Fuel cell technology: nano-engineered multimetallic catalysts
Chuan-Jian Zhong*, Jin Luo, Peter N. Njoki, Derrick Mott, Bridgid Wanjala, Rameshwori Loukrakpam, Stephanie Lim, Lingyan Wang, Bin Fang and Zhichuan Xu
Department of Chemistry, State University of New York at Binghamton, Binghamton, NY 13902, USA. E-mail: cjzhong@binghamton.edu
First published on 29th August 2008
Abstract
Fuel cells represent an attractive technology for tomorrow's energy vector because hydrogen is an efficient fuel and environmentally clean, but one of the important challenges for fuel cell commercialization is the preparation of active, robust and low-cost catalysts. The synthesis and processing of molecularly-capped multimetallic nanoparticles, as described in this report, serves as an intriguing way to address this challenge. Such nanoparticles are exploited as building blocks for engineering the nanoscale catalytic materials by taking advantage of diverse attributes, including monodispersity, processability, solubility, stability, capability in terms of size, shape, composition and surface properties. This article discusses recent findings of our investigations of the synthesis and processing of nanostructured catalysts with controlled size, composition, and surface properties by highlighting a few examples of bimetallic/trimetallic nanoparticles and supported catalysts for electrocatalytic oxygen reduction.
 From back to front, Dr Zhong to Ms Loukrakpam | Dr Zhong, a professor of chemistry, has been leading a research program aimed at developing advanced nanomaterials for applications in fuel cells, catalysis, sensors, and biosensors. |
Dr Luo, a senior scientist, has been developing methods for preparing and characterizing metal, bimetallic and trimetallic nanoparticles for fuel cell catalysis and chemical sensing. |
Dr Mott is a 2008 graduate of the PhD program, focusing on synthesis of copper and alloy nanoparticles and surface characterization of nanocatalysts. |
Dr Njoki received his PhD in 2007 and is a postdoctoral researcher, focusing on synthesis of gold and alloy nanoparticles and electrochemical characterization of nanocatalysts. |
Ms Fang is a graduate student, focusing on studying fuel cell performance of nanostructured catalysts. |
Dr Wang received her PhD in 2007 and is a postdoctoral researcher, focusing on synthesis and characterization of metal and magnetic core-shell nanoparticles and thin film assemblies as catalytic and sensing materials. |
Dr Stephanie Lim is an NSF Graduate Research Fellow and a 2008 graduate of the PhD program, focusing on synthesis and characterization of metal nanoparticles and molecularly-mediated assembly of nanoparticles as functional nanomaterials. |
Ms Wanjala is a graduate student, focusing on synthesis and characterization of bimetallic and trimetallic nanoparticles and electrocatalysts in fuel cell reactions. |
Ms Loukrakpam is a graduate student, focusing on synthesis of multimetallic nanoparticles and correlation of the nanostructures and the catalytic properties. |
Zhichuan Xu is a graduate student (who is not in the picture), focusing on synthesis and characterization of inorganic nanoparticles. |
1. Fuel cells—an alternative to tomorrow's energy vector
There are two major driving forces for the increasing interests in the development of fuel cells. One is the reality that fossil fuels are running out, and we have to find alternative energy sources. The other is the fact that the pollution from using fossil fuels has become an important issue of environmental concern to human health. Fuel cells utilizing hydrogen as fuels represent an important form of tomorrow's energy because hydrogen is an efficient fuel and is environmentally clean. Imagine that a stack of hydrogen fuel cells can create enough current to power a vehicle, a building and even an entire city, and the only product is water, which makes fuel cells the most energy-efficient and environmentally-friendly technology on the horizon. The auto industry, which relies on oil-fuelled cars, is perhaps the biggest driving force behind the massive investment in fuel cell development.1 This is understandable because the price of oil is extremely volatile and has been increasing in the past few years which is likely to continue upwards. Moreover, the harmful emissions of CO2, CO, SO2, NOx, and volatile organic compounds into the atmosphere cause serious environmental damage, increase respiratory problems in humans, and produce “greenhouse gas” that contributes to global warming.With these problems, fuel cell technology is inevitably seen as a viable alternative. Energy sources of the future will have to be cleaner and more efficient than current sources—fuel cells fulfill these requirements. The “hydrogen economy” offers an energy system based upon hydrogen for energy generation, storage, distribution, and utilization. It is an economy in which the hydrogen produced cleanly and cost-effectively from multiple sources can be used to power fuel cells and engines, which will be as common as gasoline engines being used today. While there are still challenges in the long road to the hydrogen economy to address issues including hydrogen storage, affordable cost, and infrastructure development (e.g., hydrogen fueling stations), the fact that hydrogen can be considered as an energy vector that provides power for transportation, industrial and residential needs, and emits only water to the environment is just too great a reward to pass up.
Because fuel cells generate electricity through a chemical process, they are not subject to the Carnot Limit (ηCarnot = 1 −TL/TH), a theoretical limit on the efficiency of an engine based on the flow of heat between two reservoirs (high (TH) and low (TL) temperature sources). Fuel cells can effectively extract more energy from fuel (40–70% efficiency) than traditional internal combustion engines (∼30% efficiency) (Fig. 1). Along with the hydrogen's high efficiency (from 40–70%), the possibility of utilizing both heat and electricity in fuel cells makes a significant contribution to reducing atmospheric emissions. For example, a fuel cell operating at 60% efficiency would emit 35–60% less CO2 at the fossil fuel stage and 80% less from hydrogen. This estimate is only based on efficiency comparison, which does not rule out the consideration of the CO2 emission from hydrogen generation. In comparison with batteries, fuel cells offer a reduction in weight and come in a compact package for the same amount of available energy. To increase the power in a fuel cell, more fuel is introduced into the system. To increase the power of a battery, more batteries have to be added, increasing the cost, weight and complexity of the system. A fuel cell never “runs down” because it continues to produce electricity as long as fuel is present. When a battery “runs down” it has to undergo a lengthy, inconvenient recharge time to replace the spent electricity. The wide use of today's lithium batteries in electronic devices such as laptops and cell phones is in fact showing inherent problems because of the limited lifespan. In comparison with rechargeable batteries in which the recharge is generally very slow and a fast recharge can damage the performance of the battery, the recharge of a fuel cell is very fast and the fast recharge does not compromise the performance of the fuel cell.
 | ||
| Fig. 1 Energy conversion of fuels. | ||
There are three main areas of challenges for the realization of hydrogen energy: hydrogen production, hydrogen storage, and hydrogen utilization. Fuel cells based on hydrogen fuel represents one of the most effective ways in hydrogen utilization. Fuel cells such as proton exchange membrane fuel cell (PEMFC) (Fig. 2) become attractive because of high conversion efficiency, low pollution, lightweight, high power density, and a wide range of applications from power sources in automobiles and space shuttles to power grids for buildings and factories. Fuel cells are essentially electrochemical cells and operate by the same basic mechanism as regular batteries. However, unlike batteries in which the chemicals used are contained in the cell and when the reaction is complete the battery is dead, fuel cells have a constant flow of fresh chemicals into the cell and thus potentially have an unlimited life. Hydrogen fuel cells convert flows of hydrogen and oxygen into water and produce electricity. At the anode, hydrogen is forced through a catalyst where it is ionized. At the cathode, oxygen reacts with the products from the anode (the protons and electrons) to produce water. The closed circuit of the two electrodes produces electricity and heat, and water as the only product.
 | ||
| Fig. 2 A simplified and dissected view of the basic components of a proton exchange membrane hydrogen/oxygen fuel cell. | ||
In PEMFC, electrochemical reactions occur at the surface of the catalyst at the interface between the electrolyte and the membrane. Hydrogen fed on the anode side of the membrane splits into protons and electrons. Protons travel through the membrane, while the electrons travel through the outside circuit where they perform useful work and return to the cathode side of the membrane. At the catalyst sites of the cathode oxygen is reduced, which combines with the protons, forming water. The net result of these simultaneous reactions is a current of electrons through an external circuit—direct electrical current. The basic fuel cell reactions are:
| Anode: H2→ 2H+ + 2e− |
| Cathode: ½O2 + 2H+ + 2e−→ H2O |
| Overall: H2 + ½O2→ H2O |
At a constant pressure of one atmosphere, the maximum available voltage from a fuel cell is determined by the energy differences between the initial state of reactants in the process and the final state, i.e., the Gibbs free energy change (ΔG) in the fuel cell process, which can be calculated from the reaction temperature (T), and from changes in the reaction enthalpy (ΔH) and entropy (ΔS). Under standard condition, the Gibbs free energy change is given as
ΔG0 = ΔH0−TΔS0 = −285![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 J − (298 K)(−163.2 J K−1) = −237200 J 800 J − (298 K)(−163.2 J K−1) = −237200 J |
The maximum cell voltage (E0), or the theoretical potential of fuel cell, for the hydrogen/air fuel cell reaction at a specific temperature and pressure is calculated by E = −ΔG/nF, where n is the number of electrons involved in the above reaction, and F is Faraday's constant. Since ΔG, n and F are all known, the theoretical hydrogen/oxygen fuel cell voltage can be calculated. Under standard condition (1 atm and 298 K), the maximum fuel cell voltage is given as

At 25 °C and 1 atm, the theoretical hydrogen/oxygen fuel cell potential is 1.23 V. As temperature rises from room temperature to that of an operating fuel cell (80 °C), the values of ΔH and ΔS change only slightly, but T changes by 55 °C, which leads to the decrease of the absolute value of ΔG. Thus, the maximum cell voltage decreases from 1.23 V at 25 °C to 1.18 V at 80 °C. The fuel cell voltage is in general the summation of the thermodynamic potential ENernst, the activation overvoltage ηact (from both anode and cathode overvoltages, i.e., ηact(cathode)−ηact(anode)), and the ohmic overvoltage ηohmic, which can be expressed as
| Ecell = ENernst + ηact−ηohmic. |
| ENemst = 1.229 − (8.5 × 10−4)(T− 298.15) + (4.308 × 10−5)T[ln(PH2) + 0.5ln(PO2)] |
The activation overvoltage is dependent on the electrode kinetics, which is reflected by the current flow, the oxygen and hydrogen partial pressures (PO2 and PH2) and the operation temperature (T). The ohmic overvoltage is caused by the electron flow resistance, proton flow resistance, and other contact resistances. The ohmic resistance of the membrane is proportional to the membrane thickness and inversely proportional to the membrane conductivity.3 For PEMFC, the anode overvoltage is very small when pure hydrogen is used. The major problem is the efficiency losses on the cathode electrode due to sluggish electrode kinetics for the oxygen reduction reaction (ORR).
