Performance optimization of a membrane assisted passive sampler for monitoring of ionizable organic compounds in water
L.
Chimuka
*a,
T.
Nemutandani
b,
E.
Cukrowska
a and
H.
Tutu
a
aSchool of Chemistry, University of the Witwatersrand, Johannesburg, 2050, South Africa. E-mail: luke@chem.wits.ac.za
bSchool of Environmental Sciences, University of Venda, Thohoyandou, 0950, South Africa
First published on 31st October 2007
Abstract
A thin-walled silicone rubber hollow fibre membrane has been developed as a passive sampler. The inside of the tube is filled with an aqueous solution at an appropriate pH. The tube is sealed at both ends and then immersed in a water sample. In order for the ionizable permeating compounds to be trapped in the aqueous receiving phase, the pH is adjusted such that the compounds are ionized and trapped. The major advantages are its simplicity, low cost and high selectivity, since only ionizable organic compounds are trapped. Additionally, the sampler uses no organic solvent. By adjusting the pH of the acceptor phase, it is possible to control the extraction process and whether the sampler is used in the kinetic or equilibrium regime. Since it is very selective, no further clean-up of the extract is required. The membrane assisted passive sampler has been tested for extraction of chlorophenols under laboratory conditions. The extraction process was found to be linear over a 72 h sampling period. Selectivity of the passive sampler in river water was demonstrated and the extraction process was independent of sample concentration, even at lower concentration levels of analytes. However, the sample matrix in some river water samples led to incomplete trapping, thereby reducing the amount trapped in the acceptor phase. Detection limits (three times signal to noise ratio) were dependant on sample matrix and type of detection system and ranged from 0.05 µg L–1 to about 1 µg L–1 with a UV photodiode detector in water samples from one river and 1.0 µg L–1 to 20 µg L–1 in another but with an ordinary UV detector. The enrichment factors in river water were 28 for 2-chlorophenol and 44 for 2,4-dichlorophenol over a 72 h sampling period. 4-chlorophenol was poorly extracted and its enrichment factor was 3.
Introduction
Monitoring of the presence of various organic chemicals, such as phenols, in water bodies is important as it gives vital information on the toxic risks associated with them. The presence and levels of phenols in aquatic environments is of concern because of their wide spread release as by-products in the production of plastics and dyes, pulp and paper industries. Chlorinated phenols can also form during wastewater treatment, since chlorine is added as a disinfectant. Because of the toxicity and persistent nature of these compounds, they need to be determined, even at single µg L–1 levels.The common approach is to collect part of the environmental media, which is later analyzed for potential pollutants in the laboratory. This approach, among other advantages, provides manageable control over accuracy and precision of the results. However, information obtained from grab environmental samples is only about concentration levels at the time of sampling and may fail to account for episodic contamination. This can be addressed by collecting many representative samples over a time period but the cost of analysis is increased.
An alternative and more cost effective approach is to obtain time-weighted average (TWA) concentrations of pollutants using passive samplers. Sodergren1 developed a first sampler for the monitoring of compounds in water bodies consisting of a dialysis membrane filled with an organic solvent. The major disadvantage of the system was successive loss of the organic solvent from the device through diffusion during environmental exposure and lack of selectivity. Huckins et al.,2 described the second field sampler, a semi-permeable membrane device (SPMD) for passive and integrative in situ monitoring of water and air borne contaminants. The SPMD sampler consisted of layflat polyethylene tubing containing a thin film of triolein, a high molecular weight neutral lipid. This has been commercialized.3 The other advantage, apart from being very rugged, is that high preconcentration factors of the analytes are obtained and are limited by the partition coefficients into the lipid and exposure time.4 The disadvantage of the SPMD is the laborious recovery of the analytes, usually involving large volumes of organic solvents.
Other passive samplers have recently been reported, especially those based on solid phase material as trapping media. These include the Chemcatcher that use commercially available solid phase extraction C18 Empore disks as receiving phase.5,6 The partitioning of the analytes onto the sorbent and their subsequent desorption is similar to the solid phase extraction technique. The type of Empore disk can also be chosen according to the properties of the analytes of interest. A membrane enclosed sorptive coating (MESCO) passive sampler that uses a stir bar coated with poly(dimethylsiloxane) (PDMS) enclosed in a dialysis membrane bag as receiving phase has also been described.7 The stirrer bar used as a receiving phase is similar to the one used in the stir bar sorptive extraction (SBSE) technique.8 It combines the advantages of the passive sampling approach with solventless preconcentration of organic solutes from aqueous matrices and subsequent desorption of the sequested analytes on line with capillary gas chromatography. It avoids clean up of extracts, required for other samplers, and the whole extract is injected. A new MESCO that uses a silicone collector, instead of a stirrer bar coated with PDMS, has been reported by Paschke et al.9 The silicone rods are later thermally desorbed into a gas chromatographic system, as in the MESCO. A polar organic chemical integrative sampler (POCIS) has been described by Alvarez et al.10 It consists of a solid receiving phase material enclosed in microporous polyethersulfone diffusion-limiting membrane. Jonsson’s research group11 has also reported to have developed an equilibrium sampling through membranes (ESTM) technique for measuring the free fraction of ionizable organic compounds in water that could be applied as a passive sampler. In this case, a porous polypropylene hollow fibre membrane is impregnated with a non-polar solvent. The inner side of the hollow fibre serves as the acceptor phase and is filled with appropriate aqueous solution. The same research group has recently reported an equilibrium sampling of freely dissolved organic chemicals into a thin film of 1-octanol supported on a porous hollow fibre membrane, which could also be applied as a passive sampler.12 In both of the above cases, solvent loss may be a problem when the equilibrium extraction techniques are used as passive samplers. However, this could depend on environmental conditions and sampling time. As interest in passive samplers is growing, more passive samplers for monitoring organic compounds in water bodies have been reported. These are discussed in several review papers.13–16
In this work, a simple and very selective membrane assisted passive sampler (MAPS), that does not use organic solvents, based on a silicone hollow fibre membrane for extraction of ionisable organic compounds in water bodies, is described. The potential of the sampler, especially its selectivity and simplicity, have been demonstrated in the laboratory using chlorophenols as model compounds.
Theory of the extraction process
The theory of extraction for the MAPS is similar to the one developed for the supported liquid membrane (SLM) extraction technique.17–19 In brief, in order for the chlorophenols to dissolve into the silicone membrane from the sample, they have to be non-ionized at the sample pH. The compounds then diffuse through the membrane into the acceptor phase. Once in the acceptor phase, they are ionized and trapped. The concentration of non-ionized phenols in the acceptor phase is thus kept at zero. This maintains a concentration gradient between the two phases; the donor and acceptor phases. This way the concentration of the compounds in the acceptor solution can be increased much higher than in the original sample, without experiencing a plateau or maximum, and is limited by the sample volume and/or the extraction time. This also gives selective enrichment, since only compounds that are ionized at the pH of the acceptor phase are enriched.20 Compounds that are ionized at the pH of the sample solution do not dissolve into the membrane, since they are too polar. Large molecules have slow diffusion in the membrane and are less excluded. From the developed theory,17–19 an acidic compound (like chlorophenols) needs an acceptor pH that is 3.3 units above its pKa value for it to be completely trapped and the sample pH must be 2 units below its pKa for the compounds to dissolve into the membrane. Therefore, it is possible to determine the best trapping conditions for the acceptor phase in the sampler from the pKa of the ionisable organic compound.Two important parameters that are often measured in the liquid membrane extraction techniques are the extraction efficiency (E), and enrichment factor, En. The extraction efficiency is defined as the fraction of analyte in the extracted sample that is found in the acceptor phase and is given by the equation below.17–19 It is also a measure of mass transfer between the donor and acceptor phase and is constant under specified extraction conditions.
| E = CAVA/CDVD | (1) |
| En = CA/CD | (2) |
Experimental
Chemicals
The following chlorophenol standards were used: 4-chlorophenol (>99%), 2-chlorophenol (>98%) and 2,4-dichlorophenol (>99%) were from Merck–Schuchardt (Darmstadt, Germany). Other chemicals used were trisodium phosphate (99%), proanalysis sulfuric acid (99%) and HPLC grade methanol and acetonitrile, all from Merck–Schuchardt (Darmstadt, Germany).Solutions
Stock solutions at 1000 mg L–1 were prepared in methanol and were stored in the refrigerator at 4 °C. Fresh stock solutions were prepared after every three months. Solutions for hollow fibre extraction were prepared by diluting the stock with deionised water, and the methanol concentration did not exceed 0.1%.Chromatographic conditions
The three chlorophenols were best separated with a mobile phase composition of 60% water and 40% acetonitrile, at a flow rate of 1.0 mL min–1, with the UV detector set at 280 nm. A C18 column with dimensions 25 cm × 4 mm × 5 µm was used from Supelco (Bellefonte, PA, USA). Two chromatographic systems were used. At first, a Waters HPLC system (Milford, MA, USA) with a 515 pump, Waters 996 Photodiode Array detector and Millenium32 chromatographic software was used. The mobile phase was continuously degassed by a 200 series Perkin Elmer degasser (PA, USA) and an injection volume of 100 µL was used. In some experiments, an SRI (LA, California, USA) 210 HPLC system with a UV detector (VUV-24) and peak simple chromatographic system was used. The mobile phase was degassed offline and filtered before use. The injection volume was 20 µL.Hollow fibres
Three hollow fibre membranes used for the optimization process were from Technical Products Inc., (Georgia, USA). Their dimensions are summarized in Table 1. The hollow fibres were bought as long tubes that were cut to appropriate lengths when used.| Type | Inner diameter/cm | Outer diameter/cm | Thickness/cm | Length/cm | Volume/cm3 | Surface area/cm2 |
|---|---|---|---|---|---|---|
| Smallest | 0.0635 | 0.1194 | 0.0284 | 58 | 0.187 | 21.74 |
| Medium | 0.1575 | 0.2413 | 0.0420 | 23.5 | 0.458 | 17.81 |
| Biggest | 0.4775 | 0.9525 | 0.2375 | 7.8 | 1.396 | 23.33 |
Procedures
 | ||
| Fig. 1 Schematic set-up for the passive sampling. | ||
Optimization of extraction parameters
| Length/cm | Volume/cm3 | Surface area/cm2 |
|---|---|---|
| 29 | 0.565 | 21.97 |
| 48 | 0.935 | 36.37 |
| 56 | 1.090 | 42.43 |
The detection limit was obtained from chromatograms obtained after extraction of the 10 µg L–1 mixtures of the chlorophenols spiked in river water and extracted for 72 h. The detection limit was calculated as the concentration that would give a peak height three times the baseline noise.
Results and discussion
Optimizations of the sampler
 | ||
| Fig. 2 (a) Plots of the hollow fiber thickness and (b) length of the hollow fiber against the amount extracted for 24 h from deionized water spiked with 0.5 mg L–1 mixtures of chlorophenols; (c) sample volume and (d) extraction time against enrichment factor after extraction of 0.05 mg L–1 mixtures of chlorophenols spiked in deionized water. | ||
The influence of the hollow fibre surface area on the mass transfer (Fig. 2b) was also not very conclusive. A length of 56 cm did not give much better extraction compared to the 48 cm longer fibre despite having a higher surface area. The reason for this could be positioning in the sample container that could influence the extraction process. A length of 48 cm was chosen for convenience in handling, especially putting in the sample container. A fibre that is too long would require folding several times before putting it in the sample container.
If the trapping conditions are correctly set in the acceptor phase,17,18 the extraction can go on until all the analytes in the sample are extracted. This means that a linear relationship is expected between the extraction time and the amount accumulated in the acceptor phase. Fig. 2d shows that such a relationship was obtained. Linear relationships have been observed in other passive samplers working in the kinetic regime, such as the Chemcatcher,5,6 MESCO7,21 and PDMS.9
 | ||
| Fig. 3 Comparison of enrichment factors under static and stirred conditions in river water spiked with 10 µg L–1 mixtures of chlorophenols and with extraction time of 72 h. | ||
 | ||
| Fig. 4 Plot of the different extracted concentrations against accumulated amount in river water after 72 h extraction. | ||
Fig. 5 shows a comparison of the enrichment factor in river water and deionized water extracted over a 72 h period. From the Figure, it shows that enrichment factors were much lower in river water compared to deionized water. In river water, 4-chlorophenol was not even detected, due to poor extraction. The most probable cause of this is due to co-extraction of other acidic compounds in river water that are also in high concentrations. This lowers the acceptor pH, thus leading to incomplete trapping of the target compounds. This agrees well with the results, since 2-chlorophenol, with a highest pKa, would be affected most by pH decrease in the acceptor phase, with least affected being 2,4-dichlorophenol, with a pKa of 7.6. In this case, a much stronger/concentrated buffer is recommended. Jonsson et al.,11 studied the decrease of acceptor pH due to matrix found in various water samples, such as deionized water, river water and leachate water. pH decreased most in leachate and river waters and very little in deionized water.
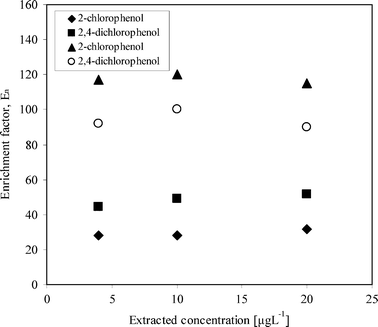 | ||
| Fig. 5 Comparison of the enrichment factors in river and deionized waters. ▲ and ○ represent enrichment factors in deionized water. ◆ and ■ represent enrichment factors in river water. Extraction time was for 72 h with optimized parameters. | ||
Table 3 shows the comparison of detection limits by direct injection and after hollow fibre passive sampling in deionized and river water. The detection limit was calculated as the concentration that would give a peak height three times the baseline noise. It clearly shows that the developed sampler can be used for passive sampling of chlorophenols in water bodies at trace levels.
| Sample type | Detection limits/µg L–1 | ||
|---|---|---|---|
| 2-Chlorophenol | 4-Chlorophenol | 2,4-Dichlorophenol | |
| a River water-2 was from a different area and was extracted for 24 h instead of 72 h. Injection volume was also 100 µL instead of 20 µL and with a photodiode array detector. | |||
| Direct injection | 60 | 65 | 60 |
| Deionised water | 0.5 | 1.5 | 0.7 |
| River water-1 | 1.5 | 20 | 1.0 |
| River water-2a | 0.05 | 1.0 | 0.85 |
Fig. 6a shows the chromatogram passive extraction of a 10 µg L–1 mixture of the chlorophenols in spiked deionized water (Fig. 6a) and river water (Fig. 6b). The clean chromatograms demonstrate the selectivity of the passive sampler.
 | ||
| Fig. 6 Chromatograms obtained after 72 h passive extraction of 10 µg L–1 mixtures of chlorophenols spiked in deionized water (a) and in river water (b). | ||
Conclusions
The potential for passive sampling of acidic compounds in MAPS has been demonstrated. The sampler is inexpensive, selective and uses no organic solvents. By changing the acceptor solution from basic to acidic conditions, the MAPS can be used to extract basic compounds. However, before the passive sampler is used for field application, the influence of environmental factors, such as temperature, turbulence and biofouling, will have to be investigated. These factors are currently being considered as part of further method development, along with enclosing the silicone hollow fibre into an enclosure, such as copper mesh.Acknowledgements
Authors would like to thank Prof. Jönsson (Lund University-Sweden) for donating the peristaltic pump used in the experimental and Prof. Rohwer (University of Pretoria-South Africa) for donating initial silicone hollow fibre tubes. The authors also thank Water Research Commission of South Africa for funding.References
- A. Södergren, Environ. Sci. Technol., 1987, 21, 855.
- J. N. Huckins, M. W. Tubergen and G. K. Manuweera, Chemosphere, 1990, 20, 533 CrossRef CAS.
- Environmental Science Technologies Inc. (USA), http://est-lab.com.
- P. Sabaliunas and A. Sodergren, Environ. Pollut., 1997, 96, 195 CrossRef CAS.
- J. K. Kingston, R. Greenwood, G. A. Mills, G. M. Morrison and L. B. Person, J. Environ. Monit., 2000, 2, 487 RSC.
- B. Vrana, G. A. Mills, E. Dominiak and R. Greenwood, Environ. Pollut., 2006, 142, 333 CrossRef CAS.
- B. Vrana, P. Popp, A. Paschke and G. Schuurmann, Anal. Chem., 2001, 73, 5191 CrossRef CAS.
- E. Baltussen, P. Sandra, F. David and C. Cramels, J. Microcolumn Sep., 1999, 11, 737 CrossRef CAS.
- A. Paschke, K. Schwab, J. Brummer, G. Schuurmann, H. Paschke and P. Popp, J. Chromatogr., A, 2006, 1124, 187–195 CrossRef CAS.
- D. A. Alvarez, J. D. Petty, J. N. Huckins, T. L. Jones-Lepp, J. P. Goddard and S. E. Manahan, Environ. Toxicol. Chem., 2004, 23, 1640 CrossRef CAS.
- J. F. Liu, J. Å. Jonsson and P. Mayer, Anal. Chem., 2005, 77, 4800 CrossRef CAS.
- J. F. Liu, X. L. Hu, J. F. Peng, J. Å. Jonsson, P. Meyer and G. B. Jiang, Anal. Chem., 2006, 78, 8526 CrossRef CAS.
- B. Vrana, G. A. Mills, I. J. Allan, E. Dominiak, K. Svensson, J. Knutsson, G. Morrison and R. Greenwood, TrAC, Trends Anal. Chem., 2005, 24, 845 CrossRef.
- Y. Lu, Z. Wang and J. Huckins, Aquat. Toxicol., 2002, 60, 139 CrossRef CAS.
- J. D. Petty, C. E. Orazio, J. N. Huckins, R. W. Gale, J. A. Lebo, J. C. Meadows, K. R. Echools and W. L. Cranor, J. Chromatogr., A, 2000, 879, 83 CrossRef CAS.
- F. Stuer-Lauridsen, Environ. Pollut., 2005, 136, 503 CrossRef CAS.
- L. Chimuka, N. Megersa, J. Norberg, L. Mathiasson and J. Å. Jonsson, Anal. Chem., 1998, 70, 3906 CrossRef CAS.
- J. Å. Jonsson and L. Mathiasson, TrAC, Trends Anal. Chem., 1999, 18, 325 CrossRef CAS.
- J. Å. Jönsson, P. Lövkvist, G. Audunsson and G. Nilvé, Anal. Chim. Acta, 1993, 277, 9 CrossRef.
- N. Megersa, T. Solomon and J. Å. Jönsson, J. Chromatogr., A, 1999, 830, 203 CrossRef CAS.
- B. Vrana, A. Paschke and P. Popp, Environ. Pollut., 2006, 144, 296 CrossRef CAS.
- M. Papantoni, N. K. Djane, K. Ndungu, J. Å. Jönsson and L. Mathiasson, Analyst, 1995, 120, 1471 RSC.
- M. Knutsson, L. Mathiasson and J. Å. Jönsson, Chromatographia, 1996, 42, 165 CAS.
- T. Miliotis, M. Knutsson, J. Å. Jönsson and L. Mathiasson, Intern. J. Environ. Anal. Chem., 1996, 64, 35 Search PubMed.
- L. Chimuka, L. Mathaiasson and J. Å. Jönsson, Anal. Chim. Acta, 2000, 416, 77 CrossRef CAS.
- N. Megersa and J. Å. Jönsson, Analyst, 1998, 123, 225 RSC.
| This journal is © The Royal Society of Chemistry 2008 |