The relationship between solvent polarity and molar volume in room-temperature ionic liquids†
Mark N.
Kobrak
*
Department of Chemistry, Brooklyn College and the Graduate Center of the City University of New York, 2900 Bedford Ave., Brooklyn, NY 11210, USA. E-mail: mkobrak@brooklyn.cuny.edu
First published on 12th November 2007
Abstract
Solvent polarity is a subject of great interest to chemists. A significant component of a solvent's polarity is its capacity for nonspecific electrostatic interactions, which is often parameterized using the dielectric constant ε or the Kamlet–Taft dipolarity/polarizability parameter π*. Recent theoretical work has established a connection between the molar volume of an ionic liquid and its capacity for nonspecific electrostatic interactions with a neutral dipolar solute. In this work, we make use of a recently-developed theoretical method to estimate the molar volume of a series of ionic liquids, and explore the variation of experimentally-measured ε and π* values with molar volume. Both variables are found to vary with molar volume, and we observe an anomaly in the behavior of π* that offers insight on the nanoscale inhomogeneity of ionic liquids. An important outcome of this work is a simple scheme for the estimation of the relative polarities of ionic liquids; while not quantitatively accurate, the scheme permits prediction of the change in solvent polarity on ionic substitution or derivitization. The approach is sufficiently simple that for most commonly-used ionic liquids it can be implemented on a pocket calculator in a matter of minutes, making it a practical aid to researchers seeking to design task-specific ionic liquids.
I. Introduction
In recent years, there has been considerable interest in a novel class of molten salts known as room-temperature ionic liquids (ILs).1–5 ILs possess many physical properties that make them useful as solvents, and have been found capable of solvating a wide range of organic and inorganic compounds. However, the nature of the solvation process in ILs remains poorly understood, and the relationship between ionic structure and solvent polarity is particularly opaque.Like all molten salts, ILs are highly structured materials. Much of the crystal structure of the solid state is retained on melting, and the best model of the liquid environment is a crystalline lattice with a high population of voids and defects.6 Simulations7,8 and experiments9 suggest that nanoscale structures emerge in ILs incorporating long-chain hydrocarbon substituents. In these systems, a three-dimensional network of ionic charge centers forms, with aliphatic tails forming separate nanoscale domains interspersed in the medium. Further simulation data10 indicates that solutes may partition preferentially into one or the other domain, suggesting that regions of multiple polarity exist.
The definition of solvent polarity is simple neither in theory nor in practice,11 but polarity is generally taken as an indicator of the combined strength of specific solute–solvent interactions (e.g.hydrogen bonding) and nonspecific (electrostatic) solute–solvent interactions. Previous studies of polarity in ILs have shown that specific interactions in ionic liquids can, with few exceptions,12 be understood from the same principles at work in molecular solvents. The binary nature of ionic materials can complicate the implementation of these principles through the creation of competing ion–solute and ion–counterion interactions,13,14 but the nature of, for example, the hydrogen bond, appears the same in both molecular and ionic liquids.
In this work, we focus on understanding electrostatic solute–solvent interactions, and consider both the macroscopic, static dielectric constant ε and the Kamlet–Taft dipolarity/polarizability parameter π*. The static dielectric constant is defined by the zero-frequency polarization response of the medium,15 and is used to estimate the electrostatic energy (or free energy) of solute–solvent interactions in dielectric continuum models such as the Polarizable Continuum Model.16 While there are many subtleties in the application of the macroscopically-measured dielectric constant to molecular solvation, it has proven highly successful in understanding solute–solvent interactions in molecular liquids. However, for many well-studied ILs, the measured dielectric constants fall in the range of 10–15.17 Characterizations of IL polarity based on molecular spectroscopy17–23 and solubility24–28 generally indicate nonspecific electrostatic interactions of ILs that are equal to those of molecular liquids possessing significantly higher macroscopically-measured dielectric constants. This may have to do with the strong wavelength-dependence of the dielectric constant of fused salts,29 as the dielectric constant measured in macroscopic (long-wavelength) experiments may not be the same as that associated with the response to a microscopic (molecular) field.
One may circumvent this possible discrepancy by using a molecular measurement of electrostatic polarization, such as the Kamlet–Taft scheme.30–32 The Kamlet–Taft scheme is a semi-empirical description of solvent polarity based on a linear free energy relationship describing the solvatochromic response of a series of dye molecules. The dyes are chosen to possess different electrostatic and hydrogen-bonding properties, permitting derivation of parameters for the solvent representing its ability to donate and accept hydrogen bonds, and its capacity for nonspecific electrostatic interactions. This last parameter is labeled π*, and is referred to as representing the dipolarity/polarizability of the solvent. In molecular liquids, it includes both the reorientation of static molecular dipoles and the polarization of the electron cloud of individual molecules. In addition to permitting us to evaluate electrostatic solute–solvent interactions at a molecular level, the value of π* is known to be an important determinant of reaction outcomes.33–36 Understanding the relationship between π* and molecular structure is thus critically important in designing IL solvents.
We recently37 derived an expression describing the electrostatic energy of interaction between a neutral, dipolar solute and an ionic solvent. While the expression was not analytically solvable, its form indicated that, in the lowest order electrostatic description of solvation, the only ionic property relevant to solute-solvent interactions is ionic volume. This prediction was confirmed by comparison to experimental data, in which the measured value of π* was plotted against the number density of the solvent. The result showed a clear trend in which π* decreased with increasing molar volume. This contrasts with the situation for molecular liquids, where the correlation between the two variables is very weak.
While the dataset was sufficient to confirm the predicted trend, experimental data on liquid density were not available for all species of known π* value. In this work, we take advantage of a model prepared by Ye and Shreeve38 that allows estimation of ionic volumes based solely on chemical structure. We use this method to estimate molar volumes for 40 ILs of known π* value, and analyze the observed trend. We find that the predictions of the theory are further born out by the data, and we also find indirect evidence of the partitioning of solute species into aliphatic regions predicted above. We also examine the relationship between the static dielectric constant and the molar volume, though the dataset is more limited. The results are of interest to researchers seeking to design task-specific ionic liquids of a given polarity.
II. Methodology
The dataset for the present study, given in Table 1, represents the result of an extensive search of the literature for IL species with known Kamlet–Taft π* and/or ε value. Ion identities are given in Table 2. Molar volumes were calculated according to the procedure in ref. 38. As we discuss below, there were some cases in which volume parameters had to be estimated, or for which the estimation of the volume in this way was ambiguous. However, the errors associated with these limitations of the model are small. The goal of the present study is to establish a qualitative rather than quantitative relationship between molar volume and solvent polarity, and the values employed are sufficiently accurate for that purpose.| Cation | Anion | Cation volume/Å3 | Anion volume/Å3 | Total volume/Å3 | π* | ε |
|---|---|---|---|---|---|---|
| Group A | ||||||
| C1MIM | DCA | 154 | 86 | 240 | 1.1151 | |
| C2MIM | DCA | 182 | 86 | 268 | 1.0751 | |
| C2MIM | BF4– | 182 | 73 | 255 | 12.917 | |
| C2MIM | C2OSO3 | 182 | 147 | 329 | 27.917 | |
| C2MIM | C4OSO3 | 182 | 203 | 385 | 17.517 | |
| C2MIM | Tf | 182 | 129 | 311 | 15.117 | |
| C2MIM | Tf2N | 182 | 230 | 412 | 12.317 | |
| 12.245 | ||||||
| Avg: 12.2 | ||||||
| C3MIM | Tf2N | 210 | 230 | 440 | 11.817 | |
| C4MIM | BF4– | 238 | 73 | 311 | 1.0513 | 11.717 |
| C4MIM | Cl– | 238 | 47 | 285 | 1.175 | |
| C4MIM | DCA | 238 | 86 | 324 | 1.0551 | |
| C4MIM | PF6– | 238 | 107 | 345 | 1.0313 | 11.417 |
| 0.9218 | ||||||
| 0.915 | ||||||
| Avg: 0.95 | ||||||
| C4MIM | SbF6– | 238 | 121 | 359 | 1.0413 | |
| C4MIM | Tf | 238 | 129 | 367 | 1.0113 | 13.217 |
| C4MIM | Tf2N | 238 | 230 | 468 | 0.9852 | 11.617 |
| 11.545 | ||||||
| Avg: 11.6 | ||||||
| C5MIM | Tf2N | 266 | 230 | 496 | 11.417 | |
| C6MIM | DCA | 294 | 86 | 380 | 1.0551 | |
| C6MIM | Tf | 294 | 129 | 423 | 0.9853 | |
| C6MIM | Tf2N | 294 | 230 | 524 | 0.9853 | |
| C6MIM | PF6– | 294 | 345 | 639 | 8.942 | |
| C8MIM | Cl– | 350 | 47 | 397 | 1.095 | |
| C8MIM | PF6– | 350 | 107 | 457 | 0.885 | |
| C8MIM | Tf2N | 350 | 230 | 580 | 0.9753 | |
| C4MMIM | BF4– | 266 | 73 | 339 | 1.0813 | |
| C4MMIM | Tf2N | 266 | 230 | 496 | 1.0153 | 11.617 |
| 11.445 | ||||||
| Avg: 11.5 | ||||||
| C6MMIM | Tf2N | 322 | 230 | 552 | 0.9953 | |
| EMP | DCA | 182 | 86 | 268 | 1.1051 | |
| MPyrim4 | Tf2N | 258 | 230 | 488 | 0.9653 | |
| MPyrim6 | Tf2N | 314 | 230 | 544 | 0.9853 | |
| MPyrim8 | Tf2N | 370 | 230 | 600 | 0.9653 | |
| MDMAPyrim6 | Tf2N | 383 | 230 | 613 | 0.9853 | |
| N2HHH | NO3– | 71 | 64 | 135 | 1.1213 | 26.217 |
| 1.2441 | ||||||
| Avg: 1.18 | ||||||
| N33HH | SCN– | 183 | 71 | 254 | 1.165 | |
| N3HHH | NO3– | 99 | 64 | 163 | 1.1741 | |
| N4HHH | SCN– | 127 | 71 | 198 | 1.2341 | |
| N(B2)HHH | SCN– | 127 | 71 | 198 | 1.2841 | |
| N4111 | Tf2N | 220 | 230 | 450 | 12.543 | |
| N444H | NO3– | 360 | 64 | 424 | 0.9741 | |
| N5222 | Tf2N | 332 | 230 | 562 | 10.017 | |
| Pyrim4 | Tf2N | 230 | 230 | 455 | 11.343 | |
| 11.517 | ||||||
| Avg: 11.4 | ||||||
| Pyrr12 | DCA | 197 | 86 | 283 | 1.0351 | 14.044 |
| Pyrr14 | Tf | 253 | 129 | 382 | 1.0252 | |
| Pyrr14 | Tf2N | 253 | 230 | 483 | 0.9552 | 11.917 |
| 11.743 | ||||||
| Avg: 11.8 | ||||||
| Pyrr15 | Tf2N | 281 | 230 | 511 | 11.117 | |
| Group B | ||||||
| N3333 | CHES | 360 | 272 | 632 | 1.0841 | |
| N3333 | MOPSO | 360 | 279 | 639 | 1.0541 | |
| N4444 | BES | 472 | 280 | 752 | 1.0741 | |
| N4444 | CHES | 472 | 272 | 744 | 1.0141 | |
| N4444 | MOPSO | 472 | 279 | 751 | 1.0741 | |
| N5555 | BES | 584 | 280 | 864 | 0.9941 | |
| N5555 | CHES | 584 | 272 | 856 | 1.0041 | |
| N5555 | MOPSO | 584 | 279 | 863 | 1.0241 | |
| Cations | |
| CnMIM | 1-Alkyl-3-methylimidazolium |
| n denotes alkyl chain length (methyl, butyl, hexyl, octyl) | |
| CnMMIM | 1-Alkyl-2,3-dimethylimidazolium |
| n denotes alkyl chain length (butyl, hexyl, octyl) | |
| EMP | 1-Ethyl-2-methylpyrazolium |
| Pyrr1n | 1-Alkyl-1-methylpyrrolidinium |
| n denotes alkyl chain length (ethyl, butyl) | |
| Pyrimn | 1-Alkylpyridinium |
| n denotes alkyl chain length (butyl) | |
| MPyrimn | 1-Alkyl-3-methylpyridinium |
| n denotes alkyl chain length (butyl, hexyl, octyl) | |
| MDMAPyrim6 | 1-Hexyl-3-methyl-4-dimethylaminopyridinium |
| Nijkl | Indicates an alkylammonium species |
| ijkl denote a hydrogen (H) or carbon chain length (1–5) | |
| (B2) denotes a sec-butyl group | |
| Anions | |
| CiOSO3 | n-Alkylsulfate; i denotes alkyl chain length (ethyl, butyl) |
| DCA | Dicyanamide |
| Tf | Trifluoromethylsulfonate |
| Tf2N | Bis(trifluoromethylsulfonyl)imide |
| CHES | 2-(Cyclohexylamino)ethane sulfonate |
| BES | 2-[Bis(2-hydroxyethyl)amino] ethanesulfonate |
| MOPSO | 2-Hydroxy-4-morpholinopropane sulfonate |
| All other anions are denoted by their chemical formula. | |
Where possible, ionic volumes were taken directly from Table I of ref. 38. For organic species, it was necessary to apply the authors' rules regarding additivity of functional groups to estimate the molar volume. The rules for imidazolium, pyrrolidinium and pyridinium species are straightforward and can be applied without ambiguity. For tetraalkylammonium species, however, one obtains slightly different results depending on whether one “builds up” from Ye and Shreeve's tetramethylammonium cation or from a simple ammonium ion. We make a simple rule: Alkylammonium species incorporating only one or two alkyl substituents are constructed from the ammonium ion, while those incorporating three or four substituents are constructed from the tetramethylammonium cation. The latter is consistent with Ye and Shreeve's calculation of tetraalkylammonium species, though those authors do not consider mono- and dialkylammonium species. Details of the construction of each ion are given in the ESI;† in the most extreme case, the difference in volumes computed between the two pathways leads to an uncertainty of 6% in the calculated RTIL volumes. This is sufficiently low that it will not obscure the underlying trend in the data that is of interest in the present study.
No parameters were available describing the ring of the 1-ethyl-2-methylpyrazolium (EMP) ion. We estimated the volume to be equal to that of the 1-ethyl-3-methylimidazolium (C2MIM) ion, on the grounds that both are composed of five-membered aromatic rings incorporating 2 nitrogen atoms and decorated with equivalent alkyl substituents. In their discussion of heterocyclic cationic species, Ye and Shreeve assign a single ionic volume to a nitrogen atom in an aromatic ring, regardless of whether it is bonded to neighboring carbon or nitrogen atoms. Their results, which include examination of triazolium and tetrazolium species, suggest this approximation is sufficiently accurate that we may assign the pyrazolium and imidazolium rings the same molar volume (and by extension, equivalent volumes for C2MIM and EMP).
For ions and functional groups not explicitly discussed in Ye and Shreeve, it was necessary to follow their example and obtain estimated volumes from thermochemical radii reported in Jenkins et al.39 Volumes for SbF6– and SCN– were obtained from this source and used without modification. A number of organoanions in the present study incorporate an SO3– substituent, the volume of which cannot be directly calculated from data in either source. We therefore took the value of the volume for S2O62– from Jenkins et al. and divided it in half, assigning a value of 76 Å for the SO3– substituent. This is similar to the value one would obtain from, for example, subtracting the molar volume of a fluoride ion (or fluorine atom) from the volume of FSO3–, also reported by Jenkins et al.39 The potential error associated with this estimate is in any case small relative to the volumes of the ILs in question.
The values of π* are taken from the experimental literature, with references included in Table 1. Experimental values of the static dielectric constant, estimated from microwave spectroscopy, are also included. Where we were aware of multiple, significantly different values reported for the same liquid, all are reported and their average used in graphical analysis.
III. Results and discussion
A. The Kamlet–Taft dipolarity/polarizability
A plot of π* vs. molar volume is shown in Fig. 1. The theoretical results that motivated this work37 predict no specific functional form for the relationship between π* and molar volume, only a general inverse relationship. The overall trend in the total dataset, indicated by the dashed line, confirms this expectation (see Table 3 for the details of the regression), and analysis of the linear correlation coefficient40 for the sample confirms a correlation between π* and molar volume to 99.9% certainty. The π* parameter accounts for both the electronic and nuclear components of the solvent polarization, so one might question whether the correlation is really with the density of charge centers in the liquid or some other phenomenon that correlates with the polarizability of the electron clouds of individual ions. However, our previous study37 explored the relationship between molar volume and π* for molecular liquids, and found only a very weak correlation. The observed pattern in Fig. 1 must therefore arise from the number density of charge centers, as predicted by theory.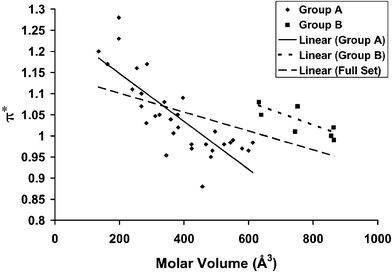 | ||
| Fig. 1 Graphical analysis of π*. See text for discussion. | ||
 | ||
| Fig. 2 Graphical analysis of ε. See text for discussion. | ||
The sharp discontinuity in π* at 620 Å3, and the relatively small scatter in the data on either side of the step suggests it is productive to look for a detailed explanation of this phenomenon. For purposes of analysis, we divide the sample into two groups: Group A corresponds to liquids with a molar volume of less than 620 Å3, and Group B corresponds to liquids with higher molar volumes. Examination of Table 1 reveals three facts:
(1) The data for all liquids in Group B is reported in a single reference.
(2) Group B contains all ILs incorporating symmetric tetraalkylammonium cations (i.e. tetraalkylammonium ions based on four equivalent substituents), and only cations of this type.
(3) Group B contains all ILs incorporating anions of molar volume greater than 250 Å3, and contains only anions of this type.
In light of (1), one might question whether the deviation is an artifact of experimental methodology. The study, ref. 41, represents an early (indeed, pioneering) study in the field, when concerns about aqueous and other impurities were not fully developed. However, the authors' experimental protocol does not appear flawed, and of the 13 ILs reported in the same study, five fall in Group A and are well within the observed trend. Further, the scatter of points within Group B is considerably narrower than the size of the step in the value of π* at 620 Å3. It is conceivable that the behavior of Group B arises from some artifact that is unique to the ILs of highest molar volume and leads to a consistent shift in the value of π* without an increase in its variance, but there is no evidence to suggest that this is the case.
It is therefore likely that the increase in π* is a physical effect associated with the ionic structures of the Group B ions. We hypothesize that the increase in π* is associated with the alteration or elimination of the nanoscale structure associated with Group A ILs. As discussed in the Introduction, the formation of nanoscale regions of aliphatic character reduces the effective polarity by creating a relatively low polarity domain into which molecular solutes may partition. Reduction of the volume of this domain, or its elimination, would therefore increase the observed (ensemble-averaged) polarity.
The high volume and symmetry of the Group B cations is the most likely culprit in such a scenario. The formation of nanoscale regions of well-defined ionic character requires the close coordination of ions, and the bulky, highly-symmetric character of the tetraalkylammonium cations may frustrate such close association. The presence of four equivalent neutral substituents prevents the formation of a well-defined aliphatic domain, and ionic volume and symmetry also hinder the close association of cation and anion charge centers that could facilitate the creation of an ionic domain. Group A includes more conventional IL structures, which include highly asymmetric cations and relatively small anions. Tf2N, the largest Group A anion, is something of an exception to this trend, but it is highly flexible and possesses a symmetric and diffuse distribution of charge; it should thus be capable of strongly coordinating to cations. In contrast, the Group B anions are not only bulky, but (as shown in Fig. 3) they are asymmetric, with charge largely centered at the sulfonate moiety. Thus, both cations and anions in Group B contain structures that hinder the close association of ionic centers, and cation symmetry hinders the formation of a well-defined aliphatic domain.
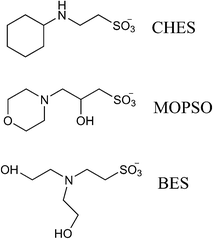 | ||
| Fig. 3 Structures of anions in Group B. See text for details, and Table 2 for full nomenclature. | ||
These arguments suggest that the relationship between ionic structure and polarity is quite complex. Derivitization of a molecular solvent by a nonpolar substituent almost inevitably leads to a reduction in solvent polarity. By contrast, such derivitization of an ionic liquid can lead to an increase in solvent polarity if it disrupts the formation of aliphatic nanostructures in the liquid. This is a useful insight for the design of task-specific ILs.
Note that while the partitioning of the solute into nanoscale domains appears to reduce the effective polarity of the solvent, the values of π* for ionic liquids are still substantially larger than those observed in molecular liquids. This implies that electrostatic interactions with the ionic domain are still of great importance in the determination of polarity. While the proper account of the structural inhomogeneity of ILs requires extension of previous theory,37 it does not invalidate the basic principles identified in that work as governing solute–solvent electrostatic interactions.
B. The static, macroscopic dielectric constant
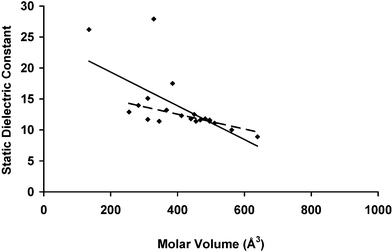
Table 1 also presents experimental data on the dielectric constants of ILs, taken from the literature. The conductive nature of ILs makes measurement of the dielectric constant challenging, and the reported values are based on microwave dielectric relaxation spectroscopy, with the static values estimated by extrapolation from the relaxation data.17,42–45 Such extrapolation is difficult, but where multiple datasets are available, results from different studies are very consistent (see Table 1).The available dataset of experimental values for ε (20 datapoints) is smaller than that for π*, and includes neither Group B ionic liquids nor any of obviously similar chemical structure. We therefore consider the entire dataset for ε as similar in character to Group A. The most obvious question is whether ε varies with molar volume, and the answer is that it does. Statistical analysis of the linear correlation coefficient40 indicates a correlation between the two variables with 99.8% certainty, and a simple linear regression of the data also indicates some variation (see Table 3). This regression analysis is applied both to the full dataset, and to a truncated dataset in which two outlying datapoints of ε > 25 ([N2HHH][NO3–] and [C2MIM][C2OSO3]) are arbitrarily dropped to eliminate their disproportionate influence on the trend.
Our previously-developed theory37 does not consider the macroscopic dielectric response, and so offers no insight on the nature of this relationship. Hydrodynamic theories based on the product of molecular (ionic) volume and viscosity are sometimes used to interpret the dynamics observed in dielectric relaxation experiments,45 but such dynamic theories are not applicable to the static dielectric constant.
Two recent papers46,47 spell out the formulation relating the dielectric response of the IL to translational and rotational ionic motion. This formalism employs the dipole moment associated with the ionic center of mass, and rotational motion represents a component of the polarization response in a manner analogous to the rotation of dipoles in molecular liquids. Many ILs in the present study are structurally analogous families synthesized by the derivitization of parent ions with successively longer alkyl substituents; such derivitization would tend to increase both the dipole moment (by moving the center of mass away from charge centers) and the molar volume. One might therefore expect an increase in molar volume to correlate with an increase in dielectric constant, yet this is the reverse of the observed trend. It is possible that ionic dynamics are more weakly correlated in systems possessing a larger molar volume, so that the collective motion responsible for the macroscopic dielectric response is weaker, decreasing the static dielectric constant.
It may be that ILs should be viewed as possessing characteristics similar to those of glasses rather than liquids. Simulation results48 are indicative of supercooled or glassy dynamics in ILs, and broadband dielectric studies49 observe secondary relaxation processes that are similar to those observed in molecular glasses. Schrödle et al.44 have noted a secondary relaxation process in their microwave dielectric relaxation studies that is at least superficially similar to that observed in molecular glasses; similar secondary relaxations are observed in other experimental studies,43 though the authors do not draw explicit analogy to glassy systems. Disordered ionic solids owe much of their dielectric response to the behavior of defects in their crystalline structure, which are known to display slow, complex relaxation dynamics.50 If similar “defects” (nonuniformities in the liquid charge distribution) are responsible for the macroscopic dielectric response in ILs, the probability for the formation of these defects could reasonably be expected to vary inversely with the molar volume, if only because of the larger number of possible defects per unit volume. It may also be true that the creation of such defects polarizes the medium on some length scale in a manner analogous to polarization by a molecular solute. In this case, the free energy associated with the formation of the defect would be reduced by the lower IL molar volume by the same mechanism at work in the response to a molecular dipole.37 However, this is speculative. We do not claim to have a clear interpretation of the relationship between the static, macroscopic dielectric constant and the molar volume of the IL, and simply lay the issue before the community for further discussion.
It would be desirable to study the relationship between π* and ε directly, since their joint variation with molar volume implies some level of correlation. However, as indicated in Table 1, we are aware of only seven liquids for which both parameters are available. While the reader can easily construct the analysis from the data provided, we find that the sample is simply too limited to support any useful conclusions. We therefore forego discussion in the interest of brevity.
IV. Conclusions
We have used the method of Ye and Shreeve38 to estimate molar volumes for a series of ionic liquids, and have confirmed our previously-derived theory that the value of the π* dipolarity/polarizability factor should vary inversely with the molar volume of an IL. In so doing, we have also uncovered evidence that the IL nanostructures identified in simulation affect the solvation properties of an IL. We have also identified a relationship between the static, macroscopic dielectric constant and the molar volume of an ionic liquid, which suggests there may be a relationship between the macroscopic dielectric response and the microscopic (molecular) electrostatic behavior of the IL, though this relationship appears quite different than that observed in molecular liquids.The results concerning π* are of interest to researchers seeking to design task-specific ILs. For example, the work demonstrates that if a high dipolarity/polarizability is desired for a certain task, the liquid should either (1) incorporate very small ions, or (2) incorporate ionic structures that frustrate the formation of nanoscale domains. Such frustration is observed in the present study for ILs incorporating large, asymmetric anions in combination with symmetric tetraalkylammonium cations, though it is not yet clear whether either or both features are necessary for this phenomenon. Further studies comparing π* and molar volume may provide additional insight and reveal alternative strategies.
A major advantage of the present theory is its ease of implementation. For common cations and anions, Ye and Shreeve's38 method for estimating molar volumes can be implemented on a pocket calculator in a matter of minutes. And while the relationship between π* and molar volume is not sufficiently robust to permit quantitative predication of π*, the relative change of polarity associated with chemical derivitization or ionic substitution can be estimated. Solvent polarity can be an important determinant of reaction outcomes, and the technique presented here is thus a simple and powerful aid to chemists seeking to design task specific ILs.
Acknowledgements
Grateful acknowledgement is made to the donors of the American Chemical Society Petroleum Research Fund for support of this research.References
- J. L. Anderson, D. W. Armstrong and G. Tzo, Anal. Chem., 2006, 78, 2893–2902 CAS.
- J. B. Harper and M. N. Kobrak, Mini-Rev. Org. Chem., 2006, 3, 253–269 Search PubMed.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667–3692 CrossRef CAS.
- P. Wasserscheid and T. Welton, Ionic Liquids in Synthesis, Wiley-VCH, Mörlenbach, Germany, 2002 Search PubMed.
- M. Blander, Some Fundamental Concepts in the Chemistry of Molten Salts, in Molten Salts: Characterization and Analysis, ed. G. Mamantov, Marcel-Dekker, New York, 1969 Search PubMed.
- Y. Wang and G. A. Voth, J. Phys. Chem. B, 2006, 110, 18601–18608 CrossRef CAS.
- J. N. Canongia-Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 3330–3335 CrossRef.
- A. Triolo, O. Russina, H.-J. Bleif and E. D. Cola, J. Phys. Chem. B, 2007, 111, 4641–4644 CrossRef CAS.
- J. N. Canongia-Lopes, M. F. C. Gomes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 16816–16818 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- C. Hanke, A. Johansson, J. Harper and R. Lynden-Bell, Chem. Phys. Lett., 2003, 374, 85–90 CrossRef CAS.
- L. Crowhurst, P. Mawdsley, J. Perez-Arlandis, P. Salter and T. Welton, Phys. Chem. Chem. Phys., 2003, 5, 2790–2794 RSC.
- V. I. Znamenskiy and M. N. Kobrak, J. Phys. Chem. B, 2004, 108, 1072–1079 CrossRef CAS.
- J. D. Jackson, Classical Electrodynamics, 2nd edn., John Wiley & Sons, New York, 1975 Search PubMed.
- C. J. Cramer and D. G. Truhlar, Chem. Rev., 1999, 99, 2161–2200 CrossRef CAS.
- H. Weingärtner, Z. Phys. Chem., 2007, 220, 1395–1405.
- S. N. Baker, G. A. Baker and F. V. Bright, Green Chem., 2002, 4, 165–169 RSC.
- S. V. Dzyuba and R. A. Bartsch, Tet. Lett., 2002, 43, 4657–4659 Search PubMed.
- K. A. Fletcher and S. Pandey, Appl. Spectrosc., 2002, 56, 266–271 CAS.
- M. J. Muldoon, C. M. Gordon and I. R. Dunkin, Perkin Trans., 2001, 2, 433–435 Search PubMed.
- S. N. V. K. Aki, J. F. Brennecke and A. Samanta, Chem. Commun., 2001, 413–414 RSC.
- A. J. Carmichael and K. R. Seddon, J. Phys. Org. Chem., 2000, 13, 591–595 CrossRef CAS.
- M. H. Abraham, A. M. Zissimos, J. G. Huddleston, H. D. Willauer, R. D. Rogers and W. E. Acree, J. Ind. Eng. Chem. Res., 2003, 42, 413–418 Search PubMed.
- J. G. Huddleston, H. D. Willauer, R. P. Swatloski, A. E. Visser and R. D. Rogers, Chem. Commun., 1998, 1765–1766 RSC.
- J. L. Anderson and D. W. Armstrong, Anal. Chem., 2003, 75, 4851–4858 CrossRef CAS.
- J. L. Anderson, J. Ding, T. Welton and D. W. Armstrong, J. Am. Chem. Soc., 2002, 124, 14247–14254 CrossRef CAS.
- D. W. Armstrong, L. He and Y.-S. Liu, Anal. Chem., 1999, 71, 3873–3876 CrossRef CAS.
- M. Rovere and M. P. Tosi, Rep. Prog. Phys., 1986, 49, 1001–1081 CrossRef CAS.
- T. Yokoyama, R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 1976, 98, 3233–3237 CrossRef CAS.
- R. W. Taft and M. J. Kamlet, J. Am. Chem. Soc., 1976, 98, 2886–2894 CrossRef CAS.
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef CAS.
- M. H. Abraham, P. L. Grellier, J.-L. M. Abboud, R. M. Doherty and R. W. Taft, Can. J. Chem., 1988, 66, 2673–2686 CAS.
- K. Dernbecher and G. Gauglitz, J. Chem. Phys., 1992, 97, 3245–3251 CrossRef CAS.
- M. Jonsson, A. Houmam, G. Jocys and D. D. M. Wayner, J. Chem. Soc., Perkin Trans. 2, 1999, 425–429 RSC.
- P. M. Mancini, G. Fortunato, C. Adam, L. R. Vottero and A. J. Terenzani, J. Phys. Org. Chem., 2002, 15, 258–269 CrossRef CAS.
- M. N. Kobrak, J. Phys. Chem. B, 2007, 111, 4755–4762 CrossRef CAS.
- C. Ye and J. M. Shreeve, J. Phys. Chem. A, 2007, 111, 1456–1461 CrossRef CAS.
- H. D. B. Jenkins, H. K. Roobottom, J. Passmore and L. Glasser, Inorg. Chem., 1999, 38, 3609–3620 CrossRef.
- R. J. Larsen and M. L. Marx, An introduction to mathematical statistics and its applications, 2nd edn, Prentice-Hall, Engelwood Cliffs, 1986 Search PubMed.
- S. K. Poole, P. H. Shetty and C. F. Poole, Anal. Chim. Acta, 1989, 218, 241–264 CrossRef CAS.
- C. Wakai, A. Oleinikova, M. Ott and H. Weingärtner, J. Phys. Chem. B, 2005, 109, 17028–17030 CrossRef CAS.
- H. Weingärtner, P. Sasisanker, C. Daguenet, P. J. Dyson, I. Krossing, J. M. Slattery and T. Schubert, J. Phys. Chem. B, 2007, 111, 4775–4780 CrossRef.
- S. Schrödle, G. Annat, D. R. MacFarlane, M. Forsyth, R. Bucher and G. Hefter, Chem. Commun., 2006, 1748–1750 RSC.
- C. Daguenet, P. J. Dyson, I. Krossing, A. Oleinikova, J. Slattery, C. Wakai and H. Weingärtner, J. Phys. Chem. B, 2006, 110, 12682–12688 CrossRef CAS.
- C. Schröder, T. Rudas and O. Steinhauser, J. Chem. Phys., 2006, 125, 244506 CrossRef CAS.
- C. Schröder, C. Wakai, H. Weingärtner and O. Steinhauser, J. Chem. Phys., 2007, 126, 084511 CrossRef CAS.
- C. J. Margulis, H. A. Stern and B. J. Berne, J. Phys. Chem. B, 2002, 106, 12017–12021 CrossRef CAS.
- A. Rivera and E. A. Rössler, Phys. Rev. B, 2006, 73(212201), 1–4.
- W. Dietrich and P. Maass, W. Dieterich and P. Maass, 2002, 284, 439–467 Search PubMed.
- Y. Yoshida, O. Baba and G. Saito, J. Phys. Chem. B, 2007, 111, 4742–2749 CrossRef CAS.
- L. Crowhurst, R. Falcone, L. Lancaster, V. Llopis-Mestre and T. Welton, J. Org. Chem., 2006, 71, 8847–8853 CrossRef CAS.
- B. R. Mellein, S. N. V. K. Aki, R. L. Ladewski and J. F. Brennecke, J. Phys. Chem. B, 2007, 111, 131–138 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Ionic volume calculations. See DOI: 10.1039/b711991g |
| This journal is © The Royal Society of Chemistry 2008 |