Remote in situ voltammetric techniques to characterize the biogeochemical cycling of trace metals in aquatic systems
Mary-Lou
Tercier-Waeber
*a and
Martial
Taillefert
b
aGroup of Analytical and Biophysical Environmental Chemistry (CABE), Department of Inorganic and Analytical Chemistry, University of Geneva, Sciences II, 30 Quai E.-Ansermet, CH-1211, Geneva 4, Switzerland
bSchool of Earth & Atmospheric Sciences, Georgia Institute of Technology, 311 Ferst Drive, Atlanta GA, 30332-0340, USA
First published on 11th December 2007
Abstract
The contamination of aquatic ecosystems by natural and anthropogenic metals has lead to a need to better characterize their impact in the environment. To a large extent, the fate and the (eco)toxicity of these elements in aquatic systems are related to their chemical speciation, which may vary continuously in space and time. Detailed measurements of the fraction of specific metal species or groups of homologous metal species and their variation as a function of the bio-physicochemical conditions of the natural media are thus of prime importance. To determine these metal fractions as well as redox chemical species regulating their distribution (dissolved oxygen, sulfides, iron and manganese oxides), new analytical tools capable of performing in situ, real-time monitoring in both water columns and sediments with minimum perturbation of the media are required. This paper reviews the challenges associated with metal speciation studies, and the progress made with state of the art voltammetric techniques to measure the speciation of metals in situ. More specifically, it summarizes the specific conceptual, analytical, and technical criteria that must be considered and/or fulfilled to develop rugged, field deployable, non-perturbing sensors and probes. Strategies used to satisfy these criteria are presented by describing the up-to-date most advanced voltammetric sensors, mini-/micro-integrated analytical systems, and submersible equipments developed for in situ measurements of trace metals and main redox species in aquatic systems. The spatial and temporal resolutions achieved by these news tools represent a significant advantage over traditional laboratory techniques, while simultaneously remaining cost effective. The application of these tools to aquatic systems is illustrated by several examples of unattended and remote in situ monitoring and/or profiling in water columns and sediments.
 Mary-Lou Tercier-Waeber Mary-Lou Tercier-Waeber | Mary-Lou Tercier-Waeber, B.Sc, is currently Senior Researcher in the Analytical and Biophysical Environmental Chemistry (CABE) group at the University of Geneva (Switzerland). For the past 20 years, her work has focused on the development of new voltammetric sensors, mini/micro-integrated analytical systems, submersible probes and analytical techniques, and the application of these tools in laboratory, in-field and in situ to study priority trace metal complexation, speciation and biogeochemical cycles to better understand the circulation, role and impact of trace metals in aquatic ecosystems. She has over 40 publications in international peer reviewed journals in Analytical Chemistry and Environmental Sciences, 3 invited book chapters, and 1 patent. |
 Martial Taillefert Martial Taillefert | Martial Taillefert, PhD, is presently an Associate Professor in the School of Earth and Atmospheric Sciences at the Georgia Institute of Technology (USA). He has long-term interests in metal cycling and the role of bacterial processes in the regulation of geochemical processes in aquatic systems. His research combines laboratory investigations with natural samples and in situ measurements at redox interfaces to investigate the mechanisms regulating the transformation of metals in aquatic systems. He has published 27 peer reviewed papers in the geochemical sciences. |
1. Introduction
The impact of human activities, in particular chemical pollution, on the environment is becoming increasingly important. Natural ecosystems are complex environments, regulated by a large number of physical, chemical and biological processes, which all together ensure the functioning of the system and its homeostasis. Ecosystem health requires that all these processes, in particular the chemical ones, are well tuned and balanced. In this context, chemical pollution may result in concentration changes of major and minor constituents, which may shift equilibria from one state to another. Ecosystems are thus functioning very much like living organisms with a capacity to acclimatise or not to significant variation in chemical species concentrations.Among these chemicals, trace elements, which are ubiquitous and diverse components of the Earth’s geochemistry, play critical roles in ecosystem function. Environmental chemistry investigations and toxicological studies have provided a wealth of information on the role of trace metals in biogeochemical processes.1–4 In particular: As, Cd, Pb, Hg, Cr, Al are key elements due to their extreme toxicity even at low concentrations, while Mn, Fe, Cu, Zn, Ni, Co, and Se are important because they may be essential or toxic depending on their concentrations and the type of organisms considered. The contamination of the environment by these metals, classified as priority metal pollutants (except Mn, Fe and Al) by the EU Commission and the US-EPA, is widespread around the world, as illustrated by the global budget of their sources into air, soils, and aquatic ecosystems reported by Pacyna et al.5 and summarized in Tables 1 and 2. This inventory clearly shows that anthropogenic activities have become the most important catalyser of the global geochemical cycling of trace metals and, thus, may present a threat for ecosystems as well as for humans who often lay at the top of the trophic chain.1
| Natural sources to the atmosphere | Anthropogenic sources to the atmosphere | Anthropogenic sources to aquatic systems | Anthropogenic sources to terrestrial systems |
|---|---|---|---|
| a Metal mentioned: contribution up to >/ = 15% of their max. total release from the various sources and in the order of importance. Modified from ref. 5. | |||
| Biogenic sources (Se, Hg, Cd, As, Pb, Zn) | Fuel combustion (coal, oil, gasoline, wood) (Ni, Pb, Hg, Se, Cr, Mn, Cu, As, Zn) | Waste disposal (Ni, Mn, Cr, Cu, As, Zn, Cd, Se) | Disposal of municipal and industrial wastes (Cd, Zn, Hg, Ni, Cu, Cr, Mn, As, Pb, Se) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Volcanic emissions (Ni, Hg, Cd, Cr, As, Ph, Zn) | Nonferrous metal industry (Cd, Cu, As, Zn, Se) | Steam electric (Hg, Se, As, Ni) | Commercial waste products (e.g. various chemicals) (Cu, Cr, As, Pb, Zn, Mn) |
|
|||
| Windborne soil particles (Cr, Mn, Ni, Ph, Zn, Cu, As) | Other industries and use (Cr, Mn, Zn, Se) | Mining, smelting, and refining (Cd, Se, Zn, As, Mn, Cu, Ni) | Coal fly ash and bottom fly ash (Se, Mn, Ni, As, Cd, Cr, Hg, Cu, Zn, Pb) |
|
|||
| Seasalt spray | Waste incineration (Hg, Mn, Cd) | Manufacturing processes (Zn, Cr, Cu, Hg, Cd, As) | Fertilizer (As, Cd, Hg, Pb) |
|
|||
| Wild forest fires | Atmospheric deposition (Pb, Hg, Cd, Ni, As, Zn) | Peat (agricultural and fuel uses) | |
| Atmospheric deposition (Hg, Cr, Pb, Cd, As) | |||
| Me | Atmospheric emissions from natural sources | Anthropogenic emissions to the atmosphere | Anthropogenic emissions to aquatic systems | Anthropogenic emissions into soils |
|---|---|---|---|---|
| As | 1.1–23.5 | 12.0–25.6 | 11.6–70.3 | 52.4–111.6 |
| Cd | 0.1–3.9 | 3.2–12.0 | 2.1–16.3 | 5.6–37.7 |
| Cu | 2.2–53.8 | 19.7–50.8 | 34.7–190.5 | 541.5–1402.8 |
| Cr | 4.5–82.8 | 7.2–53.7 | 45.6–238.8 | 484.6–1309.5 |
| Pb | 0.9–23.5 | 287.5–376.0 | 97.2–276.7 | 479.1–1039.4 |
| Hg | 0–4.9 | 0.9–6.2 | 0.2–8.8 | 1.6–15.0 |
| Ni | 2.9–56.8 | 24.2–87.2 | 33.1–194.2 | 93.3–493.8 |
| Se | 0.7–18.1 | 1.7–5.8 | 10.1–71.9 | 6.0–76.5 |
| Zn | 4.0–85.9 | 70.4–193.5 | 77.5–394.7 | 689.3–1953.7 |
A deeper understanding of metals of environmental and biological concern, their role and impacts on the environment, and the distinction between biologically useful and toxic concentrations is thus of prime importance to define appropriate strategies to maintain ecosystems in good “health”. These strategies should be based on the following criteria:
• A fundamental understanding of ecosystem functioning, especially with respect to the environmental physical chemistry of trace metals, as well as the role of biogeochemical processes and physical forcing on the regulation of trace metal distribution
• Continuous, real-time information about the ecosystems obtained through networks of submersible instruments and probes to assess any disfunctioning of an ecosystem as early as possible
• A diagnosis, followed by the recommendation of possible treatments, established by comparing the record of analytical data in impacted systems with that of pristine environments.
Even if these criteria have been recognized for more that 20 years, they have yet to be implemented in aquatic systems.
The objective of this paper is to illustrate the challenging work required to satisfy the above criteria by presenting the state of the art in the in situ monitoring of trace metals and the main redox chemical species regulating their distribution in aquatic ecosystems. It reviews the key aspects that must be considered to better understand the role and the fate of metals in aquatic systems (section 2). Based on this information, the limitations of conventional monitoring strategies and techniques are summarized (section 3), and the tools needed to monitor the speciation and fate of trace metals more reliably are presented (section 4). The characteristics of voltammetric sensors and probes for this purpose (section 4) and the up-to-date (most) advanced voltammetric systems presently developed to detect specific fractions of trace metals and the main redox species in situ are described (section 5). Finally, the potential of such systems for more efficient environmental monitoring of the trace metal speciation and the main redox species in the aquatic ecosystems is illustrated with selected examples of their applications in both the water column and sediments (section 6).
2. Importance and challenge of trace metal speciation measurements in aquatic systems
Understanding the impact of the anthropogenic release of priority trace metals on the functioning and quality of aquatic ecosystems is of particular concern for two reasons. First, the direct release of these metals in natural waters ranges typically between a few thousand and up to 250 thousand tons per year (Table 2). Average metal concentrations may thus increase to reach levels several fold higher than those in unpolluted areas.5 In addition, it is estimated that 30 to 40% of metals emitted to the atmosphere5 ultimately reach natural waters. Moreover, changes in soil (e.g. open-pit mines) and groundwater management practices have resulted in an increased mobilization of metals and in an enhanced runoff of these compounds to rivers, lakes, estuaries, and costal areas. Second, metals are inherently persistent, i.e. they are neither created nor degraded by anthropogenic and biological processes. Once they have entered aquatic ecosystems (Fig. 1A), or other environmental compartments, they are transformed by biogeochemical processes and distributed under various physico-chemical forms (Fig. 1B), i.e. particulate (>0.45 µm), colloidal (1 nm–0.45 µm) and dissolved metal species (≤1 nm µm). The latter includes free (hydrate)-metal ions, which are known to constitute most of the bioavailable fraction, as well as simple inorganic and organic complexes with anthropogenic or natural organic ligands, which are potentially bioavailable (Fig. 1B).6,7 Inorganic and organic colloidal/particulate material plays a key role in coagulation, sedimentation, and adsorption processes, which influence trace metal residence times and transport from the water column to the sediments (e.g.refs. 3 and 8–12). Metals transported to the sediment can be either buried or remobilized via various diagenetic processes (Fig. 1C), e.g.reduction of manganese and iron minerals during mineralization of natural organic matter or reaction with sulfides produced during sulfate reduction (e.g.refs. 13 and 14). As transformations among the various metal species, involving often changes in (weak) coordinate bonding and/or changes in oxidation state, may occur continuously, the fate of trace metals strongly depends on the species, which can coexist and may or may not be in thermodynamic equilibrium. These changes are normally reversible on time scales that vary depending on the element, with the important consequence that the speciation of a metal is a function of the biophysico-chemical conditions of the medium in which it is found15 and, thus, may vary as a function of time and space. Variations in the proportion of the species present affect trace metal bioavailability, their degree of adsorption on colloids and particles, their overall mobility in the water column, and their rates of transfer across the sediment–water interface.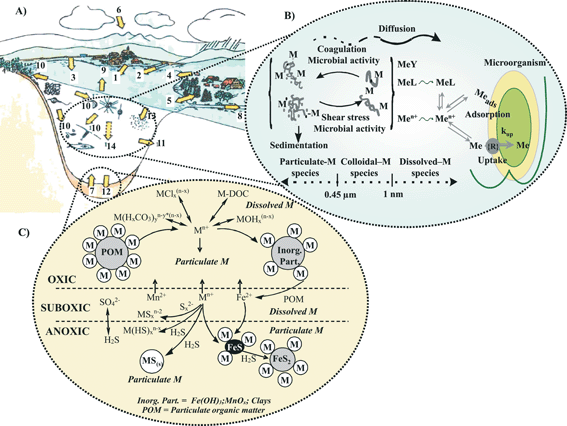 | ||
| Fig. 1 Schematic representation of the different fluxes of trace metals between aquatic ecosystems (A), with emphasis on the geochemical processes regulating metal cycling in the water column (B) and in sediments (C). Trace metals may reach aquatic systems via: (1) waste water treatment; (2) tributaries, surface runoff from (3) roads and (4) agricultural lands and soils, (5) exchange with groundwater, (6) atmospheric deposition, (7) release from sediments; they may be eliminated through: (8) discharge in effluents, (9) evaporation, (10) uptake by surrounding vegetation and organisms, (11) infiltration into subterranean waters, (12) reaction in the upper sediment layers, (13) adsorption by suspended matter, and (14) sedimentation. | ||
The complexity of trace metal transformations described above indicates that assessing the risk of metal contamination in aquatic systems is more difficult than for organic contaminants (which are largely determined by the form in which the organic compounds enters the environmental compartment and by any subsequent, generally irreversible, degradation processes).15 In particular, it is obvious that standard procedures based on the measurements of total, or total dissolved, metal concentrations alone do not yield sufficient information. The measurements of specific metal species or groups of homologous metal species are required to better understand and interpret the biological and geochemical cycling of trace elements. Concurrent measurements of the major biophysico-chemical parameters in ambient conditions (temperature, pressure, pH, conductivity, primary productivity) and main redox chemical species (oxygen, sulfides, iron and manganese oxides) are also required to assess the influence of habitat-specific constraints on the spatial distribution and temporal variations of metals species. In summary, it is necessary to characterize the chemical speciation of trace metals and the main redox chemical species with a great temporal and spatial resolution to be able to assess the risk posed by their accumulation in aquatic systems.