In addition to the most popular hydrogen/air fuel cells, i.e., PEMFC, as described above, another type of fuel cells, direct methanol fuel cell (DMFC), has also attracted increasing interests because of its high conversion efficiency, low pollution, lightweight, high power density, and applications from small power supplies for electronic devices such as PCs, notebooks, and cellular phones. The cell reactions are indicated below.
| Anode: CH3OH + H2O → CO2 + 6H+ + 6e− |
Cathode: ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) O2 + 6H+ + 6e−→ 3H2O O2 + 6H+ + 6e−→ 3H2O |
| Overall: CH3OH + O2→ CO2 +2H2O |
Methanol is a highly attractive fuel for fuel cells as it has a theoretical cell voltage of 1.2 V, and a theoretical energy density of 6094 W h kg−1 (∼5× that of the best battery couples). The thermodynamic potential of the methanol–air fuel cell is also close to that of the hydrogen–air fuel cell (1.23 V). However, its practical energy density is only 1500–3100 W h kg−1 while its operating cell voltage is 0.4 V. Currently, the operation voltages for anode and cathode at 60 °C are 0.3 and 0.7 V, respectively.4 For the methanol fuel cell, while there is a loss in the cathode side due to methanol cross over from the anode side, the main losses are in the anodic electrode kinetics because the methanol oxidation reaction is very slow.
It is clear from the above discussion of the two types of fuel cells, i.e., PEMFC and DMFC, that the slow electrode kinetics at the cathode for the oxygen reduction reaction in PEMFC, and at the anode for the methanol oxidation reaction in DMFC, are the major areas of problems. The large overpotential for oxygen reduction at the cathode represents a loss of about 20% from the theoretical maximum efficiency for the hydrogen/air fuel cells. Due to the high degree of irreversibility of the oxygen-reduction reaction, even under open-circuit conditions, the overpotential at the oxygen electrode in PEMFC is about 0.2 V. The situation is even worse with the DMFCs. The thermodynamic potential for a DMFC is 1.21 V, which is only 20 mV less than that for the PEMFC. Both the methanol oxidation and oxygen reduction reactions are highly irreversible and thus there is a loss of about 0.2 V at the anode for DMFC under open-circuit conditions, and an enhanced loss of about 0.1 V at the oxygen electrode because of the crossover of methanol from the anode to the cathode.5
The electrochemical reactions in the fuel cells are heterogeneous surface reactions and the sluggish electrode kinetics can be addressed by catalysts supported on the electrode surfaces. The high surface areas are thus required to increase reaction rate. To reduce the cost associated with using platinum in practically all fuel cells and to achieve the optimized utilization of the platinum, one of the best approaches is to disperse catalysts as nanoparticles. The development of active and robust catalysts using supported metal or alloy nanoparticles, as discussed in this article, is a key area of current research aimed at solving the above problems.
While there are many basic components in a fuel cell, including a gas diffusion electrode, polymer membrane, and catalysts, the catalyst is one of the key components. According to the cost breakdown of fuel cell components,6 the cost of catalysts in manufacturing fuel cells is the highest (∼30%) for small production volume, and remains very high with increasing production volume. Currently, low activity, poor durability and high cost of the platinum-based anode and cathode catalysts in PEMFCs and DMFCs constitute some of the major barriers to commercialization of fuel cells. In addition, the durability of Pt-based catalysts can be compromised by sintering and dissolution of the catalysts in fuel cells. There is a major gap in the development of catalyst technology between the laboratory test and the practical application in terms of abilities to engineer the size, composition and stability. A key challenge to the ultimate commercialization of fuel cells is the development of active, robust and low-cost catalysts.7,8 This article discusses the recent progress of research in fuel cell catalysts by focusing on nanotechnology-guided catalyst research and development. Specific examples are from our recent findings in the studies of multimetallic nanoparticles with organic monolayer encapsulation for the preparation of fuel cell catalysts. There follows an overview of the general background and fundamental issues in fuel cell catalysis, but additionally the approaches to the preparation and characterization of selected examples of molecularly-capped multimetallic nanoparticles and catalysts, and the findings of the studies of their electrocatalytic oxygen reduction reaction for developing active, robust and low-cost catalysts in PEM fuel cells will be discussed.
2. Fuel cell catalysts—nanoscale engineering
2.1. Overview
One of the important challenges for fuel cell commercialization is the preparation of active, robust and low-cost electrocatalysts. However, the lack of abilities in the controlled preparation of nanoscale size and composition and the prevention of the intrinsic propensity of aggregation of nanoscale materials constitutes an obstacle to exploiting their nanometre-sized catalytic properties. The ability to harness the large surface area-to-volume ratios and the unique binding sites of nanoparticles constitutes a major driving force in both fundamental research and practical applications of nanotechnology in catalysis. It is the nanoscale size range over which metal particles undergo a transition from atomic to metallic properties, leading to new electronic and catalytic properties. The synthesis of molecularly-capped metal nanoparticles as building blocks for engineering the nanoscale catalytic materials takes advantage of diverse attributes, including monodispersity, processability, solubility, stability, and self-assembly capability in terms of size, shape, composition, and surface properties.One of the serious problems for fuel cells at the cathode for PEMFC and at the anode for DMFC is the poor activity of the catalysts,9,10 leading to a loss of about one-third of the available energy. The propensity of poisoning of Pt by CO species (including trace level CO in reformed hydrogen from methanol, natural gas, or gasoline11,12) is another problem for the extensively-studied Pt catalysts. The binary PtRu on carbon support has been studied for decades because of the bifunctional catalytic capability.12,13 The kinetic limitation of the oxygen reduction at cathode catalysts is another problem for fuel cells operating at low temperature (<100 °C) because the rate of breaking of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds to form water strongly depends on the degree of the oxygen’s interaction with the adsorption sites of the catalyst, and the competition with other species in the electrolyte (e.g., CH3OH). Metalloporphyrins14,15 and Platinum Group Metal (PGM) alloys16 have been used to design catalyst for 4e− reduction of O2 to water. Many studies focused on understanding the mechanism of oxygen reduction on Pt–Fe, Pt–Ni, and Pt–Co,10f including CO or methanol tolerance.10d,e Bulk-melted PtBi, PtIn, and PtPb intermetallic phases17 and Ru nanoparticles modified with Pt18 showed some promises for fuel cell applications. The application of high throughput, combinatorial screening methods to screen a large number of catalysts using an optical fluorescence technique by detecting proton production on an array of catalyst inks with different compositions has been successfully demonstrated.8,19 Individually addressable array electrodes have also been investigated for rapid screening.20–22 Recently, simple thermodynamic principles are proposed as guidelines that assume that one metal breaks the oxygen–oxygen bond of O2 and the other metal acts to reduce the resulting adsorbed atomic oxygen.23 The high throughput combinatorial screening of catalysts is very useful for rapid screening, including our latest work.24 Because the preparation of fuel cell catalysts has been mostly based on traditional methods such as co-precipitation or impregnation,25–27 little is known about how to optimize size, composition and morphology of multimetallic catalysts. The knowledge is very important since the nanoscale synergistic effects are expected to be very different from their macroscopic counterparts, as supported by examples of bimetallic nanoparticles (PtRu, AuPt, etc.), including our recent work.28–30
O bonds to form water strongly depends on the degree of the oxygen’s interaction with the adsorption sites of the catalyst, and the competition with other species in the electrolyte (e.g., CH3OH). Metalloporphyrins14,15 and Platinum Group Metal (PGM) alloys16 have been used to design catalyst for 4e− reduction of O2 to water. Many studies focused on understanding the mechanism of oxygen reduction on Pt–Fe, Pt–Ni, and Pt–Co,10f including CO or methanol tolerance.10d,e Bulk-melted PtBi, PtIn, and PtPb intermetallic phases17 and Ru nanoparticles modified with Pt18 showed some promises for fuel cell applications. The application of high throughput, combinatorial screening methods to screen a large number of catalysts using an optical fluorescence technique by detecting proton production on an array of catalyst inks with different compositions has been successfully demonstrated.8,19 Individually addressable array electrodes have also been investigated for rapid screening.20–22 Recently, simple thermodynamic principles are proposed as guidelines that assume that one metal breaks the oxygen–oxygen bond of O2 and the other metal acts to reduce the resulting adsorbed atomic oxygen.23 The high throughput combinatorial screening of catalysts is very useful for rapid screening, including our latest work.24 Because the preparation of fuel cell catalysts has been mostly based on traditional methods such as co-precipitation or impregnation,25–27 little is known about how to optimize size, composition and morphology of multimetallic catalysts. The knowledge is very important since the nanoscale synergistic effects are expected to be very different from their macroscopic counterparts, as supported by examples of bimetallic nanoparticles (PtRu, AuPt, etc.), including our recent work.28–30
To achieve durable and active catalysts with a low cost, new concepts and strategies must be developed for the creation of size-, composition-, and morphology-controlled multimetallic nanoparticles and catalysts. In contrast to most existing catalyst preparations based on traditional co-precipitation or impregnation methods, the foundation of the nano-engineering of multimetallic alloy catalysts31–37 has many important attributes to bridge the existing gap and lead to new opportunities in creating a synergistic balance of activity and stability of the catalysts. Nanoscale structures differ from their bulk counterparts in many significant ways for various reasons, including atomic–metallic transition, possible phase-reconstitution, different melting points due to size or alloying effects, and synergistic effects due to modified electronic band structure. The understanding of whether the formation of alloy or phase-segregation in multimetallic nanoparticles is different from bulk scale materials, and how the catalytic activity and stability are influenced by size, composition, and morphology (Scheme 1) is thus very important.
 | ||
| Scheme 1 Schematic illustrations of alloying (top) or phase-segregating (bottom) binary metals (M1M2) for nanoscale and micro/larger scale systems. | ||
The recent studies of nano gold catalysts serve as a best example to illustrate the unique properties displayed by nanoparticles. Despite the intensive research into the catalytic activity of gold in a restricted nanoscale size range,38 the catalytic origin of nanosized gold and gold-based bimetallic catalysts remains elusive. One of the main problems is the lack in understanding of the nanoscale core–surface property correlation. Gold–platinum nanoparticles of 2–5 nm diameter present an intriguing system for delineating the correlation in view of recent ability in synthesizing AuPt nanoparticles in a wide range of bimetallic composition.30,39 Whether the AuPt nanocrystal core is alloyed or phase-segregated and how the surface binding properties are correlated with the nanoscale bimetallic properties are important questions for the exploitation of catalytic activity of the nanoscale bimetallic catalysts. Our XRD studies30,39 revealed alloy properties for the nanocrystal core, which is in contrast to the miscibility gap known for the bulk counterparts.40 There are also infrared spectroscopic studies of the adsorption of CO on the nanoparticles to address the surface binding properties,41–44 including our own recent work.45 AuPt nanoparticles could provide a synergistic catalytic effect that involves the suppression of adsorbed poisonous species and a change in electronic band structure to modify the strength of the surface adsorption. The decrease in activation energy to facilitate oxidative desorption or suppress CO adsorption was previously considered to lead to sufficiently-high adsorptivity to support catalytic oxidation in alkaline electrolytes.46–49 Since the alloy properties of the bimetallic AuPt nanoparticles30,39 are in sharp contrast to the bimetallic miscibility gap known for the bulk counterparts in a wide composition range (20–90% Au), the understanding of how the bimetallic nanocrystal and surface alloy properties are related to the surface binding and catalytic activities is very important. The recent report50 on stabilization of platinum oxygen-reduction electrocatalysts using gold clusters demonstrated that Pt catalysts can be stabilized against dissolution by modifying Pt nanoparticles with Au clusters.
Arrays of various binary combinations were studied using scanning electrochemical microscopy, and Pd–Co (10–30% Co) was found to exhibit activity close to that of Pt. In view of the strong adsorption of OH forming Pt–OH which causes inhibition of the O2 reduction, there are several important aspects of the recent progress in understanding the synergistic properties of PtM catalysts in ORR in terms of the role of M (metal) in the adsorption of oxygenated species (e.g., O, OH).18a,50,51
In view of the unique catalytic properties of nano-sized gold52–54 and the high catalytic hydrogenation activity of platinum, bimetallic AuPt nanoparticles of controllable composition may serve as a synergistic catalyst system. For example, in alkaline medium the presence of Au in Pt catalysts could reduce the strength of the Pt-OH formation52,53 while providing the needed adsorption sites for –OH species. While the presence of Au in Pt increases the lattice distance of Pt, the higher electronegativity of Au over Pt could cause an increase of the amount of charge being transferred from Pt to Au, which was in fact supported by high-resolution XPS data showing Au 4f7/2 binding energy 83.32 eV for Au/Pt and 83.87 eV for bulk-like Au atoms,55 and consequently an increase of the d-orbital vacancy in the PtAu.
The preparation of Pt-group catalysts, especially Pt alloyed with other transition metals, has been extensively studied for fuel cell catalytic reactions.10,56,57 Traditional approaches to preparing supported nanoparticle catalysts involve co-precipitation, deposition–precipitation, ion-exchange, impregnation, successive reduction and calcination, etc. These methods have been widely used for preparing noble metal catalysts on support materials.58 While a variety of supported Pt-group binary or ternary catalysts have been prepared by traditional methods,16,57–62 the ability to control the size and composition is limited due to the propensity of aggregation of metals at the nanoscale. Among many emerging approaches to the preparation of nanoparticles or nanostructures, one particular class of nanoparticles with core-shell type structures is beginning to attract interest for addressing some of the challenges in nanoscale catalyst preparation.63,64 The core-shell type nanomaterials can be broadly defined as core and shell of different matters in close interaction, including inorganic/organic and inorganic/inorganic combinations.63–67 There has been an increasing volume of studies in recent years aimed at synthesizing metal nanoparticles in the presence of organic capping agents.64b,66–71 While the synthesis of monometallic nanoparticles have extensively been studied using molecular encapsulation based synthesis methods (e.g., two-phase protocol), relatively limited studies of bi- or tri-metallic nanoparticles have been reported using such synthetic methods.64b,70–72
In addition to controllable nanoscale dimensions, the prevention of the intrinsic propensity of aggregation of nanoscale materials is another challenging area. Aggregation of nanoparticles leads to eventual loss of the nanoscale catalytic activity in practical applications. The use of naked metal nanoparticle catalysts on supporting materials based on traditional preparative methods has been well demonstrated for different catalytic reactions.73 Recently, nanoparticles capped in monolayers, polymers or dendrimers have been rapidly emerging, demonstrating remarkable parallels to catalytic activities for supported nanoparticles. The catalysis includes those utilizing functional groups at the capping shell74,75 and those exploiting surface sites on the nanocrystals.67,73 Polymer-mediated self-assembly of monolayer-functionalized Pd nanoparticles and SiO2 particles through a ‘bottom–up’ approach and thermal treatment have recently been demonstrated as highly reactive, recyclable heterogeneous catalysts for both hydrogenation and carbon–carbon bond formation reactions.76 The nanotechnology-guided design and fabrication of catalysts for enhancing the catalytic activity and reducing the cost of catalysts will have enormous impacts to better catalyst preparation for fuel cell application.9,77 Importantly, deliberate tailoring of nanoparticle size, shape and composition of nanoparticles ranging down to a few nm could lead to improved or new catalytic properties.
2.2. Multimetallic nanoparticles and catalysts
In contrast to traditional approaches to the preparation of supported catalysts, the molecular encapsulation based synthesis and processing strategy to the preparation of multimetallic catalysts involves a sequence of three steps: solution-phase synthesis of the nanocrystal cores with molecular encapsulation, assembly of the encapsulated nanoparticles on support materials, and thermal treatment of the supported nanoparticles. Two examples, including bimetallic gold–platinum (AuPt) alloy and core-shell nanoparticles and trimetallic platinum–vanadium–iron (PtVFe) and platinum–nickel–iron (PtNiFe) catalysts, are described here to illustrate how such nanoscale engineered materials can advance the development of fuel cell catalysts.AuPt alloy nanoparticles. The as-synthesized AumPt100-m nanoparticles with different compositions are capped with thiol/amine monolayer shells (Fig. 3A). Varying the feeding ratio of the metal precursors used in the synthesis controls the bimetallic composition and sizes of the nanoparticles. The TEM data for the carbon-supported AuPt nanoparticles after thermal treatment (e.g., Au72Pt28/C) show an average size of 4.6 nm (Fig. 3B). As shown by the high-resolution TEM image, the nanoparticles exhibit a highly crystalline morphology (Fig. 3C). The effective removal of the capping monolayers from the nanoparticles by the thermal-treatment process79 were supported by the absence of the vibrational bands characteristic of the capping molecules in the C–H stretching region detected by FTIR, and the absence of the bands associated with sulfur species after the thermal treatment detected XPS analysis.
 | ||
| Fig. 3 (A) A schematic illustration of AuPt nanoparticle core encapsulated by a monolayer shell of thiol/amine. (B) TEM and (C) HRTEM of thermally-treated AuPt/C catalysts (e.g., Au72Pt28/C).31 | ||
As shown in Fig. 4A, in contrast to the bulk Au–Pt counterparts which display a miscibility gap at 20–90% Au,40 the lattice parameters of the bimetallic nanoparticles were found to scale linearly with Pt%. Such a relationship follows a Vegard's type law typically observed with binary metallic alloys, demonstrating the alloy properties for the bimetallic AuPt nanoparticles.30 The lattice parameters were determined for each AuPt sample by carefully determining the positions of the Bragg peaks in the diffraction patterns. In addition, the values for the lattice parameter of the nanoscale AuPt are all smaller than those for the bulk AuPt. This intriguing phenomenon suggests that nanoparticles have smaller inter-atomic distances than those for the bulk counterparts,45 which was the first example demonstrating that the nanoscale AuPt nanoparticles could exhibit single-phase character and small inter-atomic distances in the entire bimetallic composition range, both which are in sharp contrast to those known for their bulk counterparts. This finding is in fact supported by a theoretical model study,80 which showed that the nanoscale alloying is thermodynamically favored for AuPt nanoparticles smaller than ∼6 nm because the heat of formation is negative.
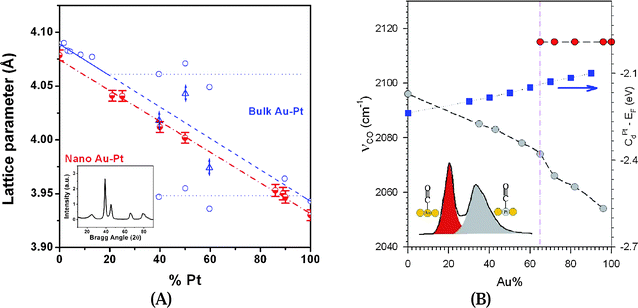 | ||
| Fig. 4 (A) The lattice parameters vs. Pt% for AuPt nanoparticles (red half-filled circles), part of the data reported recently,30 and for bulk AuPt40 (blue unfilled circles;) (triangular points represent a frozen state for bulk metals). Inset: XRD patterns for Au72Pt28/C catalyst treated at 400 °C. (B) Plots of the wavelength for Au-atop (red-filled circles, at top) and Pt-atop (grey-filled circles, at bottom) CO bands vs. the composition of Au in the alloy AuPt nanoparticles, along with the calculated trend in the d-band shift for Pt atoms in an AuPt alloy on a Au(111) surface based on calculation results in ref. 17 (blue-filled squares). Inset: schematic illustration of the band assignment for CO adsorbed to Au-atop site surrounded by Au atoms, and Pt-atop site surrounded by Pt atoms for a AuPt alloy surface. | ||
A comparison of the FTIR spectra for CO adsorption on AuPt nanoparticles over a wide range of bimetallic compositions (Fig. 4B) provides important information for assessing the surface binding properties of these bimetallic nanomaterials. By comparing CO spectra for Au/SiO2, Pt/SiO2, physical mixtures of Au/SiO2 and Pt/SiO2, and an Au72Pt28/SiO2 alloy, the CO bands for the bimetallic alloy catalyst are detected at 2115 cm−1 and 2066 cm−1, which are distinctively different from the single band feature at 2115 cm−1 for CO linearly adsorbed on atop sites of Au,41,81 and the single band feature at 2096 cm−1 for CO on atop sites for Pt.41,82 From FTIR spectra comparing CO adsorption on AuPt/SiO2 with a wide range of bimetallic compositions, two most important features can be observed from the spectral evolution as a function of bimetallic composition. First, the 2115 cm−1 band observed for Au/SiO2 (a) displays a clear trend of diminishing absorbance as Pt concentration increases in the bimetallic catalysts. It is very interesting that this band becomes insignificant or even absent at >∼45% Pt. Secondly, the lower-wavenumber CO band (∼2050 cm−1) shows a clear trend of shift towards that for the Pt-atop CO band observed for Pt/SiO2 (i) as Pt concentration increases. This trend is shown in Fig. 4B. For higher concentrations of Au, this band is strong and broad. Such a dependence of the CO bands on the bimetallic concentration is remarkable, and is to our knowledge observed for the first time. The higher-wavenumber band (2115 cm−1) is attributed to CO adsorption on Au-atop sites in a Au-rich surface environment, whereas the lower-wavenumber band and its composition-dependent shift reflect an electronic effect of the surface Pt-atop sites alloyed in the bimetallic nanocrystal. This observation is indicative of a unique synergistic surface property in which the Pt-atop CO adsorption is greatly favored over the Au-atop CO adsorption.
The understanding of the electronic effect is based on the correlation between the spectral features and findings from a previous DFT calculation on the d-band of Pt atoms in bimetallic AuPt surfaces.83 The DFT calculation showed that the d-band center of Pt atoms increases with Au concentration in the AuPt alloy on a Au(111) or Pt(111) substrate. For an AuPt alloy on Au(111), the d-band center of Pt atoms was found to show an increase from 0 to 65–70% Au, after which a slight decrease was observed. For a AuPt alloy on Pt(111), the d-band center of Pt atoms is found to increase almost linearly with the concentration of Au. Both were supported by experimental data in which the adsorption of CO showed an increased binding energy in comparison with Pt(111), due to the larger lattice constant of Au, leading to an expansion of Pt.83 Since the theoretical data for nanocrystal alloys are not yet available, the average d-band shift for Pt atoms from these two sets of DFT calculation results is included in Fig. 4B to illustrate the general trend. Interestingly, a subtle transition for the lower-wavenumber band, i.e., from a relatively-broad band feature to a narrow band feature that resembles that of the Pt-atop CO band is observed to occur at ∼65% Au, below which the Au-atop CO band basically disappeared. There exists a stronger electron donation to the CO band by a Pt-atop site surrounded by Au atoms in the bimetallic alloy surface than that from the monometallic Pt surface as a consequence of the upshift in d-band center of Pt atoms surrounded by Au atoms, which explains the preference of Pt-atop CO over the Au-atop CO adsorption. The observed decrease of the Pt-atop CO band frequency with increasing Au concentration is clearly in agreement with the d-band theory for the bimetallic system.83 Note that the observed wavenumber region of 2050–2080 cm−1 is quite close to the values found recently based on DFT calculations of CO adsorption on AuPt clusters (2030 and 2070 cm−1) depending on the binding site (Pt or Au).82
The complete disappearance of the Au–CO band for samples with a concentration below 65% Au does not necessarily imply the absence of Au on the surface of the nanoparticles; it implies rather the preferential Pt-atop CO adsorption over Au-atop CO adsorption, which is supported by the DFT calculation results.83 The observation of the maximum mass activity for electrocatalytic methanol oxidation reaction under alkaline conditions around the composition of 65–85% Au coincides remarkably with the finding of the composition of ∼65% Au for the transition of the band features for CO adsorption, suggesting a synergistic effect of the surface reactivity for Pt atoms surrounded by Au atoms. AuPt catalysts could display synergistic bifunctional properties by the reduction of the strength of Pt-OH formation, the added adsorption sites for –OH species, the increase of the lattice distance of Pt, the increase of electronegativity of Pt, or increase of the d-orbital vacancy by the presence of Au in Pt catalysts.
Based on the kinetic current extracted from the rotating disk electrode (RDE) data for ORR (Fig. 5), the mass activities were found to be strongly dependent not only on the bimetallic composition (AumPt100-m), but also on the nature of the electrolyte. The analysis of the RDE data in the kinetic region is a standard way of determining the catalyst activity. While the changes of the kinetic current appear small in Fig. 5A, they reflect the differences in the activity, which are not due to changes in particle size and specific surface area of the catalyst as evidenced by control experiments. The strong dependence of the mass activities on the bimetallic composition in the alkaline electrolyte is evident by the exhibition of a maximum in the composition region of 60–80%Au, which is higher than those for Pt/C and Au/C by a factor of 2–3. This finding is in contrast to the gradual increase of mass activity in the acidic electrolyte displaying a relatively smooth transition from the low activity of Au to the high activity of Pt (Fig. 5B). In acidic solution, Au is not capable of proving adsorption sites for –OH and the electrocatalytic activity is thus rather low.
 | ||
| Fig. 5 The RDE curves for different AumPt100-m/C catalysts (a: Au/C; b: Au72Pt28/C; c: Au56Pt44/C; d: Pt/C. 1600 rpm; catalyst loading: 11 µg AuPt/C; 10 mV s−1) in alkaline (A, 0.5 M KOH) and acidic (B, 0.5 M H2SO4) electrolytes saturated with O2. Glassy carbon working electrode's geometric surface area: 0.196 cm2. (Reprinted with permission from ref. 31a. Copyright 2006 Elsevier.) | ||
While the mass activity in the acidic electrolyte could reflect a collective effect of the activities from both Au and Pt, the concurrence of a maximized activity in the 60–80% Au region in the alkaline electrolyte suggests the operation of a remarkable synergistic effect. The possibility of an optimal fraction of Au atoms surrounding Pt could have played an important role in the observed activity maximum. These Au atoms could thus function as the sites for chemisorbed OH−ad for the dissociative adsorption of O2via interaction with OH−ad or for chemisorption of the reaction intermediate HO2−ad.84 In addition to reducing –OH on Pt by alloying Au in the Pt catalyst, similarly to those recently revealed for other metals in MxPt1-x/Pd(111)51 and Pt/M(111),18a the favorable chemisorption of oxygen on gold nanoparticles, as evidenced by the detection of AuO−, AuO2−, and AuOH on oxide-supported Au nanoparticles85 and OH−ad on Au(110) in alkaline electrolyte,84 must also have played an important role in the synergistic activity. Based on the recent DFT calculation using ‘organized alloy’ pyramid model,86 there seems to be some correlation in terms of a compromised balance between the dissociation at the Pt-site activation and the promotion at the Au-site adsorption (–OH).
AuPt core-shell nanoparticles. The examination of the electrocatalytic activity for carbon-supported core@shell nanoparticles such as Au@Pt and Pt@Au further substantiated the importance of the nanoscale surface properties.78 As revealed by the order for the increase of kinetic current and the positive-shift of the reduction potential, Au< Pt@Au < Au@Pt < Pt, the increased activity for Au@Pt catalyst is consistent with the presence of Pt on the surface.
The recent study of CO adsorption on core@shell nanoparticles (Au@Pt and Pt@Au)78 has also revealed insights into the surface binding difference of bimetallic nanoparticles. The resemblance of the observed CO bands for Au@Pt and Pt@Au nanoparticles Pt and Au nanoparticles, respectively, is consistent with the core@shell nanostructures by design. The importance of the relative surface alloying or layering arrangements of metals on single-crystal substrates has been recognized, including the “near surface alloy” model of metal adlayer on metal substrate for oxygen reduction reaction.87 Despite the extensive research in metal or oxide core@shell nanoparticles,88–91 the correlation between the synergistic catalytic properties and the composition and spatial arrangement for metal–metal and metal–oxide core@shell nanoparticles remains elusive. Built upon our recent demonstration of the alloy character of bimetallic AuPt nanoparticle catalysts,29,30 the core-shell character of Fe3O4@Au nanoparticles,92 and some recent theoretical insights,80,93 core@shell nanoparticles (Au@Pt, Pt@Au, Fe3O4@Au@Pt) were also studied for assessing their nanostructural correlation of the electrocatalytic properties for methanol oxidation reaction (MOR) and oxygen reduction reaction (ORR). Core-shell type nanoparticles can be broadly defined as core and shell of different matters in close interaction, including inorganic/organic, inorganic/inorganic, organic/organic, or inorganic/biological combinations.67 The presence of organic capping shells in each step is important for controlling the size and monodispersity of the M1@M2 nanoparticles.78
Fig. 6A shows a representative set of RDE curves for ORR in acidic electrolyte to assess the electrocatalytic activities of the carbon-supported core@shell nanoparticle catalysts. The relative changes in the RDE characteristics were compared for several different monometallic and core-shell nanoparticle catalysts. The increase of kinetic current and the positive-shift of the reduction potential followed the order of Au< Pt@Au < Au@Pt <Fe3O4@Au@Pt < Pt. The increased activity for Au@Pt and Fe3O4@Au@Pt catalysts is consistent with the presence of Pt on the surface. This is also reflected by the shift of the reduction potential of Au@Pt/C from Au to Pt, and the values obtained from Levich plots for the electron transfer number (2.2 for Au, 3.9 for Pt and 3.9 for Au@Pt). The reduction overpotential can be dramatically shifted from high (for Au) to low (for Pt) depending on the relative Au–Pt–oxide spatial configuration.
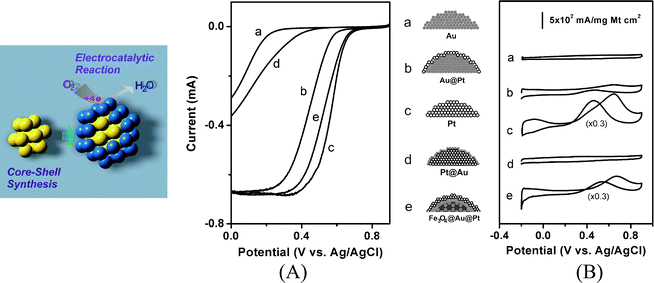 | ||
| Fig. 6 (A) RDE curves of different catalysts in 0.5 M H2SO4 saturated with O2 (metal loading 10–30%) (10 mV s−1, 1600 rpm, surface area: 0.2 cm2). The current for the curves is not normalized by the metal loading and the surface area. (B) CV curves for MOR for different catalysts in 0.5 M H2SO4 with 0.5 M MeOH. (50 mV s−1). The scheme in the middle of A and B illustrates the corresponding nanoparticle surface structures by design. The scheme on the left illustrates the core-shell synthesis and the surface electrocatalytic oxygen reduction reaction. (Reprinted with permission from ref. 78. Copyright 2008 John Wiley & Sons Inc.) | ||
Fig. 6B shows a representative set of CV data of different catalysts for MOR. In contrast to the absence of activities for Au and Au-coated nanoparticles in acidic solution, the increased activities for Pt-coated catalysts are characteristic of the electrocatalytic properties of the Pt component in Au@Pt and Fe3O4@Au@Pt catalysts. This is supported by the analysis of the methanol oxidation peak currents normalized with total metals. The catalyst treatment temperature was also found to exhibit an effect on the observed activity, which reflects the relative enrichment of metals in the core-shell nanostructure.
A remarkable finding is the high activity for Fe3O4@Au@Pt catalyst, as evidenced by the lower overpotential and the higher mass activity. The fact that the mass activity for Fe3O4@Au@Pt is close to or higher than Pt demonstrates the feasibility of producing the synergistic catalytic effect by the metal-oxide core-shell nanostructures. Preliminary XRD examination of the core-shell structure revealed phase segregation of Pt and Au, as evidenced by the assymmetric peak in the XRD peak for Fe3O4@Au@Pt/C (thermal activation at <300 °C). This observation is in sharp contrast to the single-phase character found for AuPt alloy nanoparticles.29,30 The hydrogen adsorption/evolution currents at −0.2 V and the shift of its reduction potential positively to a potential almost comparable with Pt catalysts agrees with the presence of the Pt-shell. The thermal treatment temperatures were also found to influence the relative surface distribution or alloying properties. The mass activity for Fe3O4@Au@Pt was higher than the other Au–Pt combinations (Au@Pt or AuPt), suggesting the important role played by the oxide core. Because the study of Fe3O4@Au@Pt catalyst78 is rather preliminary, the understanding of whether and how the Fe3O4 and Au underneath the Pt shell play a role for the electrocatalytic activity requires more quantitative study for a detailed correlation of the catalytic activity with various parameters in controlling the relative core-shell composition and structure. Nevertheless, the electrocatalytic activities were shown to depend on the nanoscale spatial arrangement of the metals. The relative changes in the catalytic activity should correlate with the core size, shell thickness, composition, and spatial properties.
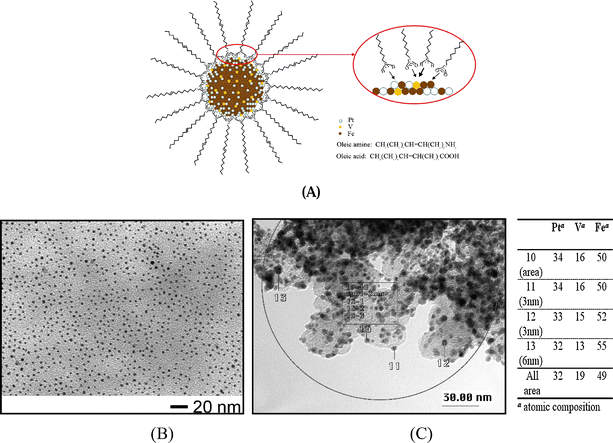 | ||
| Fig. 7 (A) A schematic illustration of PtVFe nanoparticle core encapsulated by a monolayer shell of oleic amine/oleyl amine. (B) TEM micrograph for Pt33V14Fe53 nanoparticles. (C) High resolution TEM-EDX data for the ternary nanoparticle catalyst (Pt33V14Fe53/C).24 | ||
The basic morphology of the observed nanoparticles is largely characterized by the highly-faceted nanocrystal feature, which is observable by a close examination of the shapes of the individual nanocrystals. The particles are highly monodispersed, with an average size of 1.9 ± 0.3 nm. The fact that the nanoparticles have well-defined interparticle spacing and display domains of hexagonal ordering is indicative of the encapsulation of the nanocrystal cores by organic monolayers. While the sizes of the ternary nanoparticles varied slightly depending on the actual composition, the data demonstrated the controllability over size monodispersity. Nanoparticles with average diameters ranging from 1.4 nm to 3.2 nm have been obtained. The size monodispersity in most cases was very high, ranging from ±0.2 to ±0.6 nm.
The XRD data revealed a typical fcc pattern with some insignificant features indicative of chemically-disordered structure. The calcination treatment of the nanoparticles led to rearrangement of Pt, V and Fe atoms in the nanoparticles into long-range chemically-ordered fcc structure. The formation of the alloyed nanocrystalline cores is supported by the fact that the diffraction peaks of metallic platinum shift to higher angles due to lattice shrinking resulting from the doping of smaller vanadium and iron atoms. Indeed, the diffraction peak position falls in between those for the monometallic Pt and those for monometallic V and Fe. For example, the strongest peak for Pt appears at 2θ = 40.5, slightly higher than the Pt(111) peak (2θ = 39.8).30 Diffraction peaks corresponding to V(110) (2θ = 44.7) and Fe (110) (2θ = 42.2) were not detected. The (111) peak for PtFe alloy nanoparticles (2θ = 41.270) was also not detected, suggesting the absence of PtFe nanoparticles in the PtVFe nanoparticles.
The PtVFe nanoparticles can be easily assembled on carbon support materials with controllable dispersion and mass loading. The carbon-supported alloy nanoparticles after calcination treatment were found to display high electrocatalytic activities for oxygen reduction in our recent work. The particle sizes were basically un-affected after their assembly onto carbon materials. The thermal treatments of PtVFe/C catalysts involved removal of organic shells and calcination of the ternary nanoparticles. A representative TEM-EDX image for a sample of the thermally-treated Pt32V14Fe54/C catalyst is shown in Fig. 7C. The average particle sizes after the calcination treatment were found to show a slight increase (∼0.5 nm) in comparison with that before the treatment. The subtle increase in size was found to be dependent on the calcination temperature. The particle sizes displayed a certain degree of increase (on avg., by ∼0.5 nm) after the thermal treatment, especially at higher temperatures. The metal loading data will be discussed in the next section.
From XRD spectra for Pt32V14Fe54/C treated under 550 °C, the broad peak at low angles is from carbon support materials. The diffraction peak positions for PtVFe fall between those for the monometallic Pt, V and Fe,94 which indicates that these particles are largely alloyed. No secondary phase was detected. The diffraction peaks of metallic platinum shift to higher angles due to lattice shrinking resulting from the doping of smaller vanadium and iron atoms. The as-synthesized PtVFe nanoparticles were shown to be fcc structure with little chemically-disordered structure.94 After thermal treatment, there were some indications of rearrangement of Pt, V and Fe atoms in the PtVFe nanoparticles, leading into long-range chemically-ordered fcc structure. The average sizes of the nanoparticles estimated by Scherrer correlation are slightly larger than the value determined from TEM data. The XRD data support that the ternary nanoparticles are alloy-like in character. The XRD data for nanoparticles treated at different temperatures (400 and 450 °C) were also compared. The data reveal a subtle difference in peak width which can be related to a difference in size change.
The question whether the nanoparticles are multimetallic in individual nanoparticles or in an ensemble of the nanoparticles was addressed using TEM-EDX (transmission electron microscopy-energy dispersive X-ray spectroscopy).24 TEM-EDX data (nano-composition) are also compared with DCP-AES analysis (macro-composition). By controlling the electron beam diameter and current, this measurement will yield reproducible compositions of individually isolated nanoparticles. The atomic compositions for PtVFe nanoparticles (Fig. 7C) are found to be almost identical, independent of the actual sizes, in contrast to the results observed from the traditional synthesis where large-sized particles are usually base metal rich and small particles are Pt rich. Similar compositions were found for as-synthesized PtVFe nanoparticles, indicating the composition uniformity in the individual nanoparticles. Note that the effective removal of OAM and OAC capping molecules of the nanoparticles was evidenced by FTIR data, and the metal loading (37%) was confirmed by TGA data. Similar results were obtained for PtNiFe nanoparticles. The XRD data for PtNiFe/C showed some features indicative of chemically-disordered structure before thermal treatment, but a tetragonal-type structure of the PtFe type after the thermal treatment.32
The carbon-supported trimetallic (e.g., PtVFe/C and PtNiFe/C) alloy nanoparticle catalysts are expected to be electrocatalytically active for ORR based on combinatorial studies of a series of Pt-based bimetallic alloy thin film catalysts which reveal significant increase of activities for the oxygen reduction reaction (e.g., PtFe, PtNi, and PtV thin films exhibit high activities for ORR).24 The introduction of a third metal to the alloy is expected to produce a combination of effects such as reduction of the lattice distance, the addition of surface sites for the formation of metal–oxygen bond and adsorption of OH−, and the modification of the d-band center. In the next two subsections, the results for PtVFe/C and PtNiFe/C alloy nanoparticle catalysts are described as two examples of the study. Fig. 8A shows a representative set of RDE polarization curves for ORR at Pt32V14Fe54/C. The electron transfer number (n) is close to 4, as expected for ORR at this type of catalysts.
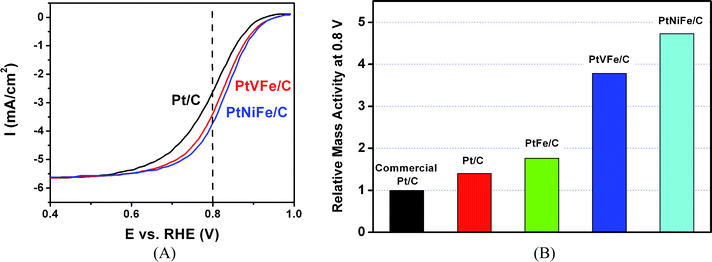 | ||
| Fig. 8 (A) RDE curves for ORR at Pt32V14Fe54/C (31% metal loading), Pt31Ni34Fe35/C (30% metal loading) and standard Pt/C (20% metal loading) catalysts on glassy carbon electrode (catalyst loading 15 µg, 0.196 cm2 geometric area) in 0.5 M H2SO4. Scan rate: 5 mV s−1, and rotating speed: 2000 rpm. (B) Comparison of the relative electrocatalytic activities for carbon-supported monometallic, binary and ternary catalysts.32 | ||
The electrocatalytic activity data were compared for the Pt32V14Fe54/C catalysts in terms of relative mass activities at 0.8 V. The relative mass-specific-activities are compared with that for standard commercial Pt/C catalyst (36.4% Pt loading). All these ternary catalysts showed increased electrocatalytic activities in comparison with the polycrystalline Pt/C catalysts. In comparison with a commercially available standard Pt/C catalyst (TKK), PtFe and PtVFe have relative activities of almost two and four times that of the commercially available Pt catalyst. Note that carbon supported platinum nanoparticles prepared in our laboratory show a 30% increase in molecular oxygen reduction in comparison with the commercial Pt/C catalyst under similar loading and test condition. The PtNiFe/C catalyst displayed a relative activity of almost five times larger than that of the commercially-available Pt/C catalyst. The activity displays the order of PtVFe/C > PtVFe/C > PtFe/C > Pt/C (Fig. 8B), demonstrating the effectiveness of the alloy composition in enhancing the electrocatalytic performance. This finding can be understood by the fact that the alloy nanoparticles are all located on the surface of the supporting carbon, whereas the traditional synthesis method cannot avoid burying some nanoparticles inside of the micropores of the carbon, which are not accessible by the molecular oxygen. The utilization of the catalytic nanoparticles was much higher on the surface than those inside the micropores.
5. Summary
In summary, the molecular encapsulation approach to the synthesis and processing of bimetallic/trimetallic nanoparticles is effective in producing alloy nanoparticles in the 2–5 nm regime with controllable composition and carbon-supported catalysts for fuel cell reactions. This approach differs from other traditional approaches to the preparation of supported catalysts in the abilities to control the nanoscale size, multimetallic composition, phase properties and surface properties. As demonstrated by the bimetallic AuPt alloy nanoparticle catalysts, synergistic activity is possible in which Au atoms surrounding Pt provide effective sites for the reaction adsorbates in the electrocatalytic reaction. The fact that this bimetallic nanoparticle system displays the unique single-phase property different from the miscibility gap of its bulk-scale counterpart serves as an important indication of the operation of nanoscale phenomena in the catalysts, which can be further exploited for the design and preparation of the nanostructured bimetallic catalysts for fuel cells.36 As demonstrated by the trimetallic nanoparticle catalysts such as PtVFe and PtNiFe nanoparticle catalysts, the size and composition of the nanoparticles can be engineered by molecularly-controlled synthesis and processing. The fact that the trimetallic catalysts exhibit electrocatalytic activities 4–5 times higher than Pt catalysts, means that upon further refinement of the activity-stability correlation, they could find applications in the development of low-cost fuel cell cathode catalysts.34,35 These examples have demonstrated the important role played by the exploration of nanotechnology for the preparation of catalysts. The preparation of nanoparticle catalysts is promising for delivering much higher catalyst utilization than those of conventional catalyst preparation methods. Further refinements of the processing parameters of the supported nanoparticle catalysts of different sizes, and composition and phase properties for optimizing activity, stability and cost have important implications for the better design of catalysts for practical applications in fuel cells. The rapid development of our fundamental understanding of the nanoscale properties and our abilities in engineering the nanoscale properties is expected to advance the research and development of active, robust, and low-cost catalysts for the commercialization of fuel cells, making an important contribution to solving some of the challenging problems in energy and the environment.Acknowledgements
This work was supported in part by the National Science Foundation (CBET-0709113 and CHE-0349040), NYSTAR, and Honda.References
- http://environmentalchemistry.com/yogi/environmental/200608hydrogenfuelcells.html.
- R. F. Mann, J. C. Amphlett, A. I. Hooper, H. M. Jensen, B. A. Peppley and P. R. Roberge, Development and application of a generalized steady-state electrochemical model for a PEM fuel cell, J. Power Sources, 2000, 86, 173–180 CrossRef CAS.
- Ryan O'Hayre, Suk-Won Cha, Whitney Colella and Fritz B. Prinz, Fuel Cell Fundamentals, John Wiley & Sons, New York, 2006 Search PubMed.
- Jiang Chu, Solid State Ionics, 2002, 148, 591–599 CrossRef CAS.
- A. K. Shukla and R. K. Raman, Annu. Rev. Mater. Res, 2003, 33, 155–68 CrossRef CAS.
- H. StoneEconomic Analysis of Stationary PEMFC Systems (DOE Hydrogen Program FY 2005 Progress Report 961), Battelle Memorial Institute, Columbus, Ohio Search PubMed.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9 CrossRef CAS.
- T. E. Mallouk and E. S. Smotkin, Combinatorial catalyst development methods, in Handbook of Fuel Cells – Fundamentals, Technology and Application, ed. W. Vielstich, A. Lamm and H. A. Gasteiger, John Wiley & Sons, 2003, p. 334 Search PubMed.
- (a) V. Mehta and J. S. Cooper, J. Power Sources, 2003, 114, 32 CrossRef CAS; (b) J. Scholta, A. Kabza, L. Jorissen and J. Garche, Battery Power Prod. Technol., 2000, 4, 21 Search PubMed; (c) X. M. Ren, P. Zelenay, S. Thomas, J. Davey and S. Gottesfeld, J. Power Sources, 2000, 86, 111 CrossRef CAS.
- (a) J. S. Spendelow and A. Wieckowski, Phys. Chem. Chem. Phys., 2004, 6, 5094 RSC; (b) H. Bonnemann and K. S. Nagabhushana, J. New Mater. Electrochem. Syst., 2004, 7, 93 Search PubMed; (c) S. Litster and G. McLean, J. Power Sources, 2004, 130, 61 CrossRef CAS; (d) A. K. Shukla and R. K. Raman, Annu. Rev. Mater. Res., 2003, 33, 155 CrossRef CAS; (e) S. B. Adler, Chem. Rev., 2004, 104, 4791 CrossRef CAS; (f) N. P. Brandon, S. Skinner and B. C. H. Steele, Annu. Rev. Mater. Res., 2003, 33, 183–213R CrossRef CAS; (g) A. E. Russell and A. Rose, Chem. Rev., 2004, 104, 4613 CrossRef CAS.
- (a) G. J. K. Acres, J. C. Frost, G. A. Hards, R. J. Potter, T. R. Ralph, D. Thompsett, G. T. Burstein and G. J. Hutchings, Catal. Today, 1997, 38, 393 CrossRef CAS; (b) S. Wasmus and A. Kuever, J. Electroanal. Chem., 1999, 461, 14 CrossRef CAS.
- (a) G. Q. Lu and A. Wieckowski, Curr. Opin. Colloid Interface Sci., 2000, 5, 95 CrossRef CAS; (b) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS.
- (a) J. W. Long, R. M. Stroud, K. E. Swider-Lyons and D. R. Rolison, J. Phys. Chem. B, 2000, 104, 9772 CrossRef CAS; (b) K. L. Ley, R. X. Liu, C. Pu, Q. B. Fan, N. Leyarovska, C. Segre and E. S. Smotkin, J. Electrochem. Soc., 1997, 144, 1543 CrossRef CAS; (c) S. A. Lee, K. W. Park, J. H. Choi, B. K. Kwon and Y. E. Sung, J. Electrochem. Soc., 2002, 149, A1299 CrossRef CAS.
- J. P. Collman, C. S. Bencosme, R. R. Durand, R. P. Kreh and F. C. Anson, J. Am. Chem. Soc., 1983, 105, 2699 CrossRef CAS.
- D. Chu and R. Jiang, Solid State Ionics, 2002, 148, 591 CrossRef CAS.
- U. A. Paulus, A. Wokaun, G. G. Scherer, T. J. Schmidt, V. Stamenkovic, V. Radmilovic, N. M. Markovic and P. N. Ross, J. Phys. Chem. B, 2002, 106, 4181 CrossRef CAS.
- E. Casado-Rivera, D. J. Volpe, L. Alden, C. Lind, C. Downie, T. Vazquez-Alvarez, A. C. D. Angelo, F. J. DiSalvo and H. D. Abruna, J. Am. Chem. Soc., 2004, 126, 4043 CrossRef.
- (a) J. L. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed, 2005, 44, 2132 CrossRef CAS; (b) K. Sasaki, J. X. Wang, M. Balasubramanian, J. McBreen, F. Uribe and R. R. Adzic, Electrochim. Acta, 2004, 49, 3873 CrossRef CAS.
- (a) Y. Sun, H. Buck and T. E. Mallouk, Anal. Chem., 2001, 73, 1599 CrossRef CAS; (b) G. Chen, D. A. Delafuente, S. Sarangapani and T. E. Mallouk, Catal. Today, 2001, 67, 341 CrossRef CAS; (c) B. C. Chan, R. X. Liu, K. Jambunathan, H. Zhang, G. Y. Chen, T. E. Mallouk and E. S. Smotkin, J. Electrochem. Soc., 2005, 152, A594 CrossRef CAS.
- R. Liu and E. S. Smotkin, J. Electroanal. Chem, 2002, 535, 49 CrossRef CAS.
- P. Strasser, Q. Fan, M. Devenney, W. H. Weinberg, P. Liu and J. K. Nørskov, J. Phys. Chem. B, 2003, 107, 11013 CrossRef CAS.
- S. Guerin, B. E. Hayden, C. E. Lee, C. Mormiche, J. R. Owen and A. E. Russell, J. Comb. Chem., 2004, 6, 149 CrossRef CAS.
- J. L. Fernandez, D. A. Walsh and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 357 CrossRef CAS.
- T. He, E. Kreidler, L. Xiong, J. Luo and C. J. Zhong, J. Electrochem. Soc., 2006, 153, A1637 CrossRef CAS.
- (a) M. Haruta and M. Date, Appl. Catal., A, 2001, 222, 427 CrossRef CAS; (b) P. C. Bismas, Y. Nodasaka, M. Enyo and Haruta, J. Electroanal. Chem., 1995, 381, 167 CrossRef CAS; (c) M. Haruta, J. New Mater. Electrochem. Syst., 2004, 7, 163 Search PubMed; (d) M. Haruta, Gold Bull., 2004, 37, 27 CAS.
- (a) M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647 CrossRef CAS; (b) M. S. Chen and D. W. Goodman, Science, 2004, 306, 252 CrossRef CAS; (c) C. T. Campbell, Science, 2004, 306, 234 CrossRef CAS.
- (a) G. C. Bond and D. T. Thompson, Catal. Rev., 1999, 41, 319 CrossRef CAS; (b) G. C. Bond, Catal. Today, 2002, 72, 5 CrossRef CAS; (c) R. J. Davis, Science, 2003, 301, 926 CrossRef CAS; (d) D. R. Rolison, Science, 2003, 299, 1698 CrossRef CAS.
- (a) J. Luo, M. M. Maye, N. N. Kariuki, L. Wang, P. Njoki, Y. Lin, M. Schadt, H. R. Naslund and C. J. Zhong, Catal. Today, 2005, 99, 291 CrossRef CAS; (b) M. M. Maye, N. N. Kariuki, J. Luo, L. Han, P. Njoki, L. Wang, Y. Lin, H. R. Naslunda and C. J. Zhong, Gold Bull., 2004, 37, 217 CAS.
- (a) N. N. Kariuki, J. Luo, M. M. Maye, S. A. Hassan, T. Menard, H. R. Naslund, Y. Lin, C. Wang, M. H. Engelhard and C. J. Zhong, Langmuir, 2004, 20, 11240 CrossRef CAS; (b) P. N. Njoki, J. Luo, L. Wang, M. M. Maye, H. Quaizar and C. J. Zhong, Langmuir, 2005, 21, 1623 CrossRef CAS.
- J. Luo, M. M. Maye, V. Petkov, N. N. Kariuki, L. Wang, P. Njoki, D. Mott, Y. Lin and C. J. Zhong, Chem. Mater., 2005, 17, 3086 CrossRef CAS.
- (a) J. Luo, P. Njoki, Y. Lin, L. Wang, D. Mott and C. J. Zhong, Electrochem. Commun., 2006, 8, 581 CrossRef CAS; (b) J. Luo, P. Njoki, Y. Lin, D. Mott, L. Wang and C. J. Zhong, Langmuir, 2006, 22, 2892 CrossRef CAS.
- (a) J. Luo, N. Kariuki, L. Han, L. Wang, C. J. Zhong and T. He, Electrochim. Acta, 2006, 51, 4821 CrossRef CAS; (b) J. Luo, L. Wang, D. Mott, P. Njoki, N. Kariuki, C. J. Zhong and T. He, J. Mater. Chem., 2006, 16, 1665 RSC.
- (a) H. Bonnemann and K. S. Nagabhushana, J. New Mater. Electrochem. Syst., 2004, 7, 93 Search PubMed; (b) N. P. Brandon, S. Skinner and B. C. H. Steele, Annu. Rev. Mater. Res., 2003, 33, 183R.
- C. J. Zhong, J. Luo, M. M. Maye, L. Han, N. Kariuki and T. He, Core-Shell Synthesis of C-supported Alloy Nanoparticle Catalysts, US Patent No 7053021, 2006 Search PubMed.
- T. He, C. J. Zhong, J. Luo, L. Han, M. M. Maye, N. Kariuki and L. Wang, Metal and Alloy Nanoparticles and Synthesis Methods Thereof, US Patent No 7335245, 2008 Search PubMed.
- C. J. Zhong, J. Luo, M. M. Maye and N. Kariuki, Gold-Based Alloy Catalysts for Use in Fuel Cell Catalysts, US Patent No 7208439, 2007 Search PubMed.
- L. Y. Wang, H.-Y. Park, S. I. Lim, M. J. Schadt, D. Mott, J. Luo, X. Wang and C. J. Zhong, J. Mater. Chem., 2008, 18, 2629 RSC.
- M. Haruta, Nature, 2005, 437, 1098 CrossRef CAS.
- D. Mott, J. Luo, P. N. Njoki, Y. Lin, L. Y. Wang and C. J. Zhong, Catal. Today, 2007, 122, 378 CrossRef CAS.
- Catalysis by Metals and Alloys, ed. V. Ponec and G. C. Bond, Elsevier, Amsterdam, 1995 Search PubMed.
- C. S. Kim and C. Korzeniewski, Anal. Chem., 1997, 69, 2349 CrossRef CAS.
- M. S. Chen, D. Kumar, C.-W. Yi and D. W. Goodman, Science, 2005, 310, 291 CrossRef CAS.
- C. Mihut, C. Descorme, D. Duprez and M. Amiridis, J. Catal., 2005, 212, 125.
- H. Lang, S. Maldonado, K. J. Stevenson and B. D. Chandler, J. Am. Chem. Soc., 2004, 126, 12949 CrossRef CAS.
- D. Mott, J. Luo, A. Smith, P. N. Njoki, L. Wang and C. J. Zhong, Nanoscale Res. Lett., 2007, 2, 12 Search PubMed.
- K. Nishimura, K. Kunimatsu and M. Enyo, J. Electroanal. Chem., 1989, 260, 167 CrossRef CAS.
- M. Morita, Y. Iwanaga and Y. Matsuda, Electrochim. Acta, 1991, 36, 947 CrossRef CAS.
- A. B. Anderson, E. Grantscharova and S. Seong, J. Electrochem. Soc., 1996, 143, 2075 CAS.
- L. D. Burke, J. A. Collins, M. A. Horgan, L. M. Hurley and A. P. O'Mullane, Electrochim. Acta, 2000, 45, 4127 CrossRef CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220 CrossRef CAS.
- J. Zhang, M. B. Vukmirovic, K. Sasaki, A. U. Nilekar, M. Mavrikakis and R. R. Adzic, J. Am. Chem. Soc., 2005, 127, 12480 CrossRef CAS.
- (a) M. Van Brussel, G. Kokkinidis, I. Vandendael and C. Buess-Herman, Electrochem. Commun., 2002, 4, 808 CrossRef CAS; (b) M. S. El-Deab and T. Ohsaka, J. Electroanal. Chem., 2003, 553, 107 CrossRef CAS.
- M. S. El-Deab and T. Ohsaka, Electrochim. Acta, 2002, 47, 4255 CrossRef CAS.
- (a) Gold 2003: New Industrial Applications for Gold, Proceeding Volume, World Gold Council, London, 2003 Search PubMed; (b) Y. D. Kim, M. Fischer and G. Gantefor, Chem. Phys. Lett., 2003, 377, 170 CrossRef CAS; (c) Y. Xu and M. Mavrikakis, J. Phys. Chem. B, 2003, 107, 9298 CrossRef CAS.
- C. Berg, H. J. Venvik, F. Strisland, A. Ramstad and A. Borg, Surf. Sci., 1998, 409, 1 CrossRef CAS.
- (a) E. Antolini, Mater. Chem. Phys., 2003, 78, 563 CrossRef CAS; (b) C. R. K. Rao and D. C. Trivedi, Coord. Chem. Rev., 2005, 249, 613 CrossRef CAS.
- H. Yang, W. Vogel, C. Lamy and N. Alonso-Vante, J. Phys. Chem. B, 2004, 108, 11024 CrossRef CAS.
- Nanoscale Materials in Chemistry, ed. K. J. Klabunde, John Wiley & Sons, Inc., New York, 2001 Search PubMed.
- Metal Nanoparticles: Synthesis, Characterization, and Applications, ed. D. L. Feldheim and C. A. Foss Jr., Marcel Dekker, Inc., New York, 2002 Search PubMed.
- (a) P. Waszczuk, G. Q. Lu, A. Wieckowski, C. Lu, C. Rice and R. I. Masel, Electrochim. Acta, 2002, 47, 3637 CrossRef CAS; (b) K. W. Park, J. H. Choi, B. K. Kwon, S. A. Lee, Y. E. Sung, H. Y. Ha, S. A. Hong, H. Kim and A. Wieckowski, J. Phys. Chem. B, 2002, 106, 1869 CrossRef CAS.
- T. J. Schmidt, H. A. Gasteiger and R. J. Behm, Electrochem. Commun., 1999, 1, 1 CrossRef CAS.
- (a) R. Raja, T. Khimyak, J. M. Thomas, S. Hermans and B. F. G. Johnson, Angew Chem., Int. Ed., 2001, 40, 4638 CrossRef CAS; (b) F. E. Jones, S. B. Milne, B. Gurau, E. S. Smotkin, S. R. Stock and C. M. Lukehart, J. Nanosci. Nanotechnol., 2002, 2, 81 CrossRef; (c) A. J. Dickinson, L. P. L. Carrette, J. A. Collins, K. A. Friedrich and U. Stimming, Electrochim. Acta, 2002, 47, 3733 CrossRef CAS.
- (a) M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC; (b) A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27 CrossRef CAS and references therein.
- (a) G. Schmid, V. Maihack, F. Lantermann and S. Peschel, J. Chem. Soc., Dalton Trans., 1996, 589 RSC; (b) U. A. Paulus, U. Endruschat, G. J. Feldmeyer, T. J. Schmidt, H. Bonnemann and R. J. Behm, J. Catal., 2000, 195, 383 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428 CAS.
- (a) M. A. El-Sayed, Acc. Chem. Res., 2001, 34, 257 CrossRef CAS; (b) C. J. Kiely, J. Fink, J. G. Zheng, M. Brust, D. Bethell and D. J. Schiffrin, Adv. Mater., 2000, 12, 640 CrossRef CAS.
- C. J. Zhong and M. M. Maye, Adv. Mater., 2001, 13, 1507 CrossRef CAS.
- (a) F. Caruso, Adv. Mater., 2001, 13, 11 CrossRef CAS and references therein; (b) J. J. Schneider, Adv. Mater., 2001, 13, 529 CrossRef CAS; (c) W. Schärtl, Adv. Mater., 2000, 12, 1899 CrossRef CAS.
- (a) J. J. Storhoff and C. Mirkin, Chem. Rev., 1999, 99, 1849 CrossRef CAS; (b) J. K. N. Mbindyo, B. D. Reiss, B. R. Martin, C. D. Keating, M. J. Natan and T. E. Mallouk, Adv. Mater., 2001, 13, 249 CrossRef CAS; (c) R. M. Crooks, M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181 CrossRef CAS.
- (a) S. H. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS; (b) S. H. Sun, E. E. Fullerton, D. Weller and C. B. Murray, IEEE Trans. Magn., 2001, 37, 1239 CrossRef CAS.
- M. Chen and D. E. Nikles, Nano Lett., 2002, 2, 211 CrossRef CAS.
- M. J. Hostetler, C. J. Zhong, B. K. H. Yen, J. Anderegg, S. M. Gross, N. D. Evans, M. D. Porter and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 9396 CrossRef CAS.
- (a) A. Crown, H. Kim, G. Q. Lu, I. R. de Moraes, C. Rice and A. Wieckowski, J. New Mater. Electrochem. Syst., 2000, 3, 275 Search PubMed; (b) J. D. Aiken III and R. G. Finke, J. Mol. Catal. A, 1999, 145, 1 CrossRef.
- (a) H. Li, Y.-Y. Luk and M. Mrksich, Langmuir, 1999, 15, 4957 CrossRef CAS; (b) R. S. Ingram and R. W. Murray, Langmuir, 1998, 14, 4115 CrossRef CAS.
- (a) X. Peng, M. C. Schlamp, A. V. Kadavanich and A. P. Alivisatos, J. Am. Chem. Soc., 1997, 119, 7019 CrossRef CAS; (b) S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243 CrossRef CAS.
- T. H. Galow, U. Drechsler, J. A. Hanson and V. M. Rotello, Chem. Commun., 2002, 1076 RSC.
- C. J. Zhong, J. Luo, M. M. Maye, L. Han and N. N. Kariuki, in Nanotechnology in Catalysis, ed. B. Zhou, S. Hermans and G. A. Somorjai, Kluwer Academic/Plenum Publishers, 2004, vol. 1, ch. 11, pp. 222–248 Search PubMed.
- J. Luo, L. Wang, D. Mott, P. Njoki, Y. Lin, T. He, Z. Xu, B. Wanjana, I.-Im S. Lim and C. J. Zhong, Adv. Mater., 2008 DOI:10.1002/adma.200703009.
- J. Luo, M. M. Maye, L. Han, N. Kariuki, V. W. Jones, Y. Lin, M. H. Engelhard and C. J. Zhong, Langmuir, 2004, 20, 4254 CrossRef CAS.
- S. Xiao, W. Hu, W. Luo, Y. Wu, X. Li and H. Deng, Eur. Phys. J. B, 2006, 54, 479 CrossRef CAS.
- (a) P. J. Hsu and S. K. Lai, J. Chem. Phys., 2006, 124, 44711 CrossRef CAS; (b) D. C. Meier and D. W. Goodman, J. Am. Chem. Soc., 2004, 126, 1892 CAS; (c) J. E. Bailie and G. J. Hutchings, Chem. Commun., 1999, 2151 RSC.
- R. Meyer, C. Lemire, Sh. K. Shaikhutdinov and H.-J. Freund, Gold Bull., 2004, 37, 72 CAS.
- M. Ø. Pedersen, S. Helveg, A. Ruban, I. Stensgaard, E. Lægsgaard, J. K. Nørskov and F. Besenbacher, Surf. Sci., 1999, 426, 395 CrossRef CAS.
- S. Strbac and R. R. Adzic, J. Electroanal. Chem., 1996, 403, 169 CrossRef CAS.
- L. Fu, N. Q. Wu, J. H. Yang, F. Qu, D. L. Johnson, M. C. Kung, H. H. Kung and V. P. Dravid, J. Phys. Chem. B, 2005, 109, 3704 CrossRef CAS.
- F. Tielens, J. Andres, M. Van Brussel, C. Buess-Hermann and P. Geerlings, J. Phys. Chem. B, 2005, 109, 7624 CrossRef CAS.
- (a) J. Zhang, M. B. Vukmirovic, K. Sasaki, A. U. Nilekar, M. Mavrikakis and R. R. Adzic, J. Am. Chem. Soc., 2005, 127, 12480 CrossRef CAS; (b) J. Zhang, M. B. Vukmirovic, Y. Xu, M. Mavrikakis and R. R. Adzic, Angew. Chem., Int. Ed., 2005, 44, 2132 CrossRef CAS.
- D. I. Garcia-Gutierrez, C. E. Gutierrez-Wing, L. Giovanetti, J. M. Ramallo-Lopez, F. G. Requejo and M. Jose-Yacaman, J. Phys. Chem. B, 2005, 109, 3813 CrossRef CAS and references therein.
- (a) C. Damle, K. Biswas and M. Sastry, Langmuir, 2001, 17, 7156 CrossRef CAS; (b) N. Toshima, M. Kanemaru, Y. Shiraishi and Y. Koga, J. Phys. Chem. B, 2005, 109, 16326 CrossRef CAS.
- (a) X. W. Teng, D. Black, N. J. Watkins, Y. L. Gao and H. Yang, Nano Lett., 2003, 3, 261 CrossRef CAS; (b) H. Zeng, J. Li, Z. L. Wang, J. P. Liu and S. H. Sun, Nano Lett., 2004, 4, 187 CrossRef CAS; (c) Z. Xu, Y. Hou and S. Sun, J. Am. Chem. Soc., 2007, 129, 8698 CrossRef CAS.
- (a) N. Toshima, Y. Shiraishi, A. Shiotsuki, D. Ikenaga and Y. Wang, Eur. Phys. J. D, 2001, 16, 209 CrossRef CAS; (b) J. I. Park, M. G. Kim, Y. W. Jun, J. S. Lee, W. R. Lee and J. Cheon, J. Am. Chem. Soc., 2004, 126, 9072 CrossRef CAS.
- (a) L. Wang, J. Luo, Q. Fan, M. Suzuki, I. S. Suzuki, M. H. Engelhard, Y. Lin, N. Kim, J. Q. Wang and C. J. Zhong, J. Phys. Chem. B, 2005, 109, 21593 CrossRef CAS; (b) L. Wang, J. Luo, M. M. Maye, Q. Fan, Q. Rendeng, M. H. Engelhard, C. Wang, Y. Lin and C. J. Zhong, J. Mater. Chem., 2005, 15, 1821 RSC; (c) H. Y. Park, M. J. Schadt, L. Wang, I.-I. S. Lim, P. N. Njoki, S. H. Kim, M. Y. Jang, J. Luo and C. J. Zhong, Langmuir, 2007, 23, 9050 CrossRef CAS.
- (a) Q. Ge, C. Song and L. Wang, Comp. Mater. Sci., 2006, 35, 247 Search PubMed; (b) C. Song, Q. Ge and L. Wang, J. Phys. Chem. B, 2005, 109, 22341 CrossRef CAS.
- J. Luo, L. Han, N. N. Kariuki, L. Wang, D. Mott, C. J. Zhong and T. He, Chem. Mater., 2005, 17, 5282 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |