Bacteria and yeasts as catalysts in microbial fuel cells: electron transfer from micro-organisms to electrodes for green electricity
Olivier Schaetzlea, Frédéric Barrière*a and Keith Baronianb
aUniversité de Rennes 1, CNRS UMR n° 6226, Sciences Chimiques de Rennes, Equipe MaCSE, France. E-mail: frederic.barriere@univ-rennes1.fr
bSchool of Applied Science, Christchurch Polytechnic Institute of Technology, P.O. Box 540, Christchurch, New Zealand
First published on 20th August 2008
Abstract
This article reviews the use of micro-organisms as catalysts at the electrodes of microbial fuel cells (MFCs). The principle of MFCs and their intended use for water treatment and clean electricity production is discussed. We address the different microbial structure and metabolic pathways found in prokaryote (bacteria) and eukaryote (yeasts) that allow the understanding of why electron transfer is possible between a microbe and an electrode. The different mechanisms of microbe–electrode electron transfer are discussed: direct electron transfer or through natural nanowires (pili), mediated electron transfer by natural or artificial redox mediator and finally direct redox transformation of excreted metabolites at the electrodes. This is followed by a review of the different bacteria that have been found and studied in MFCs mainly in the anodic compartment but also more recently in the cathodic side of the fuel cells. A perspective on the possible advantages and challenges of the use of yeasts in MFCs is provided, as this aspect has not been thoroughly studied so far. The fourth section of the review focuses on how to improve the performance and sustainability of MFCs through the functionalisation of the electrode surface, for instance with the covalent grafting of redox mediators and/or enzymes.
 Olivier Schaetzle | Olivier Schaetzle was born near Strasbourg in 1981 and graduated in Biology and Environmental Sciences from the Université Louis Pasteur in 2007. He is currently a Chemistry PhD student in Rennes working on Microbial Fuel Cells. |
 Frédéric Barrière and Keith Baronian | Frédéric Barrière was born and educated in Brest. During his PhD in Bioinorganic Chemistry (Mo nitrogenase), he worked in Brighton, Brest and Norwich with Chris Pickett and Jean Talarmin. He was a postdoctoral associate with Bill Geiger (Vermont, Molecular Electrochemistry) and with Dónal Leech (Galway, Bioelectrochemistry). He is a lecturer at the Université de Rennes I. Keith Baronian is a yeast microbiologist in CPIT and collaborates with Neil Pasco (Lincoln University), Alison Downard (University of Canterbury) and Gotthard Kunze (IPK, Germany). |
1 Introduction
Microbial fuel cells are fascinating bio-electrochemical devices that use living catalysts to draw electric energy from organic matter present naturally in the environment or in waste.1 The bio-catalysts are micro-organisms living at the surface of electrodes or in the electrolyte. The general principle of a microbial fuel cell is schematically shown on Fig. 1 and relies on the oxidation of organic matter by the microbes that are living in an anaerobic medium. In the absence of dioxygen, or other natural electron acceptors, some microbial species can, or have developed the ability to, transfer electrons to the anode of a fuel cell. The electric current/energy is then used through a load in the external circuit, and eventually reduces at the cathode an electron acceptor such as dioxygen. The anolyte and the catholyte are often separated with a membrane to avoid mixing of the two compartments and keep the anolyte as oxygen free as possible.
There are possible alternatives to this respiration mechanism such as the use of trans Plasma Membrane Electron Transport (tPMET) systems as in yeast, however the amount of energy that may be recovered is much less. In some instances that shall be discussed later, microbial catalysts can also be found at the cathode,2 where they catalyze the reduction of inorganic ions or the re-oxidation of reduced redox mediators. The first demonstration that a microbial fuel cell can produce electrical power came in 1912 with the publication of Potter.3 Since then microbial fuel cell research has been rather scarce and confidential until the renaissance of the field in the last decade or so, fuelled in part by the renewed interest into the search for alternative and clean energy sources. Another impetus for MFC research is the coupling of bio-energy production to the cleaning of waste waters. If practical, this technology could drive down the cost of wastewater treatment plants by recovering and possibly selling energy while bioremediating polluted water.4
It is a matter of debate as to whether these bio-electrochemical devices will ever provide a significantly high power source.5,6 The best currently available prototypes may deliver a power density of ca. 1 to 5 Watts per square meter of electrode or 50 to 100 Watts per cubic meter of the whole fuel cell volume. Hence, despite the recent dramatic increase of the MFCs power outputs, these absolute figures remain low and rather modest. Nevertheless, a real world application has recently appeared in the form of a powering device for a marine meteorological buoy.7 This type of microbial fuel cell is a so-called benthic cell as the anode is implanted in marine sediments where enough organic matter and depletion of oxidants allow for microbial anodic electron transfer to a non-corrosive electrode (e.g. graphite). The anode of the benthic microbial fuel cell is connected through the electrical circuit (the buoy in this case) to a non-corrosive cathode floating at the surface in an oxygen rich environment. To overcome limitations of the benthic MFC such as variation in power and limited voltage the MFC was wired to a voltage conditioner to meet the requirements of the powered electronic instruments and was able to charge a battery or a capacitor. According to the authors, their improved device is able to power instruments with an average consumption of ca. 100 mW. Although this figure might seem low, it is sufficient for this application and the main advantages of the device lie in the absence of maintenance (for example changing batteries), and the robustness and persistence of power generation (a minimum of two years operation without depletion of power is claimed for this technology).
The MFC field is hence set for great promises and challenges: much fundamental interdisciplinary research is needed to improve the power output of these devices and to widen their possible use beyond the niche applications that have been targeted up to now. The coupling of efficient power production by MFCs to practical waste water bioremediation has to be demonstrated as well. In this review, we first focus on the nature of the organisms responsible for bioelectrocatalysis in MFCs, their diversity, their structure and the mechanisms which allow these species to transfer electrons with an electrode. This discussion starts with bacteria, the prokaryote organisms that are used in virtually all of the current MFCs.8 The discussion on prokaryotes is followed by a section on yeasts. The eukaryote organisms may indeed be considered as well for MFCs despite their more complex cellular organisation (Fig. 2) that may, at first sight, appear less suitable for MFCs. The extracellular electron transfer mechanisms found in both kinds of organisms however have strong similarities. Possible advantages of using yeasts in future devices will also be discussed. Indeed, in Potter's original and pioneering study,3 it is relevant that both bacteria and yeast were tested for electricity generation. Power generation with MFC was demonstrated with one species of bacterium (Escherichia coli) and one species of yeast (Saccharomyces cerevisiae). This observation immediately raises fundamental mechanistic questions given the marked differences between prokaryotes and eukaryotes (Fig. 2). It is ironic that the two organisms successfully selected by Potter that provide the de facto proof of principle for MFC (the prokaryote E. coli bacteria and the eukaryote S. cerevisiae yeast) are apparently not the most suitable micro-organisms for that purpose. While the use of yeasts in general does not seem to have been considered further very deeply, recent progress has been made with E. coli, vide infra. Nevertheless, E. coli does not appear to be the best prokaryote species for MFC applications.
 | ||
| Fig. 2 Extracellular electron transfer paths in bacteria and yeasts. Electron transport chain (ETC) seems to play the most important role in electron transfer to the anode in bacteria. The electron transfer may be direct or mediated. The use of redox mediators, switching between an oxidized state (Medox) and a reduced state (Medred) is required with yeast in order to reach the ETC located at the mitochondrion within the cytoplasm. Another kind of transfer, involving trans Plasma Membrane Electron Transport (tPMET),9 can be considered for both organisms as a way to transmit electrons. | ||
The possible electron transfer paths between a microbe and an electrode include (a) direct electron transfer upon which the active centre of the membrane enzyme is directly connected to the electrode. In such a case, the electron transfer rate can be very low due to the insulation of the active site of the enzyme in the protein environment and the isolation of the enzyme from the electrode surface by its relative burial into the bacterial membrane. For some exoelectrogens species, however, the redox enzymes involved in electron transfer to electrodes may be located at the outer surface of the micro-organism membrane, and oriented as to the active site at the periphery of the redox enzyme is facing towards the external medium (or towards the electrode).89 In these circumstances (e.g. with Geobacter sulfurreducens), cyclic voltammetry studies show that this allows electron transfer at a rather high rate.91 (b) Another direct electron transfer path has been identified: It involves biological nanowires of 2–3 μm long called pili, made of fibrous protein structures. These thin protruding “wires” presumably facilitate direct electron transfer between the microbe and the electrode. (c) Mediated electron transfer usually proceeds at much faster rates. It consists of relying on an added or naturally occurring redox active species, stable in two redox states, which are able to quickly diffuse in and out of the enzymatic channels, hence effectively shuttling electrons from the enzyme active site to the electrode surface. Much higher electron transfer rates are obtained in this way. However, it is important to emphasize at this stage that relying on soluble additives in the anolyte is obviously not compatible with the purpose of water purification. (d) Another promising mechanism involves the direct oxidation at the anode of exported catabolites by the microbes, such as dihydrogen or formate for example.10
Analysis of the literature using the “microbial fuel cell” keyword (in ScienceFinder Scholar™, June 2008) shows that about 400 articles have been published in this field so far, with more than 360 from 2002. A number of recent reviews covering different aspects of the field are available (see references 2,4,11–15) and a book authored by Logan has been published this year.1
The last section of the present review will therefore focus only on the most recent advances and ideas that are being put forward through the current increasing interdisciplinary research effort in this field. The emphasis will be laid on advances with electrode surface modifications.
2 Bacterial biofuel cells
2.1 Bacteria at the anodic side
To improve the efficiency of microbial fuel cells, it is important to understand how and why micro-organisms exchange electrons with the fuel cell system. Indeed, it is important to have some notion of metabolism just to understand why external electron transfer is so essential for the bacterium and how the exchange with the electrode is occurring.Bacteria can use a large variety of organic compounds as carbon sources. Lipids, proteins and carbohydrates can be all processed to supply the organism with carbon and energy. These organic substrates serve as electron donors for a complex system of redox reactions that result in the production of an energy carrier molecule (ATP). Through different reactions, lipids, carbohydrates and proteins can be converted through glycolysis and related processes into the acetyl unit of acetyl–CoA. This molecule is then fed into the citric acid cycle, where oxidation reactions are coupled to the reduction of NAD+ and FAD to their electron carrier forms, NADH and FADH2 (Fig. 3).
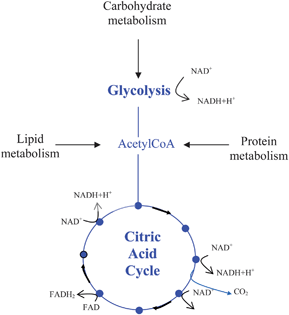 | ||
| Fig. 3 Reduction of NAD+ and FAD to their electron carrier forms (NADH and FADH2) through the Citric Acid Cycle (also known as tricarboxylic acid cycle or TCA cycle). | ||
These electron carriers then transfer electrons from the cytoplasm, where the citric acid cycle occurs, to the cell membrane. It is in the membrane where all the explanation of the need of this electron transfer resides. Indeed, before being transmitted to a terminal electron acceptor (dioxygen or any reducible inorganic compound that can be used), electrons are transferred through different membrane intermediaries, some of them pumping protons out of the cell as they are reduced. The energy of the proton gradient, mediated through the ATP synthase transmembrane protein, is used by the cell to phosphorylate ADP to produce ATP, the chemical energetic currency of living organisms (Fig. 4). The process in which ATP is produced by reduction of an inorganic terminal electron acceptor is called respiration.16
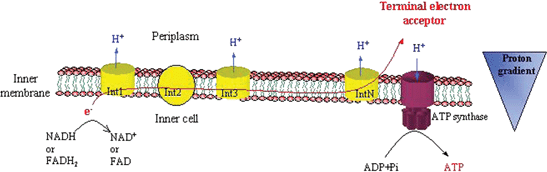 | ||
| Fig. 4 Schematic representation of bacterial membrane respiration. Note: the number of components of the electron transport chain varies with species. | ||
Bacteria are able to substitute an electrode as the terminal electron acceptor in the anodic compartment of MFCs. Investigation of the way that different strains of bacteria are “linked” to the electrode is leading to an understanding of the potentials (Fig. 5) at which electrons may be released to the electrode.
 | ||
| Fig. 5 Standard oxidation–reduction potentials (pH 7, 25 °C, vs. ENH) of some electron transport chain molecules. Electrons may leave the system at a number of places in the electron transport chain (ETC). | ||
The link between the last redox intermediary within the membrane and the terminal electron acceptor differs from one organism to another. Electron transfer to an electrode depends on where the intermediary is located in the membrane structures of the cell and if it is able to shuttle electrons out of the cell.
The microbial diversity observed in these devices showed very varied communities. Indeed, the nature and the diversity of the micro-organisms present in MFCs is a function of several factors such as the origin of the sample, the nature of the fuel, the presence of redox mediator, and the oxic conditions.13,17,18 The nature and diversity of the micro-organisms will be different if, for example, the MFC contains activated sludge or marine sediment. In most situations proteobacteria (Gram-negative) dominate the communities' composition and the ratios between α- β- γ- and δ- proteobactria are very different according to the nature of the inoculum. It was shown for example that α-proteobacteria comprised 64.5% of the communities present in a MFC fed with artificial wastewater and only 10.8% when fed by river water.17 This illustrates the sensitivity of the communities' composition to the operational parameters of the fuel cell (Fig. 6). Among proteobacteria some species showed the ability to reduce iron or manganese as terminal electron acceptors and thus have the ability to directly transfer their electrons to an electrode. The ability of Shewanella putrefaciens (γ-proteobacteria) to reduce these metals has been intensively studied.19 Some Geobacter (δ-proetobacteria) species also showed direct electron transferring properties5 and will be discussed later.
 | ||
| Fig. 6 Example of a phylogenetic tree showing 16S rDNA gene sequences of the bacterial community from MFC enriched with acetate. From: J. Lee et al.17 | ||
As well as proteobacteria, a number of other bacteria were shown to be present. The way in which species interact in the biofilm formed on the electrode however remains unknown.1 It is clear though that complex consortia are more efficient in power output than are pure cultures,1 and this is probably the result of the networks of metabolisms that function and which are yet to be clarified. Moreover, complex communities in MFCs are likely to be able to more efficiently metabolise complex fuel compositions than would a single culture.
Bacteria that are able to transfer electrons outside the cell and thus can be used in MFCs without adding soluble exogenous mediators are called “exoelectrogens”.1 Only a few of these organisms have been isolated and identified.
The way the electron transfer occurs in these bacteria is not yet completely clear. Some of them have many c-type cytochromes in their outer membrane suggesting that a direct contact with the electrode may enable electronic transfer.20 Moreover, some Shewanella and Geobacter strains showed the ability to grow pili-like structures able to reach the electron-accepting surface.21 For some other strains it was shown that a direct contact was not needed, as they were able to shuttle the electrons out of the cell by synthesising their own soluble mediators.22,23
The classification and the comparison of the micro-organisms found in MFCs is not an easy task as the actual electron transfer mechanism might not always be known and the experimental conditions may differ widely in different studies. Table 1 lists some power and current density data that were obtained with different microbes. Direct comparison is to be avoided as many parameters change from one experiment to another. Indeed, among other experimental conditions, the internal resistance, the substrate, the nature of the electrodes and the external load may be very different from one experiment to another. It would be very valuable to the field to report a systematic study using exactly the same fuel cell design as to allow a comparison between microbial species.
| Microbes | Current density/mA m−2 | Power density/mW m−2 | References |
|---|---|---|---|
| Bacteria | |||
| Aeromonas hydrophila | 120 | 18 | 24 |
| Desulfobulbus propionicus | 28 (poised electrode) | 25 | |
| Desulfuromonas acetoxidans | 125 | 14 | 26 |
| Escherichia coli | 3390 | 1300 | 22 |
| Geobacter metallireducens | 320 | 38 | 27 |
| Geobacter sulfurreducens | 65 | 49 | 28 |
| Geopsychrobacter electrodophilus | 373 | 30 | |
| Pseudomonas aeruginosa | 4310 | 23 | |
| Rhodoferax ferrireducens | 31 | 33 | 5 |
| Shewanella putrefaciens | 312 | 10 | 97 |
| Clostridium sp. EG3 | 88 | 19 | 38 |
| Geothrix fermentans | 50 | 8 | 39 |
| Yeast | |||
| Saccharomyces cerevisiae | 108 | 32 | 54 |
A summary of the different strains that have been isolated from MFCs functioning without exogenous mediators is given next starting with proteobacteria and then non-proteobacteria.
Aeromonas hydrophila. A strain initially called AP3 was isolated from a mediator-less MFC fed by artificial wastewater containing acetate.24 Its study revealed that this facultative aerobic and rod-shaped Gram-negative bacterium was closely related to A. hydrophyla (γ-proteobacteria). This bacterium showed different responses to various combinations of electron donor acceptor. Surprisingly it was not able to grow on acetate with ferric citrate as electron acceptor whereas cyclic voltammetry showed electrochemical activity only in presence of ferric citrate. Air could reversibly inactivate these electrochemical properties. An MFC was inoculated with this strain and tested under different conditions. The maximum current that could be reached with yeast extract (1260 mg L−1) and under a load of 500 Ω was 0.3 mA (120 mA m−2). Acetate supply did not increase current. The role of the bacterium in the initial MFC fed by acetate is not clear. This micro-organism might help the conversion of fuel into electricity as a member of a consortium.
Desulfobulbus propionicus. Another group of δ-proteobacteria that is significantly enriched in marine sediment MFCs is the Desulfobulbaceae, a group of sulfate-reducing bacteria.25 The ability of D. propionicus to transfer electrons to a graphite electrode was studied with a + 0.52 V/SHE poised anode. The results showed that this bacterium could use the electrode surface as an electron acceptor with lactate, propionate, pyruvate, hydrogen and sulfur as electron donors. Lactate and propionate provided a maximum current production of around 28 mA m−2 whereas hydrogen and pyruvate produced less than 8 mA m−2. However, only 21 to 27% of the electrons available from the oxidation of lactate and propionate could be transferred to the electrode. Given that the bacterium could not oxidize acetate, led to the conclusion that Desulfobulbaceae were probably not playing an important role in current generation in marine sediment fuel cells.
Desulfuromonas acetoxidans. The study of microbial composition of marine sediment MFCs showed a pronounced enrichment of δ-proteobacteria, mostly from the Geobacteraceae family.26 According to sequence analysis, the isolate was most closely related to D. acetoxidans. This anaerobe, known to grow on acetate with concomittant reduction of Fe(III), was then tested in a MFC. The device produced 14 mW m−2, which was close to the 16 mW m−2 of the initial sediment battery. It was, however, suggested that these two power densities could not really be directly compared, as the two systems may not have had similar internal resistances.1 Use of anthraquinone-2,6-disulfonate (AQDS) as electron transfer mediator only increased the current production by 24% which suggested that the power generation was close to the limits of the system.
Escherichia coli. In the first study related to the bioelectochemical properties of micro-organisms3Bacillus coli (former name of E. coli) was shown to be able to generate up to 0.534 volts. However, the way it worked in this study is not fully understood. E. coli is not known to be able to generate current without exogenous mediators. Only recently, evolved E. coli cells were used successfully in a MFC without adding mediators.22 It was demonstrated that excretion of hydroquinone derivatives able to cross the outer membrane was realised in these cells, allowing the transfer of electrons to an anode. These latest results however are unlikely to explain what happened in Potter's original experiment as no evidence of an evolution process was used in this early study.3
Geobacter metallireducens. Sediment microbial fuel cell community studies showed a large proportion of δ-proteobacteria among which the iron reducing bacterium G. metallireducens seemed to play an important role.27 An MFC working with a pure culture of G. metallireducens seemed to produce as much power as a mixed culture coming from a wastewater inoculum (40 mW m−2 for pure culture and 38 mW m−2 for the mixed culture). However, this is likely to be a consequence of the limitation by the high internal resistance of the system, again highlighting the importance of a precise knowledge of the whole system.2 Experiments were performed in a single chamber MFC to test if acetate and butyrate could be used to produce electricity. Reported power densities were 506 mW m−2 for acetate and 305 mW m−2 for butyrate. However coulombic efficiencies only reached a maximum of 31% for acetate leading to the conclusion that a substantial part of the energy is used for other processes.
Geobacter sulfurreducens. G. sulfurreducens is another iron-reducing Geobacteraceae strain that was studied in MFC devices. It has been shown that a pure culture of this strain could produce electricity from acetate with electron recovery reaching 96.8%.28 In this experiment the pure culture was tested in a two chamber MFC either with an air cathode or with a poised anode (200 mV vs. Ag/AgCl) and similar yield of electron transfer was observed in both cases. This organism showed the capacity to produce nanowires similar to those of Shewanella oneidensis.29 The structure of these nanowires were thinner than the ones observed in S. oneidensis but in both cases, these wires are conductive and may be able to connect the cell to the final electron acceptor. The nanowires (pili) may also be involved in cell to cell electron transport.
Geopsychrobacter electrodophilus. Among Geobacteraceae that were shown to be enriched in marine sediment fuel cells was a psychrotolerant bacterium (i.e. resistant to relatively low temperature), G. electrodophilus. Two rod-shaped, curved, Gram-negative strains (A1T and A2) of this species were isolated from the electrode surface.30 These bacteria could grow at low temperature (4 °C to 30 °C) and both could oxidize various electron donors and use Fe(III) nitrilotriacetic acid as an electron acceptor. They also could oxidize various electron donors (acetate, malate, fumarate and citrate) with an electrode (poised at 0.52 V/SHE) as the sole electron acceptor. The electron transfer yielded around 90% from the complete oxidation of acetate with a maximum current production of 3.73 mA cm−2. Higher current production could be achieved with fumarate (A1T produced around 8.89 mA cm−2) and malate (A2 produced up to 121.43 mA cm−2).
Pseudomonas aeruginosa. This γ-proteobacteria is well known for its production of pyocyanin (E0 = −0.034 V vs. SHE pH 7, 30 °C), a proposed naturally secreted redox mediator. Experiments were undertaken to elucidate if the bacterial community of an MFC could naturally adapt to give an increased power output.23 This pyocyanin production suggested that it could be responsible of the increase of power output. Moreover, pyocyanin was shown to be used as a mediator by other species31 but it was also demonstrated that it could inhibit the electron transfer of other bacteria.
Rhodoferax ferrireducens. Another psychrotolerant bacterium, a β-proteobacteria, was tested in a MFC.5R. ferrireducens, a dissimilatory Fe(III)-reducing bacterium, which is able to grow between 4 and 30 °C, was isolated from anoxic subsurface sediment. This bacterium is a Gram-negative, short motile rod with a single polar flagellum. In a two chamber MFC this bacterium produced a steady current density of 31 mA m−2 with a coulombic efficiency of 81% with glucose as the electron donor and ferricyanide as the electron acceptor. A comparable amount of current was produced using fructose, sucrose and xylulose.
Rhodopseudomonas palustris. This α-proteobacteria was previously known for its ability to produce hydrogen. Recently, this photosynthetic purple non sulfur bacterium demonstrated high current and power densities in a microbial fuel cell.98 Indeed, the strain DX-1 produced 2720 mW.m−2 and 0.99 mA.cm−2. It is interesting to note that this is the first example of MFC that is more efficient with a pure culture than it is with mixed cultures.
Shewanella putrefaciens and Shewanella oneidensis. S. putrefaciens IR-1 was the first bacterial strain used in a MFC working without addition of exogenous mediators.20 This facultative anaerobic, rod-shaped and motile bacteria is a member of the γ-proteobacteria group able to reduce iron and manganese and use it as terminal electron acceptor. Several strains of S. putrefaciens were studied, some of them renamed as S. oneidensis.32 Anaerobic respiration in these strains is highly documented with a lot of articles investigating membrane proteins33–35 involved in metal oxides reduction. It has been shown that the synthesis of c-type outer membrane cytochromes increased in anaerobic conditions, suggesting that these membrane intermediaries could interact with iron or manganese oxides directly by contact (these compounds being insoluble at neutral pH). An exhaustive study investigated the nature of nanowires produced by S. oneidensis MR-1.21 These wires are produced only in the absence of oxygen and seem to act as a mechanism for the bacteria to reach the closest electron-accepting compound. Scanning tunnelling microscopy was used to show that the nanowires were conductive. Mutants lacking of some genes coding for some c-type cytochromes (MtrC and OmcA, thought to be at least partially exposed to the cell surface) showed the production of less conductive nanowires. Cyclic voltammetry indicated the presence of oxidation–reduction peaks that were considered to be a manifestation of outer membrane cytochromes with electrochemically active heme groups oriented toward the cell surface.20 Some other interpretations suggest that a mediator could be produced and then be responsible of these peaks.36 One of the most efficient MFCs built with a Shewanella strain (S. oneidensis DSP10) reached 3 W m−2 (500 W m−3) power density (note however that the small volume used in the calculation did not include the volume of the culture bottle).37
Clostridium sp. EG3. An electrochemically active, Fe(III)-reducing Gram-positive bacterium was isolated from a MFC fed with starch processing wastewater. Phylogenetic analysis showed that the strain is related to Clostridium butyricum.38 This strict anaerobe could grow at pH values between 5.5 and 7.4 (with optimum of 6.8–7.4) and between 14 and 42 °C. Other Gram-positive bacteria from the Clostridium genera have been known for their Fe(III) reducing ability but EG3 is the only one known to have electrochemical activity. This strain produced up to 0.22 mA (88 mA m−2) at the beginning of the exponential growth phase but it quickly dropped to 0.08 mA (32 mA m−2) in the mid exponential phase.
Geothrix fermentans. This species is the first non-proteobacteria shown to be able to reduce insoluble iron oxides using an electron shuttle.39 This observation was made by replacing the medium in an MFC with this isolate provoking a power production drop of 50%. Such experiments with other strains known not to produce soluble mediators showed no drop in power production with a new medium, indicating that in the case of G. fermentans a soluble self-produced mediator might be involved.
2.2 Bacteria at the cathodic side
Up to now, only a few publications have reported microbial cathodes. Typically MFCs have used oxygen-reducing cathodes. However, to be efficient this reaction requires expensive catalysts such as platinum. It is also possible to use other electron acceptors like permanganate or ferricyanide but these two reduction reactions are not sustainable and will lead to the accumulation of the reduced compounds.Using biocathodes could overcome the sustainability issues and might also lead to more complete wastewater treatment strategies.40 Two main strategies are known in terms of reduction process in biocathodes: the use of micro-organisms to regenerate manganese or iron oxides42–44 or the use of cathodic electrons for other reduction reactions.40,44–48
It is only recently that cathodes have been inoculated with the same kind of samples as anodes have been.46–49 In such MFCs, biocathodes were showed to biocatalyse the oxygen reduction46 reaching up to 83 W m−3 in a batch fed system and 65 W m−3 in a continuous flow system. Complete denitrification was also obtained in MFCs working with microbial cathodes.47,48 Denitrification occurs through four successive reactions (Fig. 7). For the first time, total denitrification was obtained in a MFC not relying on H2 as electron donor.47 Even though the denitrification rate was lower than in heterotrophic systems, energy could be recovered from the substrate. Through the use of such biocathodes, a more complete wastewater treatment process is available. MFCs able to both oxidize organic compounds and reduce nitrate are promising, especially in places were nitrate contamination is an issue for drinking water quality.
 | ||
| Fig. 7 Denitrification process from nitrate to nitrogen gas using 5 mole electrons per mole of nitrogen.47 | ||
Analysis of the microbial community of a biocathode inoculated with domestic wastewater and with nitrate used as the terminal electron acceptor in the cathode showed a great diversity.48 In this study, 50% of the clones found in the cathode biofilm were β-proteobacteria with species of the genera Nitrosomonas and Azovibrio restrictus. Bacteroidetes was the second most common group of organisms (21.6%).
Geobacter metallireducens and Geobacter sulfurreducens. Already studied for their exoelectrogenic abilities these bacteria were also shown to be able to catalyse the reduction of nitrate to nitrite by using cathodic electrons.45 In this work a great enrichment of Geobacteracea was shown with a poised electrode. However, this nitrate reduction ability of pure inoculum of G. sulfurreducens was only shown in this half-cell configuration and should be tested in a complete MFC. In a more recent study, a G. sulfurreducens biofilm on stainless steel cathodes was shown to be fully responsible for the reduction of fumarate.44 The high current density (higher than 2 A m−2) obtained suggests that stainless steel electrodes could be promising materials in such bioelectrochemical devices.
Leptothrix discophora. Manganese oxidation ability by L. discophora has been known for a number of years41 but it is only recently that this was exploited in a MFC.42L. discophora was used to oxidize Mn2+ to MnO2 releasing 2 electrons for oxygen reduction; MnO2 is then abiotically reduced at the cathode to Mn2+ allowing the cycle of manganese oxidation–reduction to be sustainable. The use of this biocathode resulted in an increase in power output of more than 40-fold.
Thiobacillus ferrooxidans. A similar strategy has also been used with T. ferrooxidans.43 Regeneration of Fe3+ is coupled with its abiotic cathodic reduction. This compensates for the low abiotic oxidation rate and allows sustainable use of ferricyanide. In this study it was shown that the use of this microbial cathode decreased the energy cost of their system by 35%.
3 A perspective on yeast biofuel cells
Yeast should be ideal biocatalysts for microbial fuel cells; most are non pathogens, many have high growth rates, some display very wide substrate ranges and they are robust and easily handled. Reports on the use of yeast in microbial fuel cells are however scarce. Potter,3 using a galvanic cell comprising a glass jar containing a porous cylinder with a platinum electrode each compartment connected through a circuit, demonstrated a significant open circuit potential difference between yeast cells suspended in a nutrient fluid in the outer cylinder and the same nutrient fluid in the inner porous cylinder. Controls and a reversal of the anode and cathode demonstrated that the yeast was the source of the EMF. He also showed that different substrates resulted in different EMFs and proposed that the method could be used to study the catabolism of the various substrates.More recent investigations have used a mediator to facilitate the transfer of electrons from cells to the anode and in some cases from the cathode to an electron acceptor. Bennetto et al.,51 Wilkinson,52 Chiao et al.53 used methylene blue as the anodic mediator and potassium hexacyanