without dehydration, along with the results obtained from the DoE study (vide supra).
Application of catalyst 1 for direct amide formation
Although the trifluoromethylbenzylboronic acid 10 is the superior catalyst according (vide supra), the difference between 1 and 10 is not substantial (see Fig. 6). Hence, coupled with the commercial availability of catalyst 1 and the results of the DoE optimised reaction conditions in hand (vide supra), we needed to evaluate the scope and limitations of the direct amide condensation catalysed by 1. A variety of carboxylic acids and amines were examined refluxing fluorobenzene where possible, or if necessary in toluene if conversion to amide was slow, and over reaction times of up to 48 h. The results of the reactions generalised by eqn (2), are shown in Table 3. | (2) |
| Entry | Solvent | 1 (mol %) | Time/h | Product | Yield (%) |
|---|---|---|---|---|---|
| a Under argon. b Determined by HPLC. | |||||
| 1 | PhF | 0 | 24 |
|
16 |
| 10 | 68 | ||||
| 2 | PhF | 0 | 24 |
|
2 |
| 10 | 70 | ||||
| 3 | Toluene | 0 | 22 |
|
0a |
| 1 | 46a | ||||
| 4 | PhF | 0 | 24 |
|
4 |
| 10 | 67 | ||||
| 5 | PhF | 0 | 22 |
|
10b |
| 10 | 53 | ||||
| 6 | PhF | 0 | 24 |
|
1 |
| 10 | 50 | ||||
| 7 | PhF | 0 | 48 |
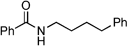
|
0 |
| 10 | 55b | ||||
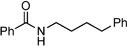
| Toluene | 5 | 24 |
|
71 | |
| 10 | 75 | ||||
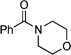
| 8 | PhF | 0 | 24 |
|
0 |
| 10 | 11 | ||||
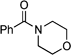
| Toluene | 0 | 24 |
|
0 | |
| 5 | 24 | 16 | |||
| 10 | 30 | 21 | |||
| 9 | PhF | 0 | 48 |
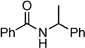
|
0b |
| 10 | 52b | ||||
| Toluene | 0 | 24 |
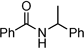
|
0 | |
| 5 | 49 | ||||
| 10 | 71 | ||||
| 10 | PhF | 0 | 24 |
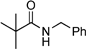
|
0 |
| 10 | 15 | ||||
| Toluene | 0 | 30 |
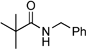
|
7 | |
| 5 | 42 | ||||
| 10 | 59 |
The results shown in Table 3 show that the majority of reactions did not proceed to 100 % completion after 48 h, though good to high isolated yields could be achieved in most cases. Fluorobenzene is certainly a suitable and practical azeotropic solvent for many of the more reactive amide formation reactions, i.e. entries 1, 2, 4, 5, 6, 7 and 9 (Table 3) using 10 mol% of catalyst 1. Importantly, at this temperature (85 °C) there is a clear advantage to using the catalyst compared to the corresponding thermal conditions. For less reactive substrates, however, for example entries 8 and 10, the use of toluene and 5 or 10 mol% catalyst loading was necessary in order to provide an improvement in the isolated yields of each of the amides (see Table 3). Although increasing the reaction temperature (i.e. refluxing toluene) increases reaction rates, the associated thermal reactions also become more significant in certain cases, making it harder to fully evaluate catalyst-derived performance alone. For example, reaction of 4-phenylbutyric acid and benzylamine (entry 1, Table 3) shows a significant thermal contribution, as previously reported.10 Interestingly, aniline reacts relatively well in toluene in the presence of only 1 mol% of catalyst 1, however, the reaction must be carried out under an inert atmosphere to prevent amine oxidation (entry 3, Table 3). The case of benzoic acid and 4-phenylbutylamine (entry 7, Table 3) demonstrates the non-linear relationship between catalyst loading and yield, as observed in the DoE, with no significant difference between a 5 and 10 mol% loading of catalyst 1. Only in the case of morpholine benzoylamide (entry 8, Table 3), could the isolated yield not be increased to a reasonable level by changing either catalyst loading or solvent boiling point. This highlights the need for further improvements in these types of direct amide formation reactions in the future, however, the majority of carboxylic acid–amine combinations are readily susceptible to this type of straightforward, clean amide formation.
Summary and conclusions
The direct reaction of amine with carboxylic acid remains the most attractive approach to preparing amide bonds in terms of avoiding reactive and toxic activated carboxylic acid derivatives, or use of in situ activating agents.5 These types of reactions are arguably best carried out without catalysis where possible, however, reaction conditions remain harsh in order to accomplish these types of direct thermal amide formation.1,2 The next most green alternative is arylboronic or boric acid catalysed direct amide formation, which offers generally more attractive reaction conditions of lower temperature reactions (refluxing fluorobenzene, toluene or xylene) and low catalyst loadings (1–10 mol%). In terms of green chemistry, the ability to recover and re-use the azeotropic solvents employed is important, and hence, the only by-product for this direct amide formation is water. Carboxylic acids and amines can be combined in a 1 : 1 stoichiometry, and with suitable reaction engineering, yields can be high, though dependent upon solvent boiling point, substrate combination and efficiency of the water removal (vide supra). Compared with, for example, an acid chloride-mediated reaction, this type of catalysed direct amide formation reaction is much more atom efficient. For example, for the reaction shown in entry 7, Table 3 in toluene, the effective mass yield (EMY)16 is 1525%, assuming that both solvent and molecular sieves are recovered and re-used efficiently, and a 5 mol% catalyst loading. This compares with preparing the same amide using, for example, thionyl chloride to prepare the acid chloride, followed by direct reaction with the amine stoichiometrically and using no additional base, giving an EMY of 87%. Clearly, the use of acylation transfer agents and bases will significantly reduce the EMY for the acid chloride-based route, hence overall, this analysis provides a graphic example of the potential of such direct amide formation processes over conventional acylation methods.The synthesis of novel substituted analogues of the bifunctional catalyst 1 has allowed us to begin to further probe the subtleties of the direct amide formation involving arylboronic acid-mediated catalysis. The addition of electron withdrawing functions to the aryl ring of 1, for example trifluoromethyl derivative 10, certainly results in increased catalytic activity for amide formation, which reinforces the view that such catalysts act by forming mixed anhydride-type analogues,10 and the electron withdrawing group increases the leaving group ability during the amide formation step. In terms of practical applications, either catalyst 1 or 10 is suitable for direct amide formation under the most ambient conditions employed to date, though commercial availability makes the use of 1 more attractive currently. Less hindered amine systems10 or more electron rich aryl systems, such as 6b, are considerably less efficient. DoE studies on the use of catalyst 1 show that catalyst loadings above 5 mol% are not required, and most interestingly, that the water-removing capacity of the solvent is an important aspect to consider during routine use of these types of catalysts. In order to optimise reactions in the most time efficient manner, higher dilutions are preferred. Clearly, alternative drying agents, perhaps employed in situ, could have a major impact upon both the rate, and hence the general applicability of these processes. Further studies along these and related directions are underway and will be reported in due course.
Experimental
N,N-Di-isopropyl-3-fluorobenzamide 4a
To a stirred solution of 3-fluorobenzoyl chloride (2.68 g, 16.9 mmol) in dry Et2O (40 mL) under Ar at 0 °C, was added dry di-isopropylamine (6.0 mL, 42 mmol) dropwise. The reaction was allowed to warm to rt, stirred for 18 h and then quenched with 5% (w/v) HCl (25 mL). The organic layer was separated and washed again with 5 % (w/v) HCl (2 × 25 mL), then brine (25 mL), 5 % (w/v) NaOH (2 × 25 mL), brine (25 mL), dried over MgSO4, and concentrated under vacuum to afford amide 4a as a white solid; yield: 3.40 g (90%); mp 74–76 °C; 1H NMR (500 MHz, CDCl3): δ = 1.17 (br s, 6H, (CH3)2CH), 1.54 (br s, 6H, (CH3)2CH), 3.55 (br s, 1H, (CH3)2CH), 3.81 (br s, 1H, (CH3)2CH), 7.01–7.11 (m, 3H, ArH), 7.37 (td, JHH = 8.0 Hz and JFH = 5.5 Hz, 1H, ArH); 13C NMR (100.6 MHz, CDCl3): δ = 20.9 (br s, (CH3)2CH), 46.2 (br s, (CH3)2CH), 51.1 (br s, (CH3)2CH), 113.2 (d, 2JFC = 22 Hz, ArC), 115.9 (d, 2JFC = 21 Hz, ArC), 121.5 (d, 4JFC = 3 Hz, ArC), 130.5 (d, 3JFC = 8 Hz, ArC), 141.1 (d, 3JFC = 7 Hz, ArC–CONiPr2), 162.9 (d, 1JFC = 247 Hz, ArC-F), 169.7 (d, 4JFC = 2 Hz, CONiPr2); 19F NMR (376.3 MHz; CDCl3): δ = 5a-112.5 (m); IR (film): νmax (inter alia) = 3072, 2971, 1629 (s), 1583, 1437, 1344 (s), 1140 cm–1; λmax(MeCN)/nm 200 (ε/dm3 mol–1 cm–1 12400), 267 (1400); MS (ES): m/z (%) = 246.1260 (100) [M + Na]+. [C19H13NOFNa]+ requires 246.1265; elemental analysis (%): calcd. for C13H18NOF: C 69.93, H 8.13, N 6.27; found: C 69.73, H 8.07, N 6.10.N,N-Di-isopropyl-3-fluoro-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzamide 5a
To a stirred solution of 4a (5.0 g, 22.4 mmol) and TMEDA (4.0 mL, 26.9 mmol) in dry THF (50 mL) under Ar at –78 °C, was added n-BuLi (16.8 mL, 1.6 M, 26.9 mmol) dropwise over 10 min. Mixture left to stir for 1 h and then trimethyl borate (3.0 mL, 26.9 mmol) was added rapidly. Mixture allowed to reach rt (2 h) and then quenched with 20% (w/v) HCl (10 mL), followed by addition of pinacol (3.2 g, 26.9 mmol). Mixture extracted into ether (3 × 20 mL) and the organic extracts washed with sat. aq. NaHCO3 (3 × 20 mL) and brine (2 × 20 mL). Extracts were dried over MgSO4 and concentrated under vacuum. Column chromatography on silica gel (hexane : EtOAc, 2 : 1) afforded pinacol boronate 5a as a white crystalline solid; yield: 5.16 g (66%); mp 104–106 °C; 1H NMR (500 MHz, CDCl3): δ = 1.19 (br s, 6H, (CH3)2CH), 1.33 (s, 12H, 4 × Me), 1.54 (br s, 6H, (CH3)2CH), 3.54 (br s, 1 H, (CH3)2CH), 3.98 (br s, 1H, (CH3)2CH), 6.98–7.04 (m, 2H, ArH), 7.32 (td, JHH = 8.0 Hz and JFH = 5.5 Hz, 1H, ArH); 13C NMR (125.7 MHz, CDCl3): δ = 20.6 (br s, (CH3)2CH), 25.0 (pinacolCH3), 46.6 (br s, (CH3)2CH), 51.2 (br s, (CH3)2CH), 83.6 (pinacolC–O), 116.0 (d, 2JFC = 25 Hz, ArC), 120.8 (d, 4JFC = 4 Hz, ArC), 131.5 (d, 3JFC = 9 Hz, ArC), 144.9 (d, 3JFC = 9 Hz, ArC–CONiPr2), 166.3 (d, 1JFC = 249 Hz, ArC-F), 170.3 (d, 4JFC = 3 Hz, CONiPr2); 11B NMR (128.4 MHz, CDCl3): δ = 26.9; 19F NMR (376.3 MHz, CDCl3): δ = –103.2 (s); IR (film): νmax (inter alia)=2973, 2930, 2361, 1617 (s), 1439, 1335 (vs), 1144 (s) cm–1; λmax(MeCN)/nm 197 (ε/dm3 mol–1 cm–1 28100), 242 (3320), 271 (1220); MS (ES): m/z (%) = 721.4 (60), 699.5 (25), 372.2 (100, [M + Na]+), 350.2295 (48, [M+H]+. [C19H30BFNO3]+ requires 350.2297); elemental analysis (%): calcd. for C19H29BFNO3: C 65.34, H 8.37, N 4.01; found: C 65.41, H 8.46, N 3.99.N,N-Di-isopropyl-3-fluorobenzylamine-2-boronic acid 6a
To a stirred solution of 5a (5.16 g, 14.8 mmol) and NaBH4 (5.6 g, 148 mmol) in dry THF (70 mL) under Ar was added TMSCl (37.6 mL, 296 mmol) and the mixture stirred at reflux for 20 h. Reaction quenched by slow addition of 5% (w/v) HCl then 20% (w/v) HCl taking the aqueous layer to pH 1, and extracted into Et2O (1 × 50 mL, 3 × 100 mL). Sat. aq. NaHCO3 was added taking the aqueous layer to pH 7 and the mixture was extracted into Et2O (3 × 100 mL). Organic extracts were combined and washed with brine (2 × 100 mL), dried over MgSO4 and concentrated under vacuum. Slow crystallisation from MeCN provided crystals suitable for single crystal X-ray analysis.‡Recrystallisation from DCM-hexane afforded boronic acid 6a as a white solid; yield: 2.71 g (72%); mp 110–111 °C; 1H NMR (500 MHz, CDCl3): δ = 1.10 (d, J = 7.0 Hz, 12H, (CH3)2CH), 3.08 (septet, J = 7.0 Hz, 2H, (CH3)2CH), 3.82 (s, 2H, ArCH2), 6.97–7.03 (m, 1H ArH), 7.07 (d, J= 7.5 Hz, 1H, ArH), 7.30 (td, JHH = 7.5 Hz and JFH = 6.5 Hz, 1H, ArH), 9.27 (br s, 2H, B(OH)2); 13C NMR (125.7 MHz, CDCl3): δ = 19.9 ((CH3)2CH), 47.7 ((CH3)2CH), 52.0 (d, 4JFC = 2 Hz, ArCH2), 115.4 (d, 2JFC = 28 Hz, ArC), 127.7 (d, 4JFC = 2 Hz, ArC), 131.7 (d, 3JFC = 11 Hz, ArC), 144.7 (d, 3JFC = 8 Hz, ArCCH2NiPr2), 168.0 (d, 3JFC = 244 Hz, ArC-F); 11B NMR (128.4 MHz, CDCl3): δ = 28.4; 19F NMR (376.3 MHz, CDCl3): δ = –104.5 (m); IR (film): νmax (inter alia) = 3307, 2971, 2539, 1604, 1565, 1450 (s), 1385 (vs), 1144 (s) cm–1; λmax(MeCN)/nm 194 (ε/dm3 mol–1 cm–1 10000), 216 (6970), 272 (1280); MS (ES): m/z (%) = 254.1722 (100, [M + H]+. [C13H22BFNO2]+ requires 246.1265); elemental analysis (%): calcd. for C13H29BFNO2: C 61.69, H 8.36, N 5.53; found: C 61.53, H 8.41, N 5.30.N,N-Di-isopropyl-3-methoxybenzamide 4b
To a stirred solution of m-anisoyl chloride (0.82 mL, 6 mmol) in dry Et2O (25 mL) under Ar at 0 °C, was added dry di-isopropylamine (2.1 mL, 15 mmol) dropwise. The reaction was allowed to warm to rt, stirred for 3 h and then quenched with 5% (w/v) HCl (25 mL). The organic layer was separated and washed again with 5% (w/v) HCl (2 × 25 mL), brine (25 mL), 5% (w/v) NaOH (2 × 25 mL), brine (25 mL), dried over MgSO4, and concentrated under vacuum to afford amide 4b as a white solid; yield: 1.38 g (98%); mp 92–93 °C; 1H NMR (500 MHz, CDCl3): δ = 1.16 (br s, 6H (CH3)2CH), 1.54 (br s, 6H, (CH3)2CH), 3.53 (br s, 1H, (CH3)2CH), 3.83 (s, 3H, OCH3), 3.86 (br s, 1H, (CH3)2CH), 6.86 (s, 1H, ArH), 6.89 (d, J = 8.0 Hz, 1H, ArH), 6.91 (d, J = 8.0 Hz, 1H, ArH), 7.61 (t, J = 8.0 Hz, 1H, ArH); 13C NMR (125.7 MHz, CDCl3): δ = 20.8 ((CH3)2CH), 46.0 (br s (CH3)2CH), 51.0 (br s, (CH3)2CH), 55.4 (OCH3), 111.1 (ArC), 114.7 (ArC), 117.8 (ArC), 129.7 (ArC), 140.3 (ArC-OMe), 159.7 (ArC-CONiPr2)), 170.9 (CONiPr2); IR (film): νmax (inter alia) = 3090, 2974, 1621 (vs), 1578, 1340 (vs), 1293, 1032 cm–1; λmax(MeCN)/nm 200 (ε/dm3 mol–1 cm–1 21300) and 280 (2290); MS (ES): m/z (%) = 236.1646 (100, [M + H]+. [C14H22NO2]+ requires 236.1645); elemental analysis (%): calcd. for C14H21NO2: C 71.46, H 8.99, N 5.95; found: C 71.45, H 9.07, N 5.81.N,N-Di-isopropyl-3-methoxy-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-benzamide 5b
To a stirred solution of 4b (1.75 g, 7.44 mmol) and TMEDA (1.56 mL, 10.4 mmol) in dry THF (25 mL) under Ar at –78 °C, was added s-BuLi (7.44 mL, 1.4 M, 10.4 mmol) dropwise over 10 min. Mixture left to stir for 90 min and then triisopropyl borate (1.89 mL, 8.18 mmol) was added rapidly. Mixture allowed to reach rt overnight (19 h) and then quenched with 20% (w/v) HCl (6 mL), followed by addition of pinacol (0.97 g, 8.18 mmol). Mixture allowed to stir for 10 min before being extracted into ether (3 × 20 mL) and the organic extracts washed with sat. aq. NaHCO3 (3 × 10 mL), and brine (20 mL). Extracts were dried over MgSO4 and concentrated under vacuum. Column chromatography on silica gel (hexane : EtOAc, 1 : 1) afforded pinacol boronate 5b as a white solid; yield: 2.05 g (76%); mp 90–91 °C; 1H NMR (500 MHz, CDCl3): δ = 1.19 (br s, 6H, (CH3)2CH), 1.35 (s, 12H, 4 × Me), 1.54 (br s, 6H, (CH3)2CH), 3.50 (br s, 1H, (CH3)2CH), 3.81 (s, 3H, OMe), 4.00 (br s, 1H, (CH3)2CH), 6.82 (d, J = 8.0 Hz, 2H, ArH), 7.28 (1H, t, J = 8.0 Hz, ArH); 13C NMR (125.7 MHz, CDCl3): δ = 20.7 (br s, (CH3)2CH), 25.0 (pinacolCH3), 46.3 (br s, (CH3)2CH), 51.0 (br s, (CH3)2CH), 55.8 (OMe), 83.5 (pinacolC–O), 110.7 (ArC), 117.3 (ArC), 130.3 (ArC), 144.0 (ArC-CONiPr2), 163.4 (ArC-OMe), 171.4 (CONiPr2); 11B NMR (128.4 MHz, CDCl3): δ = 29.0; IR (film): νmax (inter alia) = 2973, 1615 (s), 1431, 1335 (vs), 1146 cm–1; MS (ES): m/z (%) = 362.2497 (100, [M + H]+. [C20H33BNO4]+ requires 362.2497); elemental analysis (%): calcd. for C20H32BNO4: C 66.49, H 8.93, N 3.88; found: C 66.22, H 8.98, N 3.82.N,N-Di-isopropyl-3-methoxybenzylamine-2-boronic acid 6b
To a stirred solution of 5b (0.99 g, 2.74 mmol) and NaBH4 (1.04 g, 27.4 mmol) in dry THF (50 mL) under Ar was added TMSCl (6.95 mL, 54.8 mmol) and the mixture stirred at reflux for 21 h. Reaction quenched by slow addition of 5% (w/v) HCl (7 mL) and THF removed in vacuo. A further portion of 5% (w/v) HCl (15 mL) was added (aqueous layer to pH 1) and extracted into Et2O (1 × 25 mL, 2 × 15 mL). Et2O extracts washed with 5% (w/v) HCl (5 mL) and combined aqueous phase washed with Et2O (2 × 10 mL). Aqueous neutralised with NaOH (s) then 20% (w/v) NaOH until no further precipitation of white solid was observed (pH 9). The mixture was extracted into DCM (3 × 15 mL), organic extracts were combined, washed with brine (15 mL), dried over MgSO4 and concentrated under vacuum. Recrystallisation from hexane afforded boronic acid 6b as a white solid; yield: 0.36 g (50%); mp 164–165 °C; 1H NMR (400 MHz, CDCl3): δ = 1.11 (d, J = 6.8 Hz, 12H, (CH3)2CH), 3.10 (septet, J = 6.8 Hz, 2H, (CH3)2CH), 3.80 (s, 2H, ArCH2), 3.89 (s, 3H, OMe), 6.90 (d, J = 8.0 Hz, 1H, ArH), 6.93 (d, J = 7.6 Hz, 1H, ArH), 7.31 (dd, JHH = 8.4 Hz and JFH = 7.6 Hz, 1H, ArH), 9.75 (br s, 2H, B(OH)2); 13C NMR (100.6 MHz, CDCl3): δ = 19.9 ((CH3)2CH), 47.5 ((CH3)2CH), 52.4 (ArCH2), 56.0 (OMe), 110.5 (ArC), 125.6 (ArC), 131.2 (ArC), 144.8 (ArC), 164.8 (ArC-OMe); 11B NMR (128.4 MHz, CDCl3): δ = 29.3; IR (KBr): νmax (inter alia) = 3414, 2965, 1597 (s), 1466 (s), 1249 (s), 1084 cm–1; MS (ES): m/z (%) = 266.1923 (100, [M + H]+. [C14H25BNO3]+ requires 266.1922); elemental analysis (%): calcd. for C14H24BNO3: C 63.42, H 9.12, N 5.28; found: C 63.82, H 9.04, N 5.16.N,N-Di-isopropyl-3-nitrobenzamide 4c
To a stirred solution of 3-nitrobenzoyl chloride (1.41 g, 6.71 mmol) in dry Et2O (25 mL) under Ar at 0 °C, was added dry di-isopropylamine (2.7 mL, 19 mmol). The reaction was allowed to warm to rt, stirred for 18 h and then quenched with 5% (w/v) HCl (25 mL). The organic layer was separated and washed again with 5% (w/v) HCl (2 × 25 mL), then brine (25 mL), 5% (w/v) NaOH (2 × 25 mL), brine (25 mL), dried over MgSO4, and concentrated under vacuum to afford amide 4c (1.66 g, 87%) as a white solid; yield: 1.66 g (87%); mp 78–79 °C; 1H NMR (500 MHz, CDCl3): δ = 1.21 (br s, 6H, (CH3)2CH), 1.56 (br s, 6H, (CH3)2CH), 3.52 (br s, 1H, (CH3)2CH), 3.55 (br s, 1H, (CH3)2CH), 7.60 (t, J= 8.1 Hz, 1H, ArH), 7.67 (d, J = 8.1 Hz, 1H, ArH), 8.20 (s, 1H, ArH), 8.25 (d, J = 8.1 Hz, 1H, ArH); 13C NMR (125.7 MHz, CDCl3): δ = 20.9 (br s, (CH3)2CH), 46.5 (br s, (CH3)2CH), 51.5 (br s, (CH3)2CH), 121.2 ArC), 123.8 (ArC), 130.0 (ArC), 132.0 (ArC), 140.5 (ArC-CONiPr2), 148.3 (ArC-NO2), 168.4 (CONiPr2); IR (film): νmax = 2930, 1625 (s), 1527, 1438, 1334, 1153; λmax(MeCN)/nm 194 (ε/dm3 mol–1 cm–1 24000), 252 (6960); MS (ES): m/z (%) = 273.1203 (100, [M+Na]+. [C13H18N2O3Na]+ requires 273.1210); elemental analysis (%): calcd. for C13H18N2O3: C 62.38, H 7.25, N 11.19; found: C 62.34, H 7.27, N 11.18.N,N-Di-isopropyl-3-trifluoromethylbenzamide 8
To a stirred solution of 3-(trifluoromethyl)benzoyl chloride (1.78 mL, 12 mmol) in dry Et2O (30 mL) under Ar at 0 °C, was added dry di-isopropylamine (4.24 mL, 30 mmol) dropwise. The reaction was allowed to warm to rt, stirred for 2 h and then quenched with 5% (w/v) HCl (25 mL). The organic layer was separated and washed again with 5% (w/v) HCl (25 mL), then brine (25 mL), 5% (w/v) NaOH (2 x 25 mL), brine (25 mL), dried over MgSO4, and concentrated under vacuum to afford amide 8 as a white solid; yield: 3.23 g (98%); mp 58–59 °C; 1H NMR (500 MHz, CDCl3): δ = 1.17 (br s, 6H, (CH3)2CH), 1.53 (br s, 6H, (CH3)2CH), 3.57 (br s, 1H, (CH3)2CH), 3.72 (br s, 1H, (CH3)2CH), 7.47–7.54 (m, 2H, ArH), 7.57 (s, 1H, ArH), 7.63 (d, J = 8.5 Hz, 1H, ArH); 13C NMR (100.6 MHz, CDCl3): δ = 20.8 ((CH3)2CH), 46.3 (br s, (CH3)2CH), 51.3 (br s, (CH3)2CH), 122.8 (q, 3JFC = 4 Hz, ArC), 123.9 (q, 1JFC = 273 Hz, CF3), 125.6 (q, 3JFC = 4 Hz, ArC), 129.1 (ArC), 129.2 (ArC), 131.1 (q, 2JFC = 33 Hz, ArC-CF3), 139.6 (ArC-CONiPr2), 169.5 (CONiPr2); 19F NMR (376.3 MHz, CDCl3): δ = –63.2 (s); IR (film): νmax = 2970, 1621 (s), 1373, 1311 (s), 1162, 1123 cm–1; λmax(MeCN)/nm 200 (ε/dm3 mol–1 cm–1 22300), 252 (7150); MS (ES): m/z (%) = 274.1413 (100, [M + H]+. [C14H19F3NO]+ requires 274.1413); elemental analysis (%): calcd. for C14H18F3NO: C 61.53, H 6.64, N 5.13; found: C 61.24, H 6.59, N 4.96.N,N-Di-isopropyl-2-(4,4,5,5-tetramethyl-[1,3,2]dioxaborolan-2-yl)-5-trifluoromethyl-benzamide 9
To a stirred solution of 6 (1.01 g, 3.70 mmol) in dry THF (20 mL) under Ar at –78 °C, was added LDA (2.47 mL, 1.8 M, 4.44 mmol) dropwise over 15 min. Mixture left to stir for 70 min and then triisopropyl borate (0.94 mL, 4.07 mmol) was added rapidly. Mixture allowed to reach rt overnight (17 h) and then quenched with 20% (w/v) HCl (6 mL), followed by addition of pinacol (0.48 g, 4.07 mmol). Mixture allowed to stir for 15 min before being extracted into ether (2 × 20 mL, 1 × 10 mL) and the organic extracts washed with sat. aq. NaHCO3 (3 × 10 mL), and brine (20 mL). Extracts were dried over MgSO4 and concentrated under vacuum. Column chromatography on silica gel (hexane : EtOAc, 4 : 1) afforded pinacol boronate 9 as a white solid; yield 1.40 g (95%); mp 130–131 °C; 1H NMR (500 MHz, CDCl3): δ = 1.13 (d, J = 6.5 Hz, 6H, (CH3)2CH), 1.32 (s, 12H, 4 x Me), 1.58 (d, J = 6.5 Hz, 6H, (CH3)2CH), 3.53 (m, 1H, (CH3)2CH), 3.63 (m, 1H, (CH3)2CH), 7.39 (s, 1H, ArH), 7.56 (d, J = 8.5 Hz, 1H, ArH), 7.91 (d, J = 7.5 Hz, 1H, ArH); 13C NMR (125.7 MHz; CDCl3): δ = 20.3 ((CH3)2CH), 20.5 ((CH3)2CH), 25.0 (pinacolCH3), 46.1 ((CH3)2CH), 51.2 ((CH3)2CH), 84.4 (pinacolC-O), 121.4 (q, 3JFC = 4 Hz, ArC), 123.9 (q, 1JFC = 273 Hz, CF3), 124.1 (q, 3JFC = 4 Hz, ArC), 132.5 (q, 2JFC = 33 Hz, ArC-CF3), 136.2 (ArC), 145.6 (ArC-CONiPr2), 169.9 (CONiPr2); 11B NMR (160.3 MHz, CDCl3): δ = 29.8; 19F NMR (470.3 MHz; CDCl3) δ = –63.5 (s); IR (film): νmax = 2978, 1627 (s), 1312 (vs), 1132 (vs) cm–1; MS (ES): m/z (%) = 400.2265 (100, [M + H]+. [C20H30BF3NO3]+ requires 400.2265); elemental analysis (%): calcd. for C20H29BF3NO3: C 60.17, H 7.32, N 3.51; found: C 60.37, H 7.34, N 3.38.N,N-Di-isopropyl-5-trifluoromethylbenzylamine-2-boronic acid 10
To a stirred solution of 7 (1.00 g, 2.50 mmol) and NaBH4 (0.95 g, 25.0 mmol) in dry THF (50 mL) under Ar was added TMSCl (6.35 mL, 50.0 mmol) and the mixture stirred at reflux for 21 h. Reaction quenched by slow addition of 5% (w/v) HCl (7 mL) and THF removed in vacuo. A further portion of 5% (w/v) HCl (5 mL) was added (aqueous layer to pH 1) and extracted into Et2O (3 × 15 mL). Et2O extracts washed with 5% (w/v) HCl (5 mL) and combined aqueous phase washed with Et2O (2 × 10 mL). Aqueous neutralised to pH 7 with NaOH (s) then 20% (w/v) NaOH and extracted into DCM (3 × 15 mL). Combined organic extracts were washed with brine (15 mL), dried over MgSO4 and concentrated under vacuum. Recrystallisation from MeCN-H2O afforded boronic acid 10 as a white crystalline solid (crystals suitable for single crystal X-ray analysis‡); yield: 0.45 g (60%); mp 115–116 °C; 1H NMR (400 MHz, CDCl3): δ = 1.14 (d, J = 6.8 Hz, 12H, (CH3)2CH), 3.13 (septet, J = 6.8 Hz, 2H, (CH3)2CH), 3.90 (s, 2H, ArCH2), 7.47 (s, 1H, ArH), 7.55 (d, J = 7.6 Hz, 1H, ArH), 8.10 (d, J = 8.0 Hz, 1H, ArH), 10.28 (br s, 2H, B(OH)2); 13C NMR (100.6 MHz, CDCl3): δ = 19.8 ((CH3)2CH), 47.9 ((CH3)2CH), 51.9 (ArCH2), 123.8 (q, 3JFC = 4 Hz, ArC), 124.2 (q, 1JFC = 272 Hz, CF3), 127.0 (q, 3JFC = 4 Hz, ArC), 132.0 (q, 2JFC = 32 Hz, ArC-CF3), 137.3 (ArC), 143.2 (ArC); 11B NMR (128.4 MHz, CDCl3): δ = 28.5; 19F NMR (376.3 MHz, CDCl3): δ = –63.3 (s); IR (film): νmax = 3324, 2978, 1328 (s), 1166 (s), 1112 (vs) cm–1; MS (ES): m/z (%) = 304.2 (100, [M + H]+); elemental analysis (%): calcd. for C14H21BF3NO2: C 55.47, H 6.98, N 4.62; found: C 55.22, H 6.85, N 4.46.General procedure for DoE on formation of N-benzylbenzamide
Benzoic acid (1, 3 or 5 mmol) was weighed into ReactArray tubes to give appropriate reaction concentration (0.1, 0.3 or 0.5 M) followed by catalyst 1 (1, 5.5 or 10 mol%). ReactArray azeotroping condenser set assembled, naphthalene (25 mol%) as a 0.5 M standard solution, and fluorobenzene or toluene were added to give a final reaction volume of 10.6 mL. Mixture heated to reflux under nitrogen and benzylamine (1, 3 or 5 mmol) was added. Reaction stirred at reflux with sampling at 4 h intervals. Samples were subjected to the following quench/dilution protocols:• 0.1 M: 90 µL into 910 µL MeCN
• 0.3 M: 50 µL into 1650 µL MeCN
• 0.5 M: 100 µL into 900 µL MeCN, 176 µL into 824 µL MeCN
Samples were mixed and analysed by HPLC (MeCN (0.05% TFA) : water (0.05% TFA) 0 : 100 to 100 : 0 over 15 min; 1 mL min–1; tr = 9.12 min). Naphthalene was used as an internal standard.
General procedure for catalyst screen on N-benzylbenzamide formation
The appropriate catalyst (0.233 mmol, 10 mol%) was manually weighed into each reaction vessel, followed by assembly of a micro-Soxhlet apparatus loaded with activated 3Å molecular sieves under argon. Solid reagents were added using the ReactArray as standard solutions (0.5 M in fluorobenzene). Naphthalene (0.35 mmol, 15 mol%) and amine (2.33 mmol) were added to the reaction vessels at ambient temperature. The appropriate amount of fluorobenzene was then added to each reaction vessel in order to give a final reaction volume of 10 mL. After heating to reflux, carboxylic acid (2.33 mmol) was added to the stirred solution. Reactions were sampled (50 µL) at 2 or 4 h intervals (24 or 48 h reaction time, respectively). Samples were quenched with MeCN (950 µL), diluted once (50 µL in 950 µL MeCN) mixed and analysed by HPLC (gradient MeCN (0.05% TFA) : water (0.05% TFA) 0 : 100 to 100 : 0 over 15; 1 mL min–1; tr = 9.12 min). Naphthalene was used as an internal standard, with response factors calculated automatically by ReactArray DataManager.General procedure for isolation of amides (fluorobenzene)
A round-bottomed flask was equipped with stirrer bar, pressure equalising dropping funnel (in vertical neck) with a soxhlet thimble containing CaH2 (∼1 g), followed by a condenser. The appropriate carboxylic acid (5 mmol), followed by fluorobenzene (50 mL), and amine (5 mmol) were added, followed by 1 (117.6 mg, 0.5 mmol). The mixture was allowed to stir at reflux for 22, 24 or 48 h, before being concentrated under vacuum. The residue was then redissolved in DCM (25 mL), washed with brine (25 mL), 5% (w/v) HCl (25 mL), brine (25 mL), 5% (w/v) NaOH (25 mL), brine (25 mL), dried over MgSO4, and the solvent evaporated under vacuum.N-Benzyl-4-phenylbutyramide:17 yield: 0.86 g (68%)
N-4-Phenylbutyl-4-phenylbutyramide:10 yield: 1.03 g (70%)
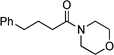
1-Morpholin-4-yl-4-phenylbutan-1-one: yield: 0.78 g (67%)
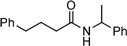
1H NMR (400 MHz, CDCl3): δ = 1.98 (quintet, J = 7.6 Hz, 2H, CH2), 2.31 (t, J = 7.6 Hz, 2H, CH2), 2.68 (t, J = 7.6 Hz, 2H, CH2), 3.35–3.39 (m, 2H, morphlino), 3.58–3.68 (m, 4H, morpholino), 7.16–7.22 (m, 3H, ArH), 7.25–7.31 (m, 2H, ArH); 13C NMR (100.6 MHz, CDCl3): δ = 26.7 (CH2), 32.2 (CH2), 35.4 (CH2), 42.0 (CH2N), 46.1 (CH2N), 66.8 (CH2O), 67.1 (CH2O), 126.1 (ArC), 128.5 (ArC), 128.6 (ArC), 141.7 (ArC), 171.6 (CON); IR (film): νmax (inter alia) = 2856, 1647 (vs), 1433 (s), 1116 (s) cm–1; MS (ES): m/z (%) = 234.2 (100) [M + H]+; elemental analysis (%): calcd. for C14H19NO2: C 72.07, H 8.21, N 6.00; found: C 71.91, H 8.24, N 6.12.N-(1-Phenylethyl)-4-phenylbutyramide:7f yield: 0.71 g (53%)
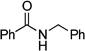
N-Benzylbenzamide:18 yield: 0.53 g (50%)
N-Phenyl-4-phenylbutyramide 7a
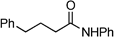
To a stirred solution of 4-phenylbutyric acid (0.821 g, 5 mmol) in toluene (50 mL), was added aniline (0.46 mL, 5 mmol) followed by catalyst 4c (11.8 mg, 0.05 mmol). The mixture was allowed to stir at reflux for 22 h, before being concentrated under vacuum. The residue was then redissolved in Et2O (25 mL), washed with brine (25 mL), 5% (w/v) HCl (25 mL), brine (25 mL), 5% (w/v) NaOH (25 mL), brine (25 mL), dried over MgSO4, and the solvent evaporated under vacuum; yield: 0.60 g (50%).N-(1-Phenyl)ethylbenzamide 19
Catalyst 1 (54.8 mg, 0.233 mmol) was weighed into a ReactArray tube, followed by assembly of a micro-Soxhlet apparatus loaded with activated 3Å molecular sieves under argon. Solid reagents were added using the ReactArray as standard solutions (0.5 M in fluorobenzene). Naphthalene (44.9 mg, 0.35 mmol), α-methylbenzylamine (301 µL, 2.33 mmol) and fluorobenzene (4.63 mL) were added to the reaction vessel at ambient temperature. After heating to reflux, benzoic acid (0.285 g, 2.33 mmol) was added to the stirred solution. Reaction was sampled (50 µL) at 4 h intervals over 48 h. Samples were quenched with MeCN (950 µL), diluted once (50 µL in 950 µL MeCN) mixed and analysed by HPLC (gradient MeCN (0.05% TFA) : water (0.05% TFA) 0 : 100 to 100 : 0 over 31 min; 1 mL min–1). Naphthalene was used as an internal standard, with response factors calculated automatically by ReactArray DataManager. Yield: 0.27 g (52%); HPLC: tr = 16.10 min.General procedure for isolation of amides (toluene)
The appropriate carboxylic acid (1 mmol) and catalyst 1 (23.5 mg, 0.1 mmol or 11.8 mg, 0.05 mmol) were weighed into ReactArray tubes followed by attachment of a ReactArray azeotroping condenser set. The appropriate amount of toluene was added to give a final reaction volume of 10 mL and the mixture heated to reflux under nitrogen. The appropriate amine (1 mmol) was added and the mixture stirred at reflux for 24 or 30 h, before being concentrated under vacuum. The residue was then redissolved in MTBE (10 mL), washed with 5% (w/v) HCl (10 mL), brine (10 mL), 5% (w/v) NaOH (10 mL), brine (10 mL), dried over MgSO4, and the solvent evaporated under vacuum.N-4-Phenylbutylbenzamide:10 yield: 0.18 g (71%)
Morpholin-4-yl-phenylmethanone:20 yield: 0.04 g (21%).
N-(1-Phenyl)ethylbenzamide:19 yield: 0.16 g (71%).
N-Benzylpivalamide:7g yield: 0.11 g (59%).
Acknowledgements
We thank the EPSRC for a research grant and DTA studentship (KA), and GlaxoSmithKline for CASE funding (KA) and the National Mass Spectrometry Service at Swansea.References
- A. W. Hofmann, Chem. Ber., 1882, 15, 977–978 Search PubMed.
- (a) J. A. Mitchell and E. E. Reid, J. Am. Chem. Soc., 1931, 53, 1879–1883 CrossRef CAS; (b) D. Davidson and P. Newman, J. Am. Chem. Soc., 1952, 74, 1515–1516 CrossRef CAS; (c) B. S. Jursic and Z. Zdravkovski, Synth. Commun., 1993, 23, 2761–2770 CAS.
- (a) Y. I. Leitman and M. S. Pevzner, Zh. Prikl. Khim., 1963, 36, 632–639 CAS; (b) W. Walter, H. Besendorf and O. Schnider, Helv. Chim. Acta, 1961, 44, 1546–1554 CrossRef.
- (a) M. P. Vazquez-Tato, Synlett, 1993, 506–506 Search PubMed; (b) L. Perreux, A. Loupy and F. Volatron, Tetrahedron, 2002, 58, 2155–2162 CrossRef CAS.
- (a) T. Ziegler, Science of Synthesis, ed. S. M. Weinreb, Thieme, Stuttgart, 2005, vol. 21, pp. 43–76 Search PubMed; (b) G. Benz, Comprehensive Organic Synthesis, ed. B. M. Trost, I. Fleming, Pergamon, Oxford, 1991, vol. 6, pp. 381–417 Search PubMed.
- (a) A. Pelter, T. E. Levitt and P. Nelson, Tetrahedron, 1970, 26, 1539–1544 CrossRef CAS; (b) R. Latta, G. Springsteen and B. Wang, Synthesis, 2001, 1611–1613 CrossRef CAS; (c) W. Yang, X. Gao, G. Springsteen and B. Wang, Tetrahedron Lett., 2002, 43, 6339–6342 CrossRef CAS.
- (a) K. Ishihara, H. Kurihara and H. Yamamoto, J. Org. Chem., 1996, 61, 4196–4197 CrossRef CAS; (b) K. Ishihara, S. Ohara and H. Yamamoto, Macromolecules, 2000, 33, 3511–3513 CrossRef CAS; (c) K. Ishihara, S. Kondo and H. Yamamoto, Synlett, 2001, 1371–1374 CrossRef CAS; (d) K. Ishihara, S. Kondo and H. Yamamoto, Org. Synth., 2002, 79, 176–185 CAS; (e) T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2007, 63, 8645–8657 CAS; (f) T. Maki, K. Ishihara and H. Yamamoto, Org. Lett., 2006, 8, 1431–1434 CrossRef CAS; (g) T. Maki, K. Ishihara and H. Yamamoto, Tetrahedron, 2006, 8, 1431–1434 CAS.
- P. Tang, Org. Synth., 2002, 81, 262–272.
- (a) R. L. Giles, J. A. K. Howard, L. G. F. Patrick, M. R. Probert, G. E. Smith and A. Whiting, J. Organomet. Chem., 2003, 680, 257–262 CrossRef CAS; (b) S. W. Coghlan, R. L. Giles, J. A. K. Howard, M. R. Probert, G. E. Smith and A. Whiting, J. Organomet. Chem., 2005, 690, 4784–4793 CrossRef CAS; (c) A. J. Blatch, O. V. Chetina, J. A. K. Howard, L. G. F. Patrick, C. A. Smethurst and A. Whiting, Org. Biomol. Chem., 2006, 4, 3297–3302 RSC.
- K. Arnold, B. Davies, R. L. Giles, C. Grosjean, G. E. Smith and A. Whiting, Adv. Synth. Catal., 2006, 348, 813–820 CrossRef CAS.
- G. Köbrich and P. Buck, Chem. Ber., 1970, 103, 1412–1419.
- (a) M. Schlosser, F. Mongin, J. Porwisiak, W. Dmowski, H. H. Büker and N. M. M. Nibbering, Chem.–Eur. J., 1998, 4, 1281–1286 CrossRef CAS; (b) M. Schlosser and M. Marull, Eur. J. Org. Chem., 2003, 1569–1575 CrossRef CAS.
- (a) For example: V. K. Aggarwal, A. C. Staubitz and M. Owen, Org. Process Res. Dev., 2006, 10, 64–69 Search PubMed; (b) R. Carlson and J. E. Carlson, Design and Optimiziation in Organic Synthesis, Elsevier, Amsterdam, 2005 Search PubMed.
- The penalty for reducing the number of experiments is one factor interactions are aliased with three factor interactions, and two factor interactions are aliased with other two factor interactions.
- The thermal reaction is not significant in either fluorobenzene or toluene in this case (0% and 5% conversion in 24 h, respectively).
- T. Hudlicky, D. A. Frey, L. Koroniak, C. D. Claeboe and L. E. Brammer, Green Chem., 1999, 57–59 Search PubMed.
- R. Verma and S. K. Ghosh, J. Chem. Soc., Perkin Trans. 1, 1998, 2377–2382 RSC.
- N. Shangguan, S. Katukojvala, R. Greenberg and L. J. Williams, J. Am. Chem. Soc., 2003, 125, 7754–7755 CrossRef CAS.
- M. Noji, T. Ohno, K. Fuji, N. Futaba, H. Tajima and K. Ishii, J. Org. Chem., 2003, 68, 9340–9347 CrossRef CAS.
- J. D. Moore, R. J. Byrne, P. Vedantham, D. L. Flynn and P. R. Hanson, Org. Lett., 2003, 5, 4241–4244 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental methods, X-ray crystallographic data for compounds 6a and 10, and 1H, 13C, 11B and 19F NMR spectra (as appropriate) for compounds 4a, 5a, 6a, 4b, 5b, 6b, 4c, 8, 9, 10 and 1-morpholin-4-yl-4-phenylbutan-1-one. See DOI: 10.1039/b712008g |
| ‡ CCDC reference numbers 632237 and 632238. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b712008g |
| This journal is © The Royal Society of Chemistry 2008 |