3. Conventional monitoring techniques and their limitations
Conventional approaches for trace metal measurements in aquatic systems depend on the type of samples collected. For the water column, discrete water samples are usually collected with peristaltic pumps or Go-Flo bottles, the dissolved metal fraction is separated by normal flow filtration on 0.45 µm (or 0.2 µm) pore size membranes, tangential flow filtration (TFF), or ultrafiltration in the field or laboratory, and the raw and filtered samples are acidified in the field or laboratory. Sometimes the filters are preserved and digested in acid. The samples are usually stored until analysis of the acidified raw, filtered, and digested fractions in the laboratory. Discrete sediment cores are usually sampled with box or individual corers. The sediments are sectioned and porewaters separated by centrifugation and filtration or porewater squeezing, generally under an inert atmosphere to prevent contaminations by oxygen of the atmosphere. Porewaters are usually stabilized using acid and/or freezing or analyzed immediately after separation. Sediment extractions are also conducted after centrifugation or squeezing of porewaters to characterize the content of the ‘wet’ sediment. The sediment can be dried before analysis if the fractions analyzed are not affected by the drying process.The main analytical techniques for trace metal analyses in laboratories are: graphite furnace atomic adsorption spectroscopy (GFAAS); inductively coupled plasma mass spectroscopy (ICP-MS), neutron activation analysis (NAA), and voltammetric techniques (in particular anodic stripping and adsorptive linear sweep voltammetric techniques). Detailed technical and analytical description of these techniques, as well as their advantages and limitations for trace metal measurements and speciation in complex media and, in particular, environmental samples, has been reported in several reviews (e.g.refs. 16 and 17). Briefly, GFAAS, ICP-MS and NAA are advantageous, compared to voltammetric techniques, because they are applicable to a larger number of elements. Their major drawbacks include their much higher cost and, above all, the fact that they allow measurements of total metal concentrations only. Consequently, speciation measurements using these analytical techniques are only achieved by coupling them with separation and extraction procedures, which drastically increase the time and cost of analyses and prevent their application for speciation measurements on large sample sets. Unfortunately, trace metal speciation has to be achieved with a high spatial and temporal resolution to address ecosystem heterogeneities and dynamics and interpret the environmental impact of metals correctly in both water column and sediments.
Other major limitations of conventional approaches to trace metal analyses, even for in-field measurements, include sample perturbations during sampling, sample handling, and possibly sample storage. As mentioned above, species distribution and properties must absolutely be preserved and known to interpret the role of metals in biogeochemical processes, in particular to assess their ecotoxicological role. Sample perturbations include contaminations of trace metals or their losses by adsorption onto the walls of containers, but also, for samples collected from depths, speciation changes due to variations in temperature, pressure, CO2, O2, and/or H2S content, and consequently in pH, redox potential, and solubility of solids known to adsorb trace metals. Perturbations of sediment samples may also occur during the collection of sediment cores and storage in the laboratory because depth-concentration profiles of trace metals display gradients at millimetre or submillimetre scale in three dimensions.18,19 Thus, preserving pH, redox and chemical gradients at that resolution within a sediment core is extremely difficult. The use of expensive clean procedures and facilities (such as clean rooms) minimize the problem of contaminations but are ineffective to solve the other problems.
4. Advanced monitoring strategies and requirements for in situ voltammetric trace metal monitoring
The aspects presented and discussed in sections 2 and 3 above clearly demonstrate that to understand and interpret the biological (bioaccumulation, bioconcentration, bioavailability, toxicity) and geochemical (transport, adsorption, precipitation, re-mobilization) cycling of trace elements in aquatic ecosystems, it is essential to:• Identify and quantify relevant specific metal species or groups of homologous metal species.
• Avoid sampling and analytical artifacts which occur with conventional techniques.
• Perform measurements with appropriate temporal and spatial resolutions.
• Measure other parameters, e.g. the major biophysico-chemical parameters (temperature, pressure, pH, dissolved oxygen, conductivity, primary productivity) and redox species (∑H2S—i.e., H2S, HS–, S2–, manganese and iron oxides or their reduced products) concurrently to assess the influence of the site-specific conditions on the spatial distribution and temporal variation of metals species.
To achieve these objectives, the development of rugged, submersible, non-perturbing sensors and probes that can be deployed in networks, together with commercially available multiparameter probes, to perform remote, in situ, long-term monitoring of trace metal analysis and speciation is required (Fig. 2). Additional advantages of a network of submersible probes include the capability of: building detailed spatial and temporal databases of complete ecosystems at low cost; sending warning/alarm signals and implementing quick remedial action in case of significant/sudden increase in pollutant concentrations; and monitoring locations difficult to access (e.g. boreholes, deep lakes and oceans).
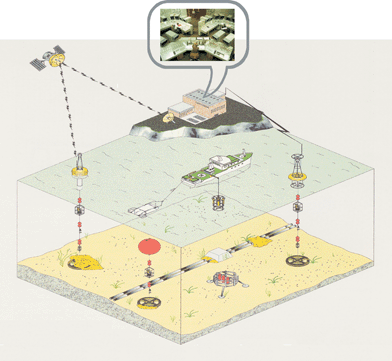 | ||
| Fig. 2 Schematic representation of “ideal” ecosystem monitoring strategies based on a network of in situ analytical probes. The detailed spatial and temporal variations of a large number of chemical compounds and bio-physicochemical parameters are monitored in real-time and continuously by a network of submersible probes remotely controlled by a land station which downloads and stores data, and provides free access to data bases through the internet.. | ||
Voltammetric techniques are very well suited to design probes for trace metal analysis (see refs. 20–22 for recent detailed reviews). In particular, using different types of electrodes and techniques: (i) a large number of trace compounds of environmental interest can be measured; (ii) several analytes can be detected in the same potential scan; (iii) low detection limits are achieved; (iv) the speciation based on the redox state, mobility, and/or lability of metal species can be measured; and (v) low cost, automated, compact equipment with low energy requirements can be built (see sub-sections 5.3 and 5.4). The applicability and usefulness of voltammetric submersible probes for in situ measurements of trace metals and main redox chemical species have been demonstrated by the first prototypes reported during the period 1990 to 1999.23–26 However, the applications of these systems were limited to short-term (typically 1 day) in situ measurements in surface waters, i.e. depth <20 m,23–25 and shallow sediments, i.e.water depths <30 m.26 Their use for long-term monitoring at greater depth was limited in particular by the following problems: (i) insufficient reliability23 and sensitivity24,25 of the voltammetric sensors used, (ii) fouling of the sensor surface due to adsorption of natural organic or inorganic matter,23,25 (iii) interferences from ill-controlled hydrodynamic conditions23,24 and dissolved oxygen,23 and/or (iv) the use of standard commercially available laboratory equipments which do not withstand pressure.23,24,26
These results have clearly highlighted that to enable reliable in situ long-term voltammetric monitoring, significant developments are required to improve conventional voltammetric devices. These improvements include:
4.1 Rugged and reliable microelectrodes
Microelectrodes have a few unique characteristics which make them particularly well suited for environmental monitoring. First of all, the metal flux at microsized electrode, measured as electrochemical current, discriminates the so-called dynamic metal species, i.e. the sum of the free metal ions and the sufficiently labile (large dissociation rate) and mobile (large diffusion rate) inorganic and organic complexes of a few nanometres in size.20,22 This fraction of metals is important as it represents the maximum fraction of metals potentially bioavailable (e.g.refs. 6, 7 and 20 and sub-section 6.1.2). Other key advantages of microelectrodes are summarized below (see e.g.refs. 27 for review): (i) their low iR drop allows direct measurements not only in sea water but also in low ionic freshwater without addition of an electrolyte. This characteristic prevents changes in the speciation that may be encountered when the ionic strength of solution is altered and is required for the measurement of some specific metal species as the dynamic metal fraction mentioned above. (ii) Radial diffusion occurs when the electrode radius, r, is much smaller than the diffusion-layer thickness, δ, (r << δ; typically r ≤ 10 µm22) and a steady-state transport is quickly established even in quiescent solution. Thus, stirring the solution is unnecessary during the pre-concentration step of stripping techniques, which greatly improves the reliability of analysis, simplifies the design and the maintenance of submersible probes and is a pre-requisite condition to perform stripping voltammetryin situ in sediment and soil porewaters. This feature is also the basis for the development of a new generation of sensors, the gel-integrated microelectrodes presented in sub-section 5.1. (iii) Thanks to their increased mass-transport combined with their lower capacitance,27 a significantly larger signal-to-noise ratio is obtained with microelectrodes, which allows the determination of sub-nanomolar concentrations with short pre-concentration times. (iv) Microsized Hg electrodes minimize the polluting capability of Hg electrodes. This is of importance as the high reliability and sensitivity required for in situ, long-term, remote voltammetric monitoring of trace metals have been achieved so far only by Hg electrodes (section 5), despite significant efforts made over the last 15 years to develop Hg-free electrodes (for more details, see ref. 22 and references cited therein).The only drawback of small microelectrodes (i.e. r ≤ 10 µm) is that the fabrication steps are more complicated and must be perfectly controlled to reach high sensitivity and reliability.28,29 In turn, larger microelectrodes can be used for direct measurements, i.e. without pre-concentration, if detection limits are sufficiently low for particular applications (e.g.ref. 30 and sub-section 5.2). Larger microelectrodes are much easier to fabricate and represent a compromise between sensitivity and cost. However, potential effects of convection on voltammetric signals must be taken into account by performing external calibrations in the flow conditions of the investigated environment,31 cross calibrations with other type of sensors,26 or internal calibrations in the studied media. The latter has been successfully achieved for oxygen measurements in the water column (e.g., ref. 32) or above the sediment–water interface (see sub-section 6.2).
4.2 Modified microelectrodes and/or analytical procedures
To: (a) determine, in addition to the dynamic metal fraction, other specific metal species or well defined groups of metal species relevant for ecotoxicity assessment and interpretation of biogeochemical cycles (e.g. sub-sections 5.1 and 5.3); (b) prevent biological, physical, and chemical interferences, including: (i) (bio)fouling of the electrode, due to adsorption of organic matter, colloids, and/or bacteria on the sensor surface, which may lead to significant perturbation and/or attenuation of voltammetric signals and, thus, inaccurate analyte concentrations; (ii) ill-controlled convection which occurs in most water bodies, may modify the sensor diffusion layer thickness and significantly affect the signals; and (iii) O2, present at concentration up to ≈3×10–4 M in saturated water, is reduced to H2O2 or OH–, depending on the applied potential, and may produce a signal 4 to 6 orders of magnitude larger than that of the analyzed trace metals, as well as a pH increase, and thus a change in metal speciation, at the electrode surface. The first interference can be minimized by using square wave anodic stripping voltammetry (SWASV) at high frequency coupled to background measurement and subtraction.23,33 This is applicable in aquatic media with a sufficiently high buffer capacity, such as sea water, where the second interference is weak. However, in oxygenated freshwaters with low buffer capacity, i.e. typical natural waters with HCO3– concentration ≤5×10–3 M, the pH at the electrode surface during the deposition step of SWASV may increase up to 11, leading to drastic changes of metal speciation with formation, and sometimes precipitation, of metal hydroxides. In these cases, in situ, on-line removal of O2 before voltammetric detection is required (see e.g. sub-section 5.3).4.3 Rugged reference electrodes
The reference electrode is the second key component of the voltammetric measurement setup. Commercially available liquid junction reference electrodes are not suitable for in situ measurements because they are pressure dependent and not easy to handle. Other types of references electrodes have been proposed for miniaturization and to avoid the use of internal solution. A detailed description of their characteristics, advantages and drawbacks for in situ applications is given in ref. 22. Some of these devices have been successfully deployed i.e.: mini-Ag/AgCl/KCl saturated Metrohm or 3% agarose gel reference electrode with a zirconium oxide ceramic junction (sub-section 5.3) as well as mini-reference based on Ag/AgCl wire inserted in a KCl–Alumina–Teflon solid state pellet have been used for in situ voltammetric and potentiometric measurements in fresh and sea waters (see ref. 22 and references cited therein); Ag/AgCl pseudo reference has been used for in situ measurements in sea water.314.4 Flow-through cells with small volume (≤1 ml)
Flow-through cells are required to perform well controlled speciation measurements in the water column and to eliminate O2 interferences (e.g. sub-section 5.3). A small volume of the cell and of its fluidic system is required to minimize the flushing volume, the dead volumes and the possible volumes of reagents (e.g. sub-section 5.3). Specific criteria must be taken into account for the fabrication of submersible mini-flow-through voltammetric cells. They are discussed in detail in ref. 22.4.5 Technical developments
Mechanical, electronic, firmware, and software improvements are also required to provide pressure resistant probes, including the sensors, cell and fluidic components; very low noise potentiostats; efficient control of the system; fast data acquisition, storage, and/or transmission from the deployed probe to the surface, the surface to a land station and vice versa.This brief overview shows that the development of voltammetric probes for in situ trace metal analysis is a challenging task. It is a multidisciplinary field which requires analytical innovations, in particular, to develop sensors providing physico-chemically rigorous speciation capability, to simplify analytical processes, and to minimize the overall complexity of these systems and their energy consumption. New concepts must be used based on both analytical and environmental knowledge. So far, only few voltammetric sensors and submersible systems, developed for in situ applications, have been systematically characterized, optimized, and applied in various environmental studies. They include gel-integrated Ir/Hg and unmodified Au/Hg microelectrodes incorporated in various type of deployable platforms which have been applied for dissolved Cu, Pb, Cd, Zn, Mn, and Fe analysis and speciation, and for the analysis of O2(aq), H2O2(aq), Fe2+, Mn2+, S2O32–, I–, ∑H2S (=S2– + HS– + H2S), S0 in S8, S0 and S2– in Sx2–, FeS(aq), and soluble organic–Fe(III) complexes, respectively. These devices are described in section 5 below and their analytical properties summarized in Table 3. The work performed for their development highlights strategies that can be used to satisfy the requirements for in situ voltammetric speciation studies mentioned above.
| Sensor/probe | Species measured | Technique | Typical tot. analysis time | LOD |
|---|---|---|---|---|
| LOD = Lower detection limit. Using:a 15 min electrochemical pre-concentration, tot. analysis t = 25 min.b 1 h chemical accumulation, 5 min electrochemical pre-concentration, tot. analysis t = 70 min.c 8 min sample pre-treatment; 5 min electrochemical pre-concentration, tot. analysis t = 20 min.d In sea water.e In freshwater. | ||||
| GIME/VIP-MPCP | Cu, Pb, Cd, Zn free ions + dynamic complexes with size of few nm | SWASV | 10 to 40 min | a Cu = 200 pM; Pb = 30 pM; Cd = 50 pM; Zn = 300 pM |
|
||||
| CGIME/MPCP | Cu, Pb, Cd free ions | Chemical pre-concentration + SWASV | 70 to 130min | b Cu = 20 pM; Pb = 10 pM; Cd = 60 pM |
| FIA-GIME/MPCP | Cu, Pb, Cd total extractable cone. | On-line sample pre-treatment + SWASV | 20 to 30 min | c Cu = 800 pM; Pb = 150 pM; Cd = 200 pM |
| GIME/VIP | Mn(II), Fe (II) | SWCSV | 10 min | Mn(II) = 0.1 uM; Fe(II) = 1 µM |
|
||||
| Au/Hg elect./ AIS ISEA | O2; H2O2 | LSV | 25 s | O2 = H2O2 = 5 µM |
|
||||
| Au/Hg elect./AIS ISEA | Mn(II), Fe(II), I–, ΣH2S, S(0), Sx–2 + S2O32 + S4O62–, HSO3– | SWCSV | 30 s | d Mn(II) = 15 uM; Fe(II) = 25 µM; eMn(II) = 5 uM; Fe(II) = 10 µM; I– = ΣH2S = S(0) = Sx–2 = 0.2 µM; S2O32– = 10 µM; S4O62– = HSO3– = 50 µM |
5. Voltammetric sensors and submersible systems applied to systematic in situ trace metal and redox species analysis
5.1 Gel-based microelectrodes for Cu, Pb, Cd, Zn, Mn and Fein situ analysis and speciation in the water column
Gel-based microsensors consist of integrated microanalytical systems coupling separation, and possibly other analytical processes, with voltammetric detection in a mm3 volume, both to eliminate interfering compounds and quantify a selected group of metal species. The simplest one reported is the gel-integrated microelectrode (GIME),34,35 which consists of a Hg-plated Ir microelectrode covered with a few micron thick agarose gel. Presently, this sensor allows simultaneous measurements of the dynamic fraction (i.e. the fraction potentially bioavailable, see section 4 and refs. 6, 7 and 20) of Cu(II), Pb(II), Cd(II), Zn(II) at picomolar levels using square wave anodic stripping voltammetry (SWASV) and of Mn(II) and Fe(II) at nanomolar levels using square wave cathodic sweep voltammetry (SWCSV) (Table 3).The GIME has been developed under two geometries: the agarose membrane-covered mercury-plated Ir single microelectrode (µ-AMMIE) (Fig. 3A) and the agarose membrane-covered mercury-plated Ir microelectrode array (µ-AMMIA) (Fig. 3B). These microsensors are produced under systematic, well-controlled steps and conditions to ensure high reliability and sensitivity of trace metal measurements in complex media.28,29,34,35 Briefly, the main steps used to produce the single Ir microelectrode include: electroetching of a Ir wire to a tip of a few µm in radius; sealing of the electroetched Ir wire in a glass capillary tubing pulled to a tip of 1 mm in diameter; soldering of the Ir wire to a copper shielded cable using either electronic bombardment under vacuum or electric arc microfusion, under inert argon atmosphere, of an intermediate gold wire; and automatic mechanical polishing of the tip using successive silicon carbide (SiC) pads of 15, 10 and 5 µm and diamond paste of 1 µm (all from Stuers) until mirror like Ir surfaces are obtained.28 The microelectrode arrays consists of 5 × 20 interconnected iridium microdiscs each of 5 µm diameter and a centre to centre spacing of 150 µm surrounded by a 300 µm thick Epon SU-8 containment ring for the gel. It is produced by means of thin film technology35,36 involving: successive deposition, on standard silicon wafer, of 2000 Å thick Si3N4, Ir, Si3N4 layers; photolithographic patterning of the Si3N4 top layer to define the interconnected Ir microelectrode array and the bonding pad; deposition and patterning of the Epon SU-8 containment ring around the 1.8 × 4 mm individual devices; mounting and wire bonding of the individual devices on printed circuit boards (PCB); and encapsulation with epoxy resin. Just before use, the Ir sensors are covered with a 1.5% LGL agarose gel layer (thickness: 300 to 600 µm for the µ-AMMIE34 and 300 µm for the µ-AMMIA35). Hg hemispheres are then plated on the Ir microdisc(s) by electrochemical reduction at –400 mV of an acidic 5 mM Hg(CH3COO)2 solution for a given period of time that varies according to the size of the Ir microdisc(s) (typically 3 to 20 min for Ir microdisc diameters of 3 to 10 µm28) and reoxidized by scanning the potential from –300 to 300 mV in KSCN for their renewal.29,33,34 Both processes are performed through the gel layer; in both cases, currents are recorded as a function of time and the radius of the mercury hemisphere(s) determined from the corresponding electrical charge. Electrochemical yields for Hg hemisphere formation are close to 100%, which ensures high reproducibility of drop size and surface area (variability ≤5%).28,29,33,34 A given agarose membrane can be used over an extended period of time of more than one month, and it has been shown that diffusion through the membrane is independent of pressure up to 600 bar.35
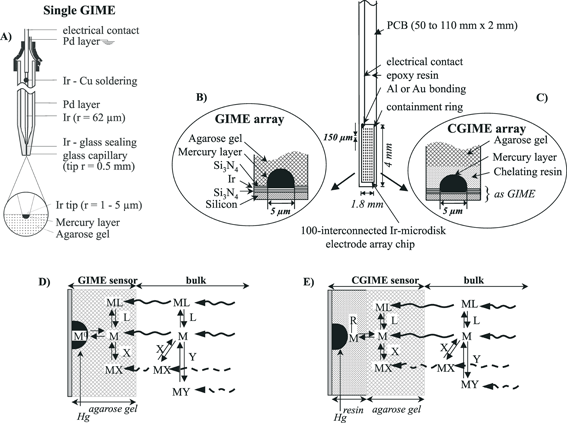 | ||
| Fig. 3 Schematic diagrams of: (A) a single gel-integrated microelectrode (GIME); (B) an array of GIME; (C) the complexing gel-integrated microsensor array (CGIME); (D) the chemical processes occurring at the GIME–solution interface; and (E) the chemical processes taking place at the CGIME–solution interface. | ||
Measurements with the GIME are performed in two successive steps: equilibration of the agarose gel with the test solution (typically 5 to 10 min for a membrane thickness of 300 µm) and voltammetric analysis inside the gel. GIME are sensitive and reliable, thanks to the characteristics of microsized electrodes mentioned in section 4 and a perfect control of their fabrication steps.28,29,34,35 Detection limits for SWASV measurements of Cu(II), Pb(II), Cd(II), Zn(II) using a pre-concentration time of 15 min are 200, 30, 50 and 300 pM, respectively (Table 3), and can be improved by increasing the pre-concentration time (i.e.GIME peak current intensities are directly proportional to the concentration of the analytes in solution and to the pre-concentration time).34,37,38 Minimum detection limits for SWCSV measurements of Mn(II) and Fe(II) are 0.1 and 1 µM, respectively (Table 3, refs. 34 and 39). The standard deviation of the average calibration slopes obtained for systematic calibrations of GIME sensors by standard additions of the target analytes in 10–3 to 0.1 M NaNO3, synthetic freshwater, and sea water at various salinity37,38 is typically ≤10%. These results show that signals measured with GIME are not influenced by the sample matrix (i.e. ionic strength and chloride concentrations) and that the GIME is applicable to all types of surface natural waters. They also show that a complete calibration before each application is not required and measurements of one or two standard solutions are sufficient. This is of particular interest for field applications. Other important features of the GIME for in situ measurements are linked to the agarose gel membrane, which enhances the performance of Hg-plated Ir microelectrodes because: (i) it acts as a dialysis membrane and allows efficient exclusion of biopolymers, colloids and macromolecules with size typically >35 nm40 which may adsorb on the sensor surface and interfere with the voltammetric measurements (i.e. minimisation of the fouling problem34,35,39); (ii) it forms a layer in which transport is controlled exclusively by molecular diffusion, i.e. ill-controlled hydrodynamic conditions of the outside water body do not influence the voltammetric signal;34,35 (iii) it stabilizes the Hg hemisphere(s) plated onto Ir; and (iv) it forms a “chamber” where specific reactions/processes can be performed to improve the selectivity or enlarge the field of application of the sensor.20 Characteristics (i) to (iii) are essential to obtain a sensor that is reliable and robust enough to be used for in situ autonomous, continuous, long-term monitoring of trace metals in complex media, such as environmental samples. More specifically, it has been shown that the GIME can be used to perform continuous replicate measurements during several days without renewal of the Hg hemispheres (variability <10% for measurements up to 14 days).34,41,42
Characteristic (iv) of the GIME has been used to develop another type of gel-integrated microsensor, the complexing gel-integrated microelectrode (CGIME) (Fig. 3C). The CGIME is a novel sensor which allows in situ simultaneous SWASV measurement of the true free concentrations of Cu(II), Pb(II), and Cd(II) at sub-nanomolar levels in natural waters (Table 3, refs. 42 and 43). Determination of free ion concentrations is crucial for the assessment of the ecotoxicological impact of trace metals as this fraction is often the preferred fraction captured by microorganisms (Fig. 1B).6,7 Detailed description of the CGIME preparation is given in ref. 43. Briefly, a few µm thin layer of a polystyrene immobilized iminodiacetate resin, with an average bead size of 0.2 µm (Microchelex resin, CETAC Technologies –USA), is deposited at the surface of the interconnected Ir microdisc chip by dropping 1 µl of a 2.5% (w/v) Microchelex aqueous suspension solution in the Epon SU-8 containment ring. The sensor is gently rotated by hand until water evaporates and results in a white deposit covering the sensor surface, then stored in a closed plastic container for at least 12 h to ensure complete drying of the resin. The gel layer is then deposited on the chelating resin by filling the containment ring with 1.5% LGL agarose gel at 80 °C. After cooling and equilibration of the agarose gel, Hg is deposited and reoxidised, for its renewal, through both layers using similar conditions to those reported above for the GIME.
Measurements with the CGIME are performed in three steps. First, the resin is left to equilibrate with the sample; because the number of sites in the resin is very small compared to the reservoir of metal ions in natural waters, metals accumulate on the resin in proportion to their bulk free-ion concentrations. In a second step, the sample is exchanged with a suprapur pH 1 acidic 0.1 M NaNO3 solution to release trace metal accumulated on the resin, and the deposition step of the SWASV technique is immediately started to reduce and pre-concentrate the metals released in the Hg hemispheres. After a 5 min deposition, the stripping step is performed to reoxidize and record the current of the electrochemically pre-concentrated metal, which chemically accumulated on the resin.
Selectivity of the CGIME to the free metal ions in the presence of small labile metal complexes has been validated by tests performed in model, well-characterized, complexing media, as well as in marine and fresh waters by intercomparison with results obtained by hollow fiber based permeation liquid membrane (HF-PLM) coupled to ICP-MS detection.44,45 HF-PLM coupled to ICP-MS is a very sensitive technique which selectively separates, pre-concentrates, and detects free ions in the presence of a large number of ligands.44,45 Precision for calibration slopes obtained by standard additions in 0.1 M NaNO3 and replicate measurements up to 8 days (max. time tested) without renewal of the Hg hemispheres are typically ≤10% for all sensors tested.42,43 Detection limits for Cu(II), Pb(II), and Cd(II) are 20 pM, 10 pM, and 60 pM, respectively, using an accumulation time of 1 h (Table 3, ref. 43) and can be improved by increasing the accumulation time (typical accumulation time used for in situ measurements: 2 h42,43).
5.2 Au/Hg micro-electrodes for in situ measurements of Mn, Fe and other redox species
Au/Hg microelectrodes usually consist of either a 13, 25, or 100 µm diameter gold wire that is soldered directly to a copper conductor with a silver-containing solder.30 The gold wire has been enclosed in different casing according to the application: capillary tubing that is pulled to a tip of 100–500 µm diameter for the depth profiling of chemical species in biofilms46 or the two-dimensional dissolution of minerals by scanning electrochemical microscopy;47 polyetherketone (PEEKR) tubing of 3 mm diameter and a few centimeters long filled with epoxy is preferred for the measurement of chemical species in deep-sea hydrothermal vents,48,49 the water column of marine and freshwater environments,32,50–52 and laboratory applications;53,54 and glass tubing pulled to a tip of 500–750 µm diameter and approximately 5 cm long and filled with epoxy is generally used for the depth profiling of chemical species in freshwater and marine sediments.12,13,19,26,30,55–59Au/Hg microelectrodes are usually prepared by first sanding the tip of the gold electrode on a fine grit sand paper and polishing with emulsified diamond-pastes of 15, 6, 1, and 1/4 µm (Buehler, Inc.) to make a mirror-like gold surface. Mercury is then plated on the gold substrate by electroreduction of an acidic 0.01 M Hg(NO3)2 solution and the Au/Hg film conditioned at –9 V in 0.1 M NaOH for periods of time that vary according to the size of the gold substrate.30,47 This last step seems to allow mercury to form a strong amalgam with gold and ensures good reproducibility and robustness.
Typically, linear sweep voltammetry (LSV) is employed from –0.1 to –1.85 V with Au/Hg microelectrodes to quantify O2 and H2O2 in natural waters, while direct square wave cathodic sweep voltammetry (SWCSV) from –0.1 to –1.75 V is used to detect, Fe2+, Mn2+, S2O32–, I–, ∑H2S (=S2– + HS– + H2S), S0 in S8, S0 and S2– in Sx2– (Table 3, refs. 30 and 60), FeS(aq),56,61 and soluble organic–Fe(III) complexes.62 To maintain reproducibility and integrity of Au/Hg electrodes, a conditioning potential is applied for a given time between each scan to remove any previously deposited electroactive species from the electrode surface. For cathodic measurements, a conditioning period of 10 s at –0.1 V removes Mn2+ and Fe2+ from the electrode surface between measurements. When soluble organic Fe(III) complexes or dissolved sulfide are present, a conditioning step at –0.9 V for 10 s removes these species from the electrode surface before the next measurement. Eventual electrode fouling by organic matter, usually evidenced by peak shifts towards positive potentials and/or decrease in sensitivity30,31 are avoided by scanning the electrode potential to –2 V. At such negative potential, sodium ions are reduced and react subsequently with water to produce H2 which reduces and desorbs organic matter.30 When the concentration of dissolved sulfide is high, square wave anodic stripping voltammetry (SWASV) is necessary to avoid formation of HgS double films.63 Scan rates of 200 mV/s are typically applied for all measurements in sediments, but higher scan rates are preferred in strong hydraulic current conditions.64,65
The redox species determined by direct SWCSV are usually calibrated using the pilot ion method with Mn2+ as pilot ion,26,30 while dissolved oxygen is calibrated independently.19 Dissolved oxygen is also calibrated in situ using a single point calibration when saturation with the atmosphere can be safely assumed and temperature and salinity are known, or when the concentration of dissolved oxygen is known from an independent method.65 The minimum detection limits in seawater range from 15 and 25 µM for Mn2+ and Fe2+,64 <0.2 µM for I–, ΣH2S, S(0), and Sx2–, to 5 µM for dissolved oxygen, 10 µM for S2O32–, 50 µM for S4O62– and HSO3–(Table 3). In freshwater, detection limits for Mn2+ and Fe2+ decrease to approximately 5 and 10 µM (Table 3), respectively, mainly because the concentration of sodium is low enough to minimize interferences. The chemical composition of soluble organic–Fe(III) complexes and FeS(aq) are still unknown and cannot be quantified directly using external calibrations. These species are therefore reported in voltammetric current intensities.61,62 The precision of replicates in seawater is usually better than ±1% with conditioning of the electrode; ±5% in shelf/slope and subtidal sediments, and ±10% in salt marsh sediments.
5.3 Submersibles probes based on gel-integrated microelectrodes: VIP and MPCP systems
The voltammetric in situ profiling system (VIP™) is the first submersible probe developed by taking into account all the criteria mentioned in section 4.33 It is the only system commercially available (Idronaut Srl, Italy; http://www.Idronaut.it) with the capacity for in situ trace metal monitoring and profiling, down to 500 m, with sub-nanomolar sensitivity (Table 3). The heart of the voltammetric probe is the GIME, either single or array, microsensor described above (Fig. 3A, 3B). The system consists of several units: a submersible voltammetric probe, a submersible on-line O2 removal module,41 a submersible multiparameter probe (Ocean Seven 316, Idronaut-Milan), a calibration deck unit, a surface deck unit, and a laptop computer. A detailed description is given elsewhere.33 The important characteristics of the sub-units are briefly summarized. The submersible voltammetric probe has been designed in two different models: model 1 with a Delrin housing for in situ measurements in surface waters (Fig. 4A); model 2 with a titanium housing for in situ measurements of trace elements in groundwater and mining boreholes (Fig. 4B). The voltammetric probe is comprised of several modules (Fig. 4A): an electronic probe housing (upper part), a pressure compensated mini-flow-through plexiglas voltammetric cell (internal volume = 1 ml), a pressure case base incorporating the preamplifier for the voltammetric microsensor, and a submersible peristaltic pump (lower part). The voltammetric cell consists of two parts: an internal flow-through cell and an external cell, held together by means of a cover. The GIME and the counter electrode, which consists of a built-in platinum ring, are located in the internal flow-through cell. The compartment between the internal and the external cell is completely filled with 0.1 M NaNO3 in 1.5% LGL agarose gel, which plays several important roles. It acts as a double bridge, with two ceramic porous junctions in contact with the working solution, for the in-house manufactured Ag/AgCl/KCl saturated 3% LGL agarose gel reference electrode located at the bottom of the external cell. It also shields both the microsensor and the counter electrode. Most importantly, it acts as a pressure equalizer through a rubber pressure compensator. Pressure compensation of the cell allows in situ measurements at great depth and solves liquid junction problems with the reference electrode. The cell is screwed, with o-ring seals, to the cover of the pressure case base. The pressure case base is mechanically assembled to the electronic housing via two titanium rod connectors, through which pass the electrical connections of the three electrodes, the microsensor preamplifier, and the motor of the submersible pump. The electronic housing contains all the hardware and firmware necessary to manage: the voltammetric measurements (available techniques: LSV, CV, SWCSV, ASV, SWASV, chronoamperometry); the data acquisition, storage in an internal non-volatile memory with its own battery to guaranty high data retention and protection, and transfer, on operator request, by RS-232 (max. depth 100 m) or telemetry (depth > 100 m); the interfacing of the multiparameter probe, the submersible peristaltic pump, and a laptop computer. The submersible on-line oxygen removal module is connected between the sampling pump and the inlet of the flow-through voltammetric cell. It consists of a silicone tubing surrounded by an enzymatic cross-linked O2 scavenging gel.41 As water is pumped through the silicone tubing, oxygen diffuses through the tubing wall and is consumed on the other side by the scavenging gel. This module is required only for in situ trace element monitoring in oxygen-containing freshwaters (see section 4). In sufficiently buffered natural media, as sea and estuarine waters, interference of O2 is eliminated by combining SWASV with high frequency and subtractive voltammetry.33 The submersible multiparameter probe controls the exact depth of the voltammetric probe model 1 (Fig. 4A) and the simultaneous measurements of the complementary master variables (T, pH, O2, conductivity, salinity, turbidity) which are necessary for both speciation and environmental interpretation. In the case of the probe model 2 (Fig. 4B), a temperature and depth sensor have been incorporated into the submersible voltammetric probe. The calibration deck unit is used in laboratory or ex situ in the field to plate or renew the Hg layers, to calibrate the probe, and to measure standards or chemically modified collected natural samples, e.g. acidified raw or filtered samples for the measurements of the colloidal and particulate forms. The surface deck unit powers and interfaces, by telemetry, the measuring system with a laptop computer. This unit provides an autonomy of about 35 h and can be recharged either in continuous mode using a solar captor or after use. A Windows management software allows the user to configure the operating conditions for the voltammetric techniques, to calibrate the voltammeric and multiparameter probes, to control the data transfer from the internal probe memory to the PC, to perform the automatic data treatment and maintenance operations, and thus to make maximum use of the probe capabilities, albeit in a user friendly manner. The system can be controlled either by an operator on board or in automatic mode following pre-programmed instructions. | ||
| Fig. 4 (A) Standard version of the VIP system for in situ monitoring and profiling in freshwater and marine water columns: a. voltammetric probe model 1; b. multiparameter probe; c. on-line O2 removal system. (B) VIP voltammetric probe model 2 for groundwater monitoring. (C) The MPCP system, a complete submersible mini-labotratory for in situ trace metal analysis and speciation, coupled to the simultaneous measurements of master bio-physicochemical variables, in the water column. | ||
A more sophisticated system, called multi physical chemical profiler (MPCP™), has been recently developed to extend the capability of the VIP to in situ monitoring of trace metal speciation42 (Fig. 4C). This system, which is a completely submersible mini-laboratory (lab-on-cable system), is in its final optimization prior to commercialization. Presently, the MPCP™ allows the simultaneous in situ monitoring and profiling (down to 150 m) of three major fractions of Cu, Pb, and Cd species coupled to the master biogeochemical variables (pressure, temperature, pH, oxygen, conductivity, salinity, redox E, turbidity and chlorophyll a).42 The three metal fractions measured by SWASV include: (i) the free metal ion concentration, which are known to be related to bio-uptake, using a CGIME (Fig. 3C); (ii) the concentration of the dynamic metal species, i.e. the potentially bioavailable species, using a GIME (Fig. 3B); and (iii) the total extractable metal concentration, i.e. the reservoir of metal in the test medium. Subtracting (ii) from (iii) provides the concentration of metal bound to particles and colloids.
The MPCP™ probe consists of an improved version of the VIP™ probe with three independent potentiostats and voltammetric pressure-compensated flow-through cells with their own fluidic systems and specially-designed submersible peristaltic pumps based on optical encoders for precise control of the head pump speed. One cell includes the CGIME, whereas the other two include a GIME. The second one is coupled to a submersible flow-injection analysis (FIA) system for automatic on-line sample pre-treatment prior to GIME measurements of the total extractable metal concentration. Metal extraction is performed by complexation with the strong ligand triethylenetetramine, followed by acidification and heating of the solution. Reagents for CGIME measurements and the FIA on-line pre-treatment of the samples are stored in soft bags for medical use. Similar bags are used to collect waste from CGIME and GIME-FIA measurements. The MPCP™ probe has been completed with the integration of a multiparameter probe (Idronaut Srl, Italy) into the voltammetric probe and the connection of an external fluorescence probe (Seapoint INC. USA) to monitor simultaneously depth, temperature, pH, oxygen, conductivity, salinity, turbidity, and chlorophyll a (as a proxy for biological productivity). The electronic housing contains all the hardware and firmware necessary to manage: (i) the simultaneous control of the fluidic system and the SWASV measurements of each cell; (ii) the interfacing of the integrated multiparameter and external fluorescence probes; and (iii) the data acquisition, storage (into a non-volatile memory with its own battery to guarantee high data retention and protection) and transfer via either RS232 (depth ≤100 m) or high speed FSK telemetry (depth > 100 m). During field trials, the system can be connected to either ship board stabilized 220 V or a 24 V battery pack (located at the surface or at depth in a pressure resistance housing) that can be re-charged after use or in continuous mode using a solar panel system. The interface between the laptop computer and the MPCP™ probe is accomplished via a custom and user friendly Windows management software. This software allows the user, through menus and memo-technique commands, to control and setup the following MPCP™ probe operating parameters and functions: (i) fluidic and electrochemical parameters, (ii) measurements with the three voltammetric channels and the multiparameter and fluorescences probes either independently or simultaneously, (iii) data “smart” processing (i.e. graphical display, curve smoothing, metal peak current and potential searching, correction of temperature effect, calculation of metal concentration using pre-configured calibration curves), (iv) calibration, and (v) diagnostic and maintenance operations of the probe components. As the VIP™, the MPCP™ can be controlled by an operator on board or in automatic mode based on pre-programmed parameters, but also in remote mode when connected to a specially designed remote controller.42 The remote controller consists of a buoy controller module (BCM), a computer-driven motorized winch, and a control land station based on cellular phone link. The control land station, through its custom-written Windows management software, allows the user to control the MPCP™ from a shore station. Three different autonomous pre-defined time-scale monitoring/profiling modes can be performed: (i) at fixed depth; (ii) at programmed depths (up to 6); and (iii) linear profiling using a pre-selected depth increment. For the last two measuring cycles, the motorized winch acts as a lift, moving up and down the MPCP™ at the next monitoring position. Transfer of the data from the MPCP™ to the land station can be performed on user request or in an automatic way at pre-defined time intervals. The land station management software also allows the user to modify the MPCP™ operating parameters and the remote measuring mode, as well as to check the status of the MPCP™ and remote controller components via diagnostic commands. The BCM, which can be installed on board ship, on a platform, on the bank of a water body, or in a moored buoy, contains all the hardware and firmware necessary to: (i) supervise the monitoring activities of the MPCP™; (ii) collect and store the data of the voltammetric, multiparameter, and fluorescence probes in a non-volatile memory; (iii) send stored data to the control land station; (iv) upgrade operating and measuring cycle parameters received from the control land station; and (v) diagnose and report failures of the MPCP™ or remote controller components.
5.4 Submersible systems based on Au/Hg sensors
Four systems, based on commercially available potentiostats (Analytical Instrument Systems Inc. USA (AIS); http://www.aishome.com) and/or a computer in pressure resistant housing, have been developed for in situ measurements with Au/Hg electrodes.64 The first instrument is designed for water depths of 30 to 50 m26 and consists of a transmitter (AIS Model DLK-LCT-1) enclosed in a pressure housing, which is connected to the working, reference, and counter electrodes though a metre long shielded cable. In this system, data are transmitted to a standard laboratory DLK-100 Electrochemical Analyzer (Analytical Instrument Systems, Inc.) onboard ship through a long cable receiver (Model DLK-LCR-1) made of three shielded wires for the electrodes as well as a ±15 V DC power unit and a ground wire. The second instrument, the in situ Electrochemical Analyzer I™ (AIS ISEA I™) is designed for real-time measurements in both shallow and deep waters and consists of a potentiostat, a multiplexer to monitor up to four electrodes successively, and an internal computer to communicate through a standard RS-232 cable with a laptop computer (Fig. 5A). The instrument is a stand-alone package that can be deployed from ships, remotely operated vehicles (ROV), or deep-sea submersibles and configured differently depending on the application. For ROV or submersibles,66 the AIS ISEA I™ can be powered externally by the vehicle. For CTD-rosette deployments,32,51,67 the instrument can be powered by an internal battery. For ROV and CTD-rosette deployments, the instrument can be tethered up to 1500 m without the need for signal amplification. The third instrument, the AIS ISEA II™, is similar in design but adapted for unattended measurements. In this generation of instrument, the internal computer is programmed to run a sequence of experiments and store data on a memory card until the analyzer is recovered. This instrument has so far been deployed on a free benthic lander (Fig. 5B) for sediment depth profiles and benthic chamber measurements68 and in a salt marsh for the long-term monitoring of sediment porewaters.59,64 For long-term deployments in shallow environments, a solar panel system can be used to recharge batteries, and communication devices are currently developed to communicate with the instrument remotely through VHF radio or cellular modem technology. The latest generation of ISEA, the AIS ISEA III™, is capable of functioning in both real-time and programmable mode and is now available from Analytical Instrument Systems, Inc.69 All ISEA instruments are controlled by a Labview software (National Instruments) to program voltammetric experiments in real-time or in a programmable sequence. The software allows the user to run linear sweep, cyclic, normal pulse, differential pulse and square wave voltammetry, including stripping techniques, between +2 and –2 V at scan rates ranging between 1 and 10000 mV s–1 and sensitivities between pA and mA. In addition, triggers can be included between experiments to switch electrodes or actuate external devices, such as the AIS model MAN-2in situmicromanipulator for depth profiles in sediments (Fig. 5B). All ISEA instruments also include two auxiliary inputs to monitor temperature and pH during each voltammetric scan simultaneously, with inputs from a multiparameter MicroCAT probe (Seabird Electronics, Inc.). Generally, single voltammograms or a sequence of voltammograms can be run in real-time by the AIS ISEA I™, while a complete deployment has to be pre-programmed for AIS ISEA II™ deployments. Data are stored on the ISEAs internal memory card and can be downloaded to a computer via an external communication cable. Alternately, the memory card can be physically removed from the electronic housing after deployments. Data can be processed with the Analytical Instruments Systems, Inc. Data Analysis software which is part of the instrument package. Alternately, large data sets can be processed with a semi-automated Matlab™-based software which also deconvolutes peaks that overlap.70
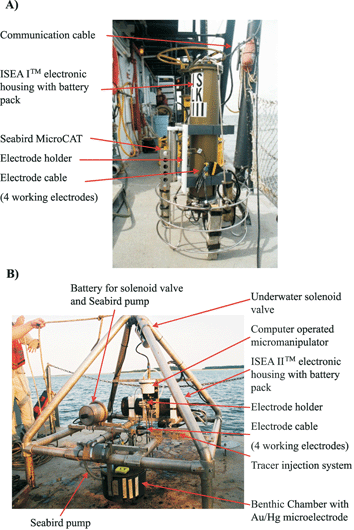 | ||
| Fig. 5 (A) Rosette with ISEA I™ system (Analytical Instrument Systems, Inc.) for real-time measurements as a function of depth in water columns. The instrument includes an internal battery pack and is equipped with a MicroCAT (Seabird Electronics, Inc.). Up to four Au/Hg microelectrodes can be positioned on the rosette. (B) Free benthic lander with an ISEA II™ system, an underwater micromanipulator (Analytical Instrument Systems, Inc.) controlled by the instrument, and a benthic chamber. During deployments, the chamber lid is maintained in the open position by a solenoid valve controlled by a timer. An underwater pump (Seabird Electronics, Inc.) mixes overlying waters in the chamber in a gentle fashion to avoid perturbing the sediment–water interface. The ISEA system runs in an automatic data acquisition mode. An Au/Hg microelectrode is positioned across the chamber lid to monitor redox chemical species composition in the benthic chamber. Up to three Au/Hg microelectrodes can be positioned on the micromanipulator to lower electrodes in the sediment. | ||
6. Applications
6.1 In situ trace metal analysis and speciation using the VIP™ and MPCP™ systems
The VIP™, Fig. 4A–4B, has been successfully deployed in situ to monitor the concentration of the dynamic fraction of trace metals (i.e.Cu(II), Pb(II), Cd(II), Zn(II), Mn(II)) and their temporal and/or spatial evolution in freshwater,33,39,41 groundwater,71 fjord waters,37 estuarine and coastal marine waters.33,38 Autonomous, semi-continuous in situ measurements, at time intervals of typically 30 min to 1 h, were performed up to one week without renewal of the sensor Hg hemispheres. The MPCP™, Fig. 4C, has been successfully applied for attended and remote in situ monitoring and profiling in various estuaries and coastal sea waters42 and more recently in freshwater (sub-section 6.1.2). Presently, a monitoring program based on the deployment of a network of VIP™ and MPCP™ probes is underway to study the temporal evolution of trace metal speciation and the spatial variation of the specific metal species or group of metal species in the upper part of the Lot River watershed in France, which is characterized by chronic polymetallic pollution resulting from now abandoned mining and Zn ore extraction activities.During each field trial, the VIP™ and MPCP™ probes are prepared in laboratory or on board ship. Hg hemisphere deposition on the gel-based Ir microelectrodes and pre-calibrations of the systems are performed the day before the deployment. In addition, measurements of two standards every two days and/or post-calibrations are performed after the systems are retrieved. Less than 10% variations were observed in standard solutions during field surveys and pre- and post-calibrations, even for a continuous application of the probes during up to 8 days without renewal of the sensor mercury layers (max. time tested,39,42). These results confirm those previously reported for the reliability and the long-term stability of the GIME and CGIME sensors obtained in laboratory measurements38,41,43 and the absence of memory effects from in situ measurements. Intercomparison of results obtained from in situ voltammetric measurements using gel-based microsensors, in-field voltammetric measurements with and without gels,39 other sets of speciation sensitive techniques,39,42,72,73 and laboratory based-techniques,33,36,39,72,73 as well as speciation modeling73 has been conducted. These systematic tests have: (i) demonstrated the capability the VIP™ and MPCP™ as a tools for in situ, reliable, real-time monitoring of environmental relevant fractions of metal species;39,42,73 (ii) demonstrated the importance and efficiency of the agarose gel as antifouling membrane;39 and (iii) confirmed laboratory results35,37,39 which showed that pressure has no effect on voltammetric signals and that a metal-dependent temperature correction (typically 3–8% per °C depending on the metal and the techniques and conditions used) should be applied to correctly quantify concentrations, as calibration are usually performed at 20 °C, whereas the temperature of natural waters may vary between, typically, 24 and 4 °C.74 Furthermore, these field measurements have clearly demonstrated the advantages of such probes, compared to traditional procedures, for biogeochemical studies of trace metals. These findings are illustrated below by the report of data obtained during two selected field studies.
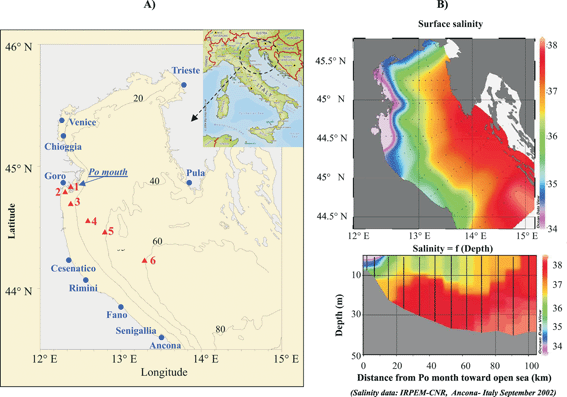 | ||
| Fig. 6 (A) Geographic location of the Po River mouth and positions of 6 stations in the Adriatic Sea where the MPCP profiling has been performed. (B) Salinity of the Adriatic Sea showing the export of freshwater to the continental shelf. (C) Vertical profiles of salinity from the Po River mouth to the shelf. | ||
The concentrations of the three distinct fractions of Cu and Pb obtained by MPCP™ in situ measurements at the first four stations during the cruise in the fall of 2002 are reported in Fig. 7. The first observations that could be made from these data were that: (i) the concentrations of the free ion and dynamic species, i.e. the most potentially toxic forms, of both metals are a small percentage of the total extractable metal concentrations; and (ii) the ratios of these species to total extractable concentrations vary, in various proportions for the two metals, as a function of depth and distance from the river mouth and period of the year (not shown). These results clearly demonstrate that the measurements of total (dissolved) metal concentrations alone are not appropriate for ecotoxicological assessment. Another observation was that while a decrease of the total extractable and dynamic concentrations of Cu and Pb was observed as a function of both distance from the Po mouth and depth (in particular between the 0.2 m surface water and the deepest layers), the variations observed for the Cu and Pb free concentrations were apparently independent of these parameters. Similar results were observed for the data collected during the cruise of spring 2004, even though the concentrations of the various Cu and Pb metal species were generally significantly higher, especially in the Po mouth (see metal species concentrations at the lowest salinity value in Figs. 8 and 9), due to the higher Po discharge generally observed at this period of the year. Comparison of the Cu and Pb speciation data, determined during both cruises (Figs. 8 and 9), with those of the master variables measured simultaneously reveals that the decrease in concentration of the colloidal/particulate metal species with distance from the mouth of the Po river is mainly related to the increase in salinity and decrease in turbidity (Fig. 8-A1, B1; Fig. 9-A1, B1). These results show that a significant proportion of these metal species is rapidly eliminated in coastal areas due to fast coagulation and sedimentation processes associated with the increase in salinity, i.e. the ionic strength of the media. The similar behaviour observed for the dynamic Cu and Pb species (Fig. 8-A2 and B2; Fig. 9-A2 and B2) suggests that a significant proportion of these species adsorbs on freshly formed particulate species and is thus eliminated by the same process. The free Cu and Pb concentrations show different trends. The free Cu concentrations were found to be strongly correlated to chlorophyll a, i.e. to the primary productivity (Fig. 8-A6 and Fig. 9-A6), whereas Pb free ion concentrations were found to be very low (typically between 0.01 and 0.02 nM) and relatively constant (Fig. 8-B6 and Fig. 9-B6). These results suggest that a significant proportion of Cu2+ is either assimilated by the phytoplankton or complexed by their exudates, or even both. The role of biota is supported by the fact that Pb2+, which is not easily assimilated, does not follow the same trend as Cu2+. These findings demonstrate that the MPCP™, by performing real-time, simultaneous measurements of environmentally-relevant fractions of metals and master variables, is unique and can help understand the biogeochemical cycles of trace metals and assess their fate. Indeed, all these information could not be obtained using traditional procedures based on laboratory measurements of total dissolved metal concentrations in discrete samples.
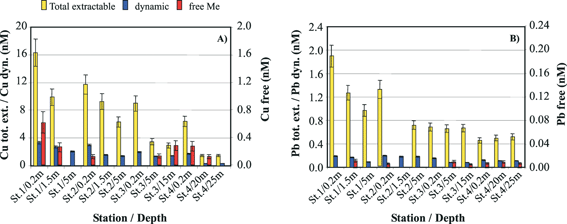 | ||
| Fig. 7 (A) Cu and (B) Pb speciation monitored in situ with the MPCP™ at various stations and depths in the Po River plume (Adriatic cruise—Italy, 27 October to 2 November 2002). Concentrations of total extractable and dynamic Me concentrations reported are average of three FIA-GIME and GIME, respectively, replicate measurements. Concentrations of free metal ions are average of two replicate CGIME measurements. | ||
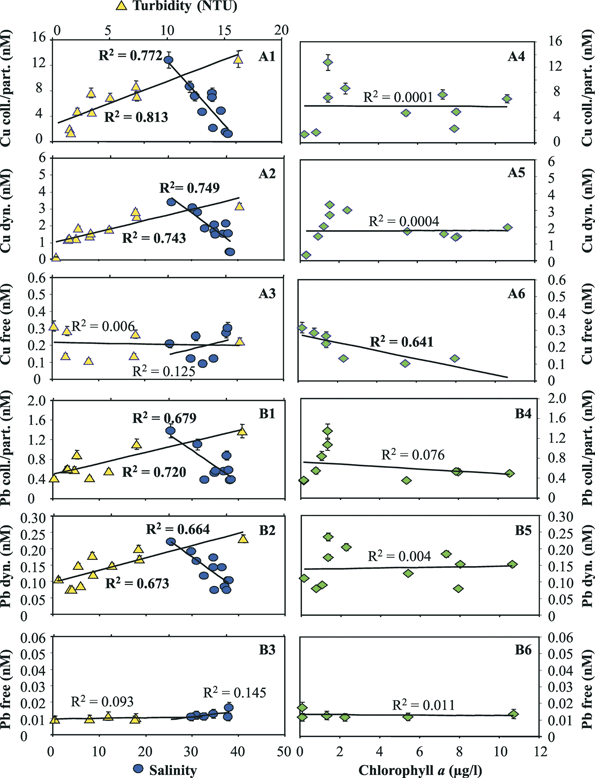 | ||
| Fig. 8 Concentrations of Cu (A) and Pb (B) species measured in situ as a function of salinity and turbidity (1–3) as well as chlorophyll a (4–6) monitored simultaneously with a MPCP™ at various stations and depths in the Po plume (First Adriatic cruise—Italy, 27 October to 2 November 2002). | ||
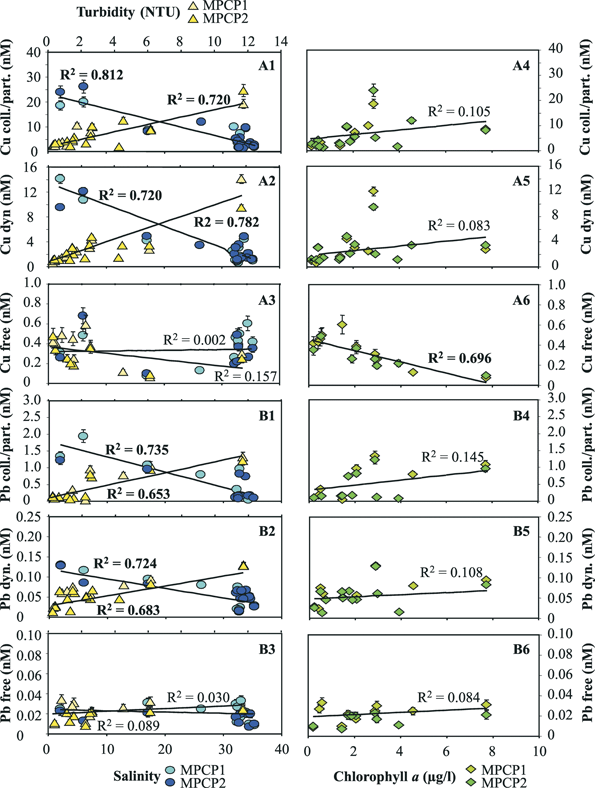 | ||
| Fig. 9 Concentrations of Cu (A) and Pb (B) species measured in situ as a function of salinity and turbidity (1–3) as well as chlorophyll a (4–6) monitored simultaneously with two MPCPs at various stations and depths in the Po plume (Second Adriatic cruise—Italy, 29 March to 3 April 2004). | ||
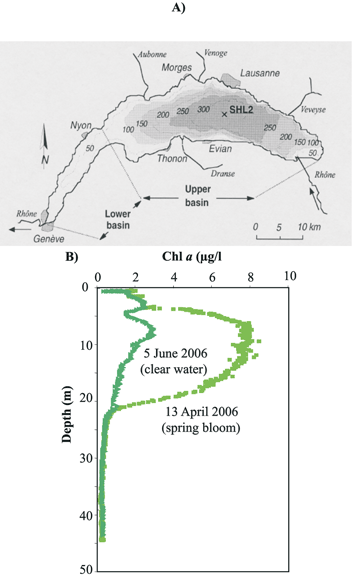 | ||
| Fig. 10 (A) Bathymetric map of Lake Geneva, one of the largest lakes in Western Europe, situated on the border between Switzerland and France, and location of the monitored station (SHL2). (B) Chlorophyll a profiles monitored with the MPCP™ during the spring phytoplankton bloom and at the clear water phase of the year 2006. | ||
Based on this information, the MPCP™ was deployed in 2006 at station SHL2 (Fig. 10A) during the spring bloom and the clear water phase (Fig. 10B), to obtain in situ profiles of trace metal speciation in the epilimnion and the upper part of the hypolimnion coupled to biophysico-chemical master variables. Profiles obtained for the various species of Cu and Pb are reported in Fig. 11A–D. First it can be seen that, as observed in the Po plume, the free and the dynamic fractions always represent a small fraction of the total concentration. But most importantly, the concentration profiles of the three Pb fractions measured simultaneously with the MPCP™ were similar during the spring bloom and the clear water phase period (Fig. 11 C–D). In contrast, a significant decrease, within analytical errors, of the concentrations of both the free Cu and the dynamic Cu species was observed in the epilimnion during the spring bloom (Fig. 11A–B). As observed in the Po plume, changes in free Cu concentrations were mainly correlated to chlorophyll a (Fig. 11E). The main difference with Lake Geneva, however, was that the variation of the dynamic fraction of Cu was found to be also mainly correlated to changes in chlorophyll a (Fig. 11E). The slope of the curve Cu dyn = f (Chl a) was found to be greater than those of the curve Cu free = f (Chl a) (Fig. 11E). Similar results were obtained previously from VIP™ profiling in Lake Geneva41 and MPCP™ profiling in the Adriatic Sea42 during summer phytoplankton blooms. These results suggest that, as also predicted by theoretical computations, small labile complexes of essential trace metals, such as Cu, may be assimilated by the biota, whereas similar species of toxic metals, such as Pb, are not (Fig. 10F). These results also show that the GIME and CGIME are not only integrated microanalytical systems coupling separation and chemical pre-concentration of selected metal species prior to highly sensitive voltammetric detection, but also behave as biological sensor analogs, i.e. sensors allowing to quantify relevant metal species or group of metal species that are directly involved in biological uptake mechanisms.20 This was expected from the very beginning of their development, as chemical processes occurring at the sensor surface–sample interface (Fig. 3D and E) are similar to those taking place at the microorganism–water interface (Fig. 1B) (see ref. 20 for more details). Such sensors should provide better insights into the biological cycles of trace metals and improve our understanding of their (eco)toxicological impact.
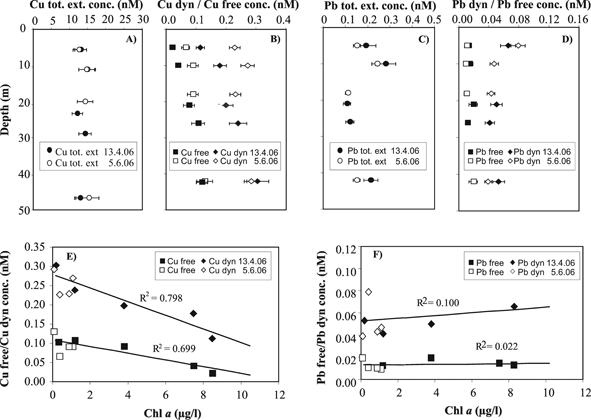 | ||
| Fig. 11 Concentration profiles of Cu (A–B) and Pb (C–D) specific fractions measured simultaneously with the MPCP in lake Geneva during the April spring phytoplankton bloom (black symbols) and the June clear water phase (open symbols) in 2006. Concentrations of the Cu (E) and Pb (F) free ion and dynamic species as a function of chlorophyll a. | ||
6.2 In situ measurements of Mn, Fe and other redox species on Au/Hg microelectrodes
Au/Hg microelectrodes have been deployed successfully in deep-sea hydrothermal vents,48,49,64 where they were used over long periods of time in highly turbid waters with high temperature, high sulfide concentrations, and strong flows. In these extreme environments, these electrodes have revealed temporal variations in the concentrations of redox chemical species that have never been observed before in real-time.48 In addition, the speciation capabilities of these electrodes revealed that the extremely complex temperature variations near the diffuse flow along vents affect the speciation of sulfur and iron over very small time scales.49 These results indicate that sample collection of hydrothermal vent fluid using conventional high pressure bottles cannot prevent changes in the speciation of chemical species during cooling after collection. Present research aims at deploying Au/Hg voltammetric microelectrodes at hydrothermal vents for long periods of time using the AIS ISEA III™ to understand how hydrothermal vent chemistry influences vent ecology.64 In addition, Au/Hg voltammetric microelectrodes are currently deployed in deep-sea boreholes to monitor deep hydrothermal vent fluid chemistry during microbial sampling to help define natural media conditions in an attempt to culture new extremophiles.Simultaneously, Au/Hg voltammetric microelectrodes have been deployed in the water column of several freshwater and marine environments including lakes,50 shallow estuaries,52 the Chesapeake Bay,67,76 and the Black Sea32,51 to investigate redox processes across oxic–anoxic interfaces. The real-time capability of the AIS ISEA I™ system coupled with the short acquisition times at fast voltammetric scans provide a spatial and temporal resolution that rivals with conventional dissolved oxygen, temperature, and conductivity probes mounted on CTD rosettes. These studies have demonstrated the dynamics of iron and sulfur cycling at oxic–anoxic transitions in stratified waters with a unique temporal resolution,52 episodic intrusions of oxygenated dense waters from the Mediterranean Sea in the reduced zone of the Black Sea,51 the tidally-driven mixing of the Chesapeake Bay chemocline,67 and its effect on the distribution of manganese and other trace metals.76
Au/Hg voltammetric microelectrodes have also been deployed extensively in freshwater and marine sediments after core collection,12,13,19,30,56,57,77,78 biofilms, and microbial mats.46,79 These studies have provided new insights into the cycling of iron and sulfur in these environments and into the role of photosynthetic processes on the oxidation of reduced chemical species, by measuring depth profiles with millimetre to submillimetre spatial resolutions in one dimension (most references above) or three dimensions19,77 and over temporal scales varying from dial cycles to seasons.13,57 Less frequently, in situ measurements in sediments have been performed with ROV and benthic landers.59,65,77 The first in situ deployments of Au/Hg voltammetric microelectrodes have been performed using the long cable receiver (Model DLK-LCR-1) coupled to a computer-controlled DLK-100 potentiostat (AIS, Inc.) onboard ship for real-time profiling in continental shelf sediments with an underwater micromanipulator.26,65 Because these measurements involved a tether, ships had to be maintained at the same position using three-point anchoring. These studies were able to investigate the main diagenetic processes responsible for the transformation of organic matter26 and the advective transport of solutes65 in continental shelf sediments.
Recently, a free benthic lander carrying a benthic chamber for flux measurements and an in situmicromanipulator for profiling in sediments has been developed (Fig. 5B, ref. 68). These measurements can be used to study diagenetic processes in marine sediments, estimate rates of natural organic matter remineralization, measure the flux of analytes across the sediment–water interface, and investigate the role of bioturbation and bioirrigation on diagenetic processes. Four-legged for stability (Fig. 5B), the lander was built according to the design of Jahnke and Christiansen.80 It can carry a cylindrical benthic chamber of 30 cm diameter and different height depending on the duration of the deployment and the intensity of benthic fluxes. The chamber is mounted on struts, such that it extends under the lander’s feet by 5 cm to ensure penetration in the sediment. Near natural chemical gradients in the chamber are maintained by gently stirring the water with an underwater pump (Seabird Electronics, Inc.). The chamber lid, made of clear acrylic plastic to maintain light conditions on the seafloor, is kept in the open position during deployments by a string terminated with a metallic pin that is held by a solenoid valve controlled by a timer. The chamber is also equipped with a device that injects iodide, a tracer measurable by voltammetry at Au/Hg microelectrodes, to calculate the exact volume of water enclosed by the benthic chamber and obtain more accurate benthic fluxes.81 After a resting period to allow for deployment on the seafloor, the pin is released to close the chamber lid and inject the tracer, while measurements begin at a time selected by the user before deployment. The ISEA instrument has been adapted on this lander to measure total oxygen uptake rates (TOU) and the depth profiles of the main terminal electron acceptors involved in natural organic matter remineralization processes simultaneously. An Au/Hg voltammetric microelectrode is fixed across the benthic chamber lid to measure chemical changes in the benthic chamber over time. Up to three other Au/Hg voltammetric microelectrodes can be mounted on the micromanipulator for microprofiles. The AIS ISEA II™ also has the capability to record pH and temperature. A long-needle pH microelectrode (Microelectrodes, Inc.) has been deployed on the micromanipulator to obtain pH depth profiles, while a temperature probe is positioned on the lander to record the temperature of the overlying water over time. Because the lander is free, it can be presently deployed anywhere to a depth of 300 m. However, for the same reasons, the data acquisition is a little more difficult to handle. The system has to be run in a data logger mode, whereby a sequence of voltammetric scans is developed for each electrode carried by the instrument. This sequence has to be consistent with the biogeochemical conditions encountered during deployments, such that artifacts (e.g., memory effects, electrode poisoning) from the experimental conditions do not interfere with the measurements.68 Ideally, benthic fluxes and microprofiles should be collected in the same sediment volume to minimize artifacts from sediment heterogeneity. Microprofiling, however, may require long deployments with the risk of depleting oxygen in the overlying water and altering the sediment’s biogeochemistry.82 To avoid this potential problem, depth profiles are collected within less than 50 cm from the center of the benthic chamber (Fig. 5B). The electrochemical equipment is designed to be adaptable on any benthic lander, such that the technology can be shared with other researchers.
The last generation of AIS ISEA III™ system has been fitted with batteries recharged by a series of solar panels and a charger for long-term deployments in coastal environments. In the near future, a telemetric system will be adapted to communicate with the instrument, program long-term sequences of measurements, download data, and monitor in live the quality of the measurements. Two selected examples of in situ measurements in sediments are given below to illustrate the feature of submersible systems based on the Au/Hg microelectrodes.
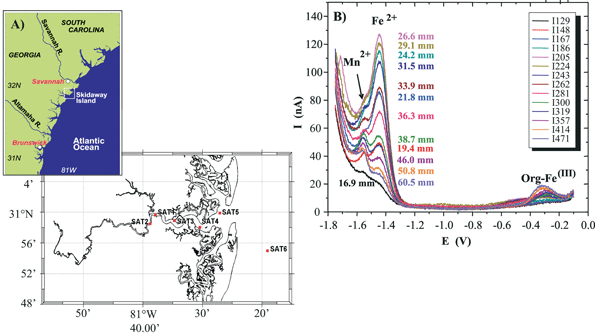 | ||
| Fig. 12 (A) Location of the Satilla River estuary and the six stations along the river and on the continental shelf. Salinities are provided in parenthesis next to the stations. (B) Example of in situvoltammograms measured in the porewaters of SAT1 as a function of depth. Square wave cathodic sweep voltammetry displays soluble organic–Fe(III) complexes (ca. –0.3 V), Fe2+ (ca. –1.43 V) and Mn2+ (ca. –1.55 V) recorded at 200 mV s–1 after a conditioning period of 10 s at –0.9 V and 10s at –0.1 V. Note that Mn2+ remains relatively constant but that soluble organic–Fe(III) and Fe2+ grow then decrease as a function of depth. Deconvolution of Mn2+ and Fe2+ currents was obtained with a home-made software.69 | ||
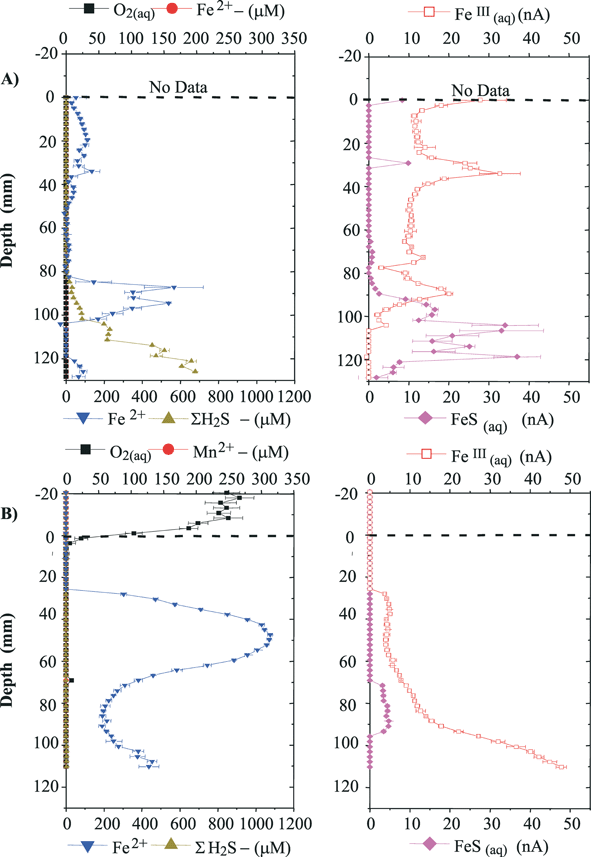 | ||
| Fig. 13 In situ depths profiles of dissolved O2, Mn2+ and Fe2+, ∑H2S (left panel) and soluble organic–Fe(III) complexes and FeS(aq) (right panel) at SAT 4 in (A) September 2005 and (B) October 2005. Dissolved O2 measurements were obtained by linear sweep voltammetry at 2 V s–1. Except for ∑H2S in September, measured anodically, species were detected by square wave cathodic sweep voltammetry at 200 mV s–1 after a conditioning period of 10 s at –0.9 V and 10 s at –0.1 V. | ||
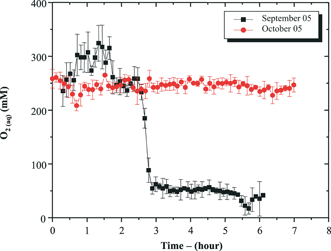 | ||
| Fig. 14 Dissolved O2 as a function of time in the benthic chamber during both September and October 2005 deployments at SAT 4. Dissolved O2 was measured as reported in Fig. 13. For these measurements, the benthic chamber Au/Hg microelectrode was calibrated in situ using the concentration of dissolved O2 in the overlying waters. The latter was calculated from the temperature measured in situ and the salinity from a separate sample assuming O2 is at saturation just above the sediment. | ||
The geochemistry at these sites has been studied extensively since 2000 using a combination of sediment core analyses with Au/Hg voltammetric microelectrodes, solid phase extractions, conventional porewater analyses, and sediment incubations.19,53,88–90 Generally, data show that the creek bank site is more oxidized than the mud flat site. Simultaneously, hydrologies measured at both sites indicate that tidal variations affect porewater transport significantly over the first 30 cm of creek bank sediments and moderately over the same depth in mud flat sediments.59 To study the effect of tidal forcing on the biogeochemistry of both sediments, a combination of water level measurements with level loggers (Solinst) deployed in monitoring wells and in situ voltammetric measurements were obtained at different depths in the sediment. At each site, a set of four monitoring wells with screens located at different heights from the well bottoms were positioned within a metre from each other, such that water entering or leaving the wells represented the porewater advecting in the sediment at depth. Four Au/Hg voltammetric microelectrodes were deployed at different depths at both sites and left in place over a tidal cycle59 and, more recently, over several tidal cycles (e.g., Fig. 15). The later deployments were obtained using an ISEA III™ instrument with external batteries recharged continuously with four solar panels. At the mud flat site, level loggers show that the tides affected the transport of porewaters to a depth of 30 cm only (Fig. 15A). In addition, the slight delays between water level changes detected in the wells at rising tide clearly show that porewaters advected initially at 15 cm deep, then at the sediment–water interface, and finally at 30 cm in the sediment. In our previous study,59 we proposed that such behavior results from an increase in hydrostatic pressure above the sediment–water interface some distance away from the site which pushes porewaters away from the incoming waters through preferential layers of high porosity. In situ voltammetric measurements obtained simultaneously show interesting features (Fig. 15B–E). First, ∑H2S and FeS(aq) were the main species found in the porewaters, which confirm our previous findings using conventional ex situ measurements that mud flat sediments are dominated by sulfate reduction.19,59,89 Second, dissolved oxygen was never detected in these sediments while ∑H2S concentrations and FeS(aq) intensities were extremely high, even at the sediment–water interface (Fig. 15B), indicating that porewater advection brought reduced porewaters to the sediment–water interface at rising tide. Third, sulfate reduction generally increased linearly during the course of these measurements and was accompanied by production of FeS(aq) at all depths except 1 cm below the sediment–water interface. Finally, small variations in ∑H2S concentrations and FeS(aq) intensities in phase with tidal cycles were observed, especially at 6 and 10 cm deep in these sediments, away from the high permeability layers near the sediment–water interface. These measurements demonstrate that heterogeneities in sediment porosities affect the transport of porewaters during tidal cycles and suggest that lateral transport may be significant in these environments if sand layers deposited during spring tides accumulate alternatively with fine grain sediments. In addition, these measurements clearly show that sulfate reduction dominates mud flat sediments. As a result, dissolved sulfide produced in large concentrations probably reduces iron oxides and precipitates FeS(s), as indicated by the large current intensities of aqueous intermediate FeS(aq) complexes observed in these sediments. Similar results were observed in creek bank sediments (not shown), though these sediments are dominated by iron and manganese reduction.19,59,90 As a result, variations in porewater manganese and iron concentrations are much more drastic, in part because hydrostatic pressures observed in confined creek bank sediments are higher than in mud flat sediments59 and in part because biogeochemical processes are driven by mineral dissolution reactions. This study demonstrates that long-term in situ measurements are necessary to characterize the complex interplay between biogeochemical processes regulating the transformation of natural organic matter and hydrological processes in intertidal salt marsh environments. Ultimately, these data could be coupled with dynamic reactive transport models to estimate net rates of organic carbon remineralization in these environments.
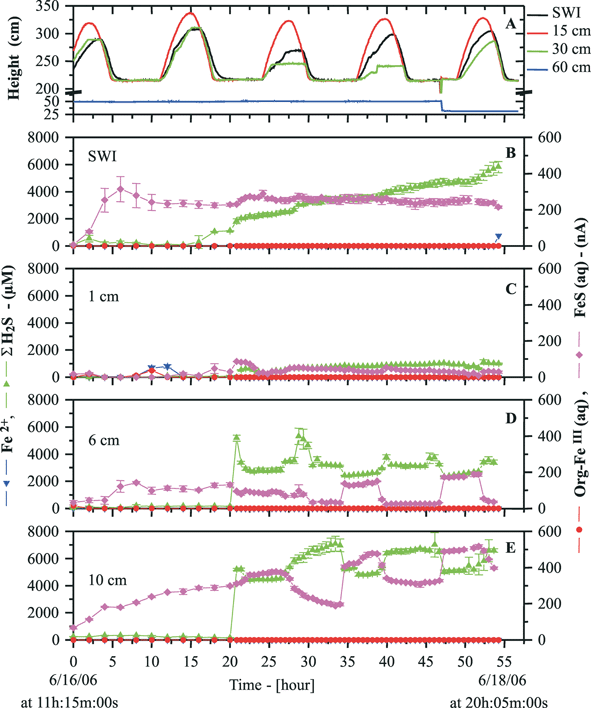 | ||
| Fig. 15 (A) In situ levels and (B)–(E) porewater voltammetric measurements of Fe2+, ∑H2S, soluble organic–Fe(III) complexes, and FeS(aq) at different depths in mud flat sediments as a function of time for more than two days in June 2006. Water levels were measured with level loggers (Solinst) in monitoring wells with screens positioned at the sediment–water interface (SWI), 15, 30 and 60 cm below the SWI. Voltammetric measurements were measured with four different Au/Hg microelectrodes positioned at the SWI, 1, 6 and 10 cm below the SWI. Dissolved O2 was measured by LSV but never detected in these anoxic sediments. Other species were monitored using conditions reported in Fig. 13. | ||
7. Summary and perspective
This review demonstrates that combining analytical expertise with environmental knowledge and breakthrough in microtechnology, micromechanics, electronics, fluidic automation, and data processing is needed to develop innovative voltammetric sensors and submersible analytical systems for in situ applications in aquatic systems. These systems should be able to measure distinct relevant species and/or fractions of priority trace metals, as well as other important environmental parameters remotely and reliably over long periods of time and with a high spatial resolution. Field applications performed so far have clearly demonstrated the key advantages of voltammetric systems compared to conventional approaches based on analyses of discrete samples in the laboratory. These advantages include: (i) direct measurements with no or minimum sample perturbations, which eliminate the large number of analytical artifacts occurring during sampling, samples handling, and storage, while simultaneously quantifying the various relevant fractions of trace metals reliably; (ii) the capability to collect detailed spatial and temporal data of trace metal speciation, as well as other important biogeochemical species, at minimum cost. Data collected so far have provided new insights in the biogeochemical processes regulating the distribution of trace metals. Future applications of these in situ instruments will undoubtedly extend our understanding of the biological (bioaccumulation, bioconcentration, bioavailability, toxicity) and geochemical (transport, adsorption, precipitation, re-mobilization) transformations of trace elements in aquatic systems, while simultaneously providing information about eventual contamination problems.Most of the technological and analytical platforms developed and presented in this paper are generic and thus can be used in the future to incorporate other types of voltammetric sensors (e.g. individually addressable GIME91,92) techniques and/or probes (e.g.ref. 91) to further improve measurement efficiency and/or extend the capability of in situ voltammetric measurements to other inorganic and organic compounds and/or environmental media (e.g.ref. 74). In addition, development of deployable instruments based on other types of chemical, optical, and electrochemical sensors for in situ measurements of different chemical compounds are underway (e.g.refs. 21 and 93). The long-term objective is thus to provide, through multidisciplinary approach, geographically dispersed networks of submersible instruments (Fig. 2) that incorporate as many sensors as possible to perform remote, simultaneous, and continuous monitoring/profiling of chemical pollutants or essential compounds coupled to master biogeochemical parameters in various aquatic systems. Such networks will be key tools for cost-effective pollution control and protection of aquatic systems. They will provide, in a timely manner, appropriate warnings or alarms for remedial action in case of significant or sudden increases in priority pollutant levels. The large databases collected with these networks should also help to improve existing environmental models and/or develop new models that predict changes related to anthropogenic pressures on a global scale. These models are required by policy makers to define appropriate strategies for protection, restoration, and/or maintenance of water quality. For example, comparison of in situ trace metal speciation data from an impacted ecosystem with reported health hazards due to exposure to contaminated waters will help establish safety limits. Finally, due to the complexity of environmental media, a thorough laboratory characterization of any novel analytical tools similar to those presented here is absolutely required before field deployments. Indeed, a fully reliable system and a detailed understanding of processes occurring in the bulk solution and at the sensor surface are prerequisite to convert signals into concentrations reliably and collect valid environmental data.
Acknowledgements
The works presented were mainly funded by: the Swiss Federal Office for Education and Science (OFES) and the European Union (European projects: VAMP, MAST-III program, contract n° MAS3-CT95-0033; IMTEC, EESD program, contract n° EVK3-CT-2000-00036; ECODIS, FP6, Contract No. 518043) to MLTW; and the US National Science Foundation CAREER (OCE-023937) and Biocomplexity (EAR-0308398) programs to MT.References
- J. O. Nriagu, Environment, 1990, 32, 6–11 Search PubMed.
- Metal Ecotoxicity: Concepts and Applications, ed. M. C. Neuman and A. W. McIntosh, Lewis Publishers, Chelsea, MI, 1991 Search PubMed.
- Chemical Processes in Lakes, ed. W. Stumm, John Wiley, New York, 1985 Search PubMed.
- Chemical Speciation in The Environment, ed. A. M. Ure and C. M. Davidson, Blakie Academic Press and Professional, Glasgow, UK, 1995 Search PubMed.
- J. M. Pacyna, M. T. Scholtz and Y.-F. Li, Environ. Rev., 1995, 3, 145–159 CAS.
- P. G. C. Campbell, O. Errécalde, C. Fortin, V. P. Hiriart-Baer and B. Vigneault, Comp. Biochem. Physiol., C, 2002, 133, 185–202.
- K. Wilkinson and J. Buffle, in Physicochemical Kinetics and Transport at Biointerfaces, ed. H. P. van Leeuwen and W. Köster, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, John Wiley, Chichester, 2004, vol. 9, pp. 445–533 Search PubMed.
- (a) Environmental Particles, ed. J. Buffle and H. P. van Leeuwen, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, Lewis Publishers INC., Chelsea, 1992, vol. 1 Search PubMed; (b) Environmental Particles, ed. J. Buffle and H. P. van Leeuwen, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, Lewis Publishers INC., Chelsea, 1993, vol. 2 Search PubMed.
- F. J. Doucet, J. R. Lead and P. H. Santschi, in Environmental Colloids and Particles, ed. K. J. Wilkinson and J. R. Jamie, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, John Wiley Chichester, 2007, vol. 10, pp. 95–157 Search PubMed.
- D. Dong, L. Liu, X. Hua and Y. Lu, Microchem. J., 2007, 85, 270–275 CrossRef CAS.
- A. Koschinsky, A. Winkler and U. Fritsche, Appl. Geochem., 2003, 18, 693–710 CrossRef CAS.
- M. Taillefert, B. MacGregor, J. F. Gaillard, D. Perret and D. A. Stahl, Environ. Sci. Technol., 2002, 36, 468–476 CrossRef CAS.
- M. Taillefert, V. C. Hover, T. F. Rozan, S. M. Theberge and G. W. Luther, III, Estuaries, 2002, 25, 1088–1096 Search PubMed.
- M. Taillefert, T. F. Rozan, B. T. Glazer, J. Herszage, R. E. Trouwborst and G. W. Luther, III, in Environmental Electrochemistry Analyses of Trace Element Biogeochemistry, ed. M. Taillefert, T. F. Rozan, American Chemical Society, Washington, DC, 2002, vol. 811, pp. 247–264 Search PubMed.
- P. G. C. Campbell, P. M. Chapman and B. A. Hale, Risk assessment of metals in the environment, Issues in, Environ. Sci. Technol., 2006, 22, 102–131 CAS.
- Analytical Chemistry of metals and metal compounds, ed. E. Merian, Verlag Chemie, Weinheim, 1991 Search PubMed.
- P. M. Bersier, J. Howell and C. Bruntlett, Analyst, 1994, 119, 219 RSC.
- M. P. Harper, W. Davison and W. Tych, Environ. Sci. Technol., 1999, 33, 2611–2616 CrossRef CAS.
- D. C. Bull and M. Taillefert, Geochem. Trans., 2001, 13, 1–8 Search PubMed.
- J. Buffle and M. L. Tercier-Waeber, TrAC, Trends Anal. Chem., 2005, 24, 172–191 CrossRef CAS.
- Environmental Electrochemistry Analyses of Trace Element Biogeochemistry, ed. M. Taillefert and T. F. Rozan, American Chemical Society, Washington, DC, 2002, vol. 811 Search PubMed.
- J. Buffle and M. L. Tercier-Waeber, in In situ monitoring of aquatic systems Chemical analysis and speciation, ed. J. Buffle and G. Hoarvai, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, Wiley, Chichester, 2000, vol. 6, pp. 279–405 Search PubMed.
- M.-L. Tercier, J. Buffle and A. Zirino, Anal. Chim. Acta, 1990, 237, 429–457 CrossRef CAS.
- J. Wang, N. Fostner, S. Armalis, D. Larson, A. Zirino and K. Olsen, Anal. Chim. Acta, 1995, 310, 223–231 CrossRef CAS.
- J. Herdan, R. Feeney, S. P. Kounaves, A. F. Flannery, C. W. Storment and G. T. A. Kovacs, Environ. Sci. Technol., 1998, 32, 131–136 CrossRef CAS.
- G. W. Luther III, C. E. Reimers, D. B. Nuzzio and D. Lovalvo, Environ. Sci. Technol., 1999, 33, 4352–4356 CrossRef.
- A. M. Bond, Analyst, 1991, 119, R1–R21 Search PubMed.
- M.-L. Tercier, N. Parthasarathy and J. Buffle, Electroanalysis, 1995, 7, 55–63 CAS.
- C. Belmont, M.-L. Tercier, J. Buffle, G. C. Fiaccabrino and M. Koudelka-Hep, Anal. Chim. Acta, 1996, 329, 203–214 CrossRef CAS.
- P. J. Brendel and G. W. Luther, III, Environ. Sci. Technol., 1995, 29, 751–761 CAS.
- G. W. Luther, III, A. B. Bono, M. Taillefert and S. C. Cary, in Environmental Electrochemistry: Analyses of Trace Element Biogeochemistry, ed. M. Taillefert and T. F. Rozan, American Chemical Society, Washington, DC, 2002, vol. 811, pp. 54–72 Search PubMed.
- S. K. Konovalov, G. W. Luther III, G. E. Friederich, D. B. Nuzzio, B. M. Tebo, J. W. Murray, T. Oquz, B. T. Glazer, R. E. Trouwborst, B. Clement, K. J. Murray and A. S. Romanov, Limnol. Oceanogr., 2003, 48, 2369–2376 CAS.
- M.-L. Tercier, J. Buffle and F. Graziottin, Electroanalysis, 1998, 10, 355–363 CrossRef CAS.
- M.-L. Tercier and J. Buffle, Anal. Chem., 1996, 68, 3670–3678 CrossRef CAS.
- C. Belmont-Hébert, M.-L. Tercier, J. Buffle, G. C. Fiaccabrino and M. Koudelka-Hep, Anal. Chem., 1998, 70, 2949–2956 CrossRef CAS.
- G. C. Fiaccabrino, N. F. de Rooij, M. Koudelka-Hep, J. Hendrise and A. van den Berg, in In situ monitoring of aquatic systems; Chemical analysis and speciation, ed. J. Buffle and G. Hoarvai, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, Wiley, Chichester, 2000, vol. 6, pp. 571–610 Search PubMed.
- M.-L. Tercier-Waeber, J. Buffle, F. Graziottin and M. Koudelka-Hep, Sea Technol., 1999, 40, 74–79 Search PubMed.
- K. A. Howell, E. P. Achterberg, C. B. Braungardt, A. D. Tappin, D. R. Turner and P. J. Worsfold, Analyst, 2003, 128, 734–741 RSC.
- M.-L. Tercier-Waeber, C. Belmont-Hébert and J. Buffle, Environ. Sci. Technol., 1998, 32, 1515–1521 CrossRef CAS.
- N. Fatin-Rouge, K. Startchev and J. Buffle, Biophys. J., 2004, 86, 2710–2719 CrossRef CAS.
- M.-L. Tercier-Waeber and J. Buffle, Environ. Sci. Technol., 2000, 34, 4018–4024 CrossRef CAS.
- M.-L. Tercier-Waeber, F. Confalonieri, G. Riccardi, A. Sina, S. Noël, J. Buffle and F. Graziottin, Mar. Chem., 2005, 97, 216–235 CrossRef CAS.
- S. Noël, M.-L. Tercier-Waeber, L. Lin, J. Buffle, O. Guenat and M. Koudelka-Hep, Electroanalysis, 2006, 18, 2061–2069 CrossRef CAS.
- N. Parthasarathy, M. Pelletier and J. Buffle, Anal. Chim. Acta, 1997, 350, 183–195 CrossRef CAS.
- J. Buffle, N. Parthasarathy, N.-K. Djane and L. Mathiasson, in In situ Monitoring of Aquatic Systems: chemical Analysis and Speciation, ed. J. Buffle and G. Horvai, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, John Wiley, Chichester, 2000, vol. 6, pp. 407–493 Search PubMed.
- K. Xu, S. C. Dexter and G. W. Luther III, Corrosion, 1997, 300, 1–18 Search PubMed.
- D. Rudolph, S. M. U. Neuhuber, C. Kranz, M. Taillefert and B. Mizaikoff, Analyst, 2004, 129, 443–448 RSC.
- G. W. Luther III, T. F. Rozan, M. Taillefert, D. B. Nuzzio, C. Di Meo, T. M. Shank, R. A. Lutz and S. C. Cary, Nature, 2001, 410, 813–816 CrossRef.
- D. B. Nuzzio, M. Taillefert, S. C. Cary, A. L. Reysenbach and G. W. Luther, III, in Environmental Electrochemistry: Analyses of Trace Element Biogeochemistry, ed. M. Taillefert and T. F. Rozan, American Chemical Society, Washington, DC, 2002, vol. 811, pp. 40–53 Search PubMed.
- G. W. Luther, III, B. T. Glazer, S. Ma, R. E. Trouwborst, B. R. Shultz, G. Druschel and C. Kraiya, Aqu. Geochem., 2003, 9, 87–110 Search PubMed.
- B. T. Glazer, G. W. Luther III, S. K. Konovalov, G. E. Friederich, D. B. Nuzzio, R. E. Trouwborst, B. M. Tebo, B. Clement, K. Murray and A. S. Romanov, Deep-Sea Res., Part II, 2006, 53, 1740–1768 CrossRef.
- S. Ma, A. Noble, D. Butcher, R. E. Trouwborst and G. W. Luther III, Estuarine, Coastal Shelf Sci., 2006, 70, 461–472 CrossRef CAS.
- E. A. Carey and M. Taillefert, Limnol. Oceanogr., 2005, 50, 1129–1141 CAS.
- M. Taillefert, J. S. Beckler, E. A. Carey, J. L. Burns, C. M. Fennessey and T. J. DiChristina, J. Inorg. Biochem., 2007, 101, 1760–1767 CrossRef CAS.
- G. W. Luther III, B. Sundby, B. L. Lewis, P. J. Brendel and N. Silverberg, Geochim. Cosmochim. Acta, 1997, 61, 4043–4052 CrossRef.
- D. Rickard, A. Oldroyd and A. Cramp, Estuaries, 1999, 22, 693–701 Search PubMed.
- A. B. Hebert and J. W. Morse, Mar. Chem., 2003, 81, 1–9 CrossRef CAS.
- B. Sundby, M. Caetano, C. Vale, C. Gobeil, G. W. Luther, III and D. B. Nuzzio, Environ. Sci. Technol., 2005, 39, 2080–2086 CrossRef CAS.
- M. Taillefert, S. M. U. Neuhuber and G. Bristow, Geochem. Trans., 2007, 8, 1–15 Search PubMed.
- T. F. Rozan, S. M. Theberge and G. W. Luther III, Anal. Chim. Acta, 2000, 415, 175–184 CrossRef CAS.
- S. M. Theberge and G. W. Luther III, Aqu. Geochem., 1997, 3, 191–211 Search PubMed.
- M. Taillefert, A. B. Bono and G. W. Luther III, Environ. Sci. Technol., 2000, 34, 2169–2177 CrossRef CAS.
- W. Davison, J. Buffle and R. De Vitre, Pure Appl. Chem., 1988, 60, 1535–1543 CrossRef CAS.
- G. W. Luther III, B. T. Glazer, S. Ma, R. E. Trouwborst, C. M. Moore, E. Metzger, C. Kraiya, T. J. Waite, G. Druschel, B. Sundby, M. Taillefert, D. B. Nuzzio, T. M. Shank, B. L. Lewis and P. J. Brendel, Mar. Chem., 2007 DOI:10.1016/j.marchem.2007.03.002.
- C. E. Reimers, H. A. Stecher III, G. L. Taghon, C. M. Fuller, M. Huettel, A. Rusch, N. Ryckelynck and C. Wild, Cont. Shelf Res., 2004, 24, 183–201 CrossRef.
- D. B. Nuzzio, M. Taillefert, S. C. Cary, A. L. Reysenbach and G. W. Luther III, in Environmental Electrochemistry: Analyses of Trace Element Biogeochemistry, M. Taillefert, T. F. Rozan, American Chemical Society, Washington DC, 2002, vol. 811, pp. 40–53 Search PubMed.
- B. L. Lewis, B. T. Glazer, P. J. Montbriand, G. W. Luther III, D. B. Nuzzio, T. Deering, S. Ma and S. M. Theberge, Mar. Chem., 2007, in press Search PubMed.
- D. Meiggs, G. Bristow, J. Schur, D. B. Nuzzio and M. Taillefert, Limnol. Oceanogr., 2007, submitted Search PubMed.
- S. Moore, D. B. Nuzzio, T. W. Deering, M. Taillefert and G. W. Luther III, Electroanalysis, 2007, 19, 2110–2116 CrossRef.
- G. Bristow and M. Taillefert, Comp. Geosci., 2007, in press Search PubMed.
- H. Pauwels, M.-L. Tercier-Waeber, M. Arenas, R. Castroviejo, Y. Deschamps, A. Lassin, F. Graziottin and F.-J. Elorza, J. Geochem. Explor., 2002, 75, 17–41 CrossRef CAS.
- L. Sigg, F. Black, J. Buffle, J. Cao, R. Cleven, W. Davison, J. Galceran, P. Gunkel, E. Kalis, D. Kistler, M. Martin, S. Noël, Y. Nur, N. Odzak, J. Puy, W. van Riemsdijk, E. Temminghoff, M.-L. Tercier-Waeber, S. Toepperwien, R. M. Town, E. R. Unsworth, K. Warnken, L. Weng, H. Xue and H. Zhang, Environ. Sci. Technol., 2006, 40, 1934–1941 CrossRef CAS.
- E. R. Unsworth, K. W. Warnken, H. Zhang, W. Davison, F. Black, J. Buffle, J. Cao, R. Cleven, J. Galceran, P. Gunkel, E. Kalis, D. Kistler, H. P. van Leeuwen, M. Martin, S. Noël, Y. Nur, N. Odzak, J. Puy, W. van Riemsdijk, L. Sigg, E. Temminghoff, M.-L. Tercier-Waeber, S. Toepperwien, R. M. Town, L. Weng and H. Xue, Environ. Sci. Technol., 2006, 40, 1942–1949 CrossRef CAS.
- M.-L. Tercier-Waeber, F. Confalonieri, M. Koudelka-Hep, J. Dessureault-Rompré, F. Graziottin, and J. Buffle, Electroanalysis, 2007, in press Search PubMed.
- O. Annevile, V. Ginot, J.-C. Druart and N. Angeli, J. Plankton Res., 2002, 24, 993–1007 Search PubMed.
- S. S. Chow, D. B. Nuzzio, G. W. Luther III and M. Taillefert, Aqu. Geochem., 2007, submitted Search PubMed.
- G. W. Luther III, P. J. Brendel, B. L. Lewis, B. Sundby, L. Lefrançois, N. Silverberg and D. B. Nuzzio, Limnol. Oceanogr., 1998, 43, 325–333.
- B. Sundby, C. Vale, I. Cacador, F. Catarino, M. J. Madureira and M. Caetano, Limnol. Oceanogr., 1998, 43, 245–252 CAS.
- K. S. Sell and J. W. Morse, Aqu. Geochem., 2006, 12, 179–198 Search PubMed.
- R. A. Jahnke and M. B. Christiansan, Deep-Sea Res., 1989, 36, 625–637 Search PubMed.
- A. M. F. Rao and R. A. Jahnke, Limnol. Oceanogr., 2004, 80, 75–90.
- B. Sundby, L. G. Anderson, P. O. J. Hall, A. Iverfeldt, M. M. R. Van Der Loeff and S. F. G. Westerlund, Geochim. Cosmochim. Acta, 1986, 50, 12.
- M. Alber, C. R. Alexander, J. O. Blanton, A. Chalmers and K. Gates, The Satilla River estuarine system: The current state of knowledge, Georgia Sea Grant College Program and South Carolina Sea Grant Consortium, 2003 Search PubMed.
- F. Wenzhofer and R. N. Glud, Deep-Sea Res., Part I, 2002, 49, 1255–1279 CrossRef CAS.
- W. Davison and G. Seed, Geochim. Cosmochim. Acta, 1983, 47, 67–79 CrossRef CAS.
- K. N. Buck, M. C. Lohan, C. J. M. Berger and K. W. Bruland, Limnol. Oceanogr., 2007, 52, 843–855 CAS.
- W.-J. Cai, Z. A. Wang and Y. Wang, Geophys. Res. Lett., 2003, 30, 1849 CrossRef.
- J. M. Caffrey, Estuaries, 2004, 27, 90–101 Search PubMed.
- S. M. U. Neuhuber, MS Thesis, Georgia Institute of Technology, 2003.
- J. D. Newton, MS Thesis, Georgia Institute of Technology, 2006.
- M.-L. Tercier-Waeber, J. Pei, J. Buffle, G. C. Fiaccabrino, M. Koudelka-Hep, G. Riccardi, F. Confalonieri, A. Sina and F. Graziottin, Electroanalysis, 2000, 12, 27–34 CrossRef CAS.
- J. Pei, M.-L. Tercier-Waeber, J. Buffle, G. C. Fiaccabrino and M. Koudelka-Hep, Anal. Chem., 2001, 73, 2273–2281 CrossRef CAS.
- In situ monitoring of aquatic systems: Chemical analysis and speciation, ed. J. Buffle and G. Hoarvai, IUPAC Series on Analytical and Physical Chemistry of Environmental Systems, Wiley, Chichester, 2000, vol. 6 Search PubMed.
| This journal is © The Royal Society of Chemistry 2008 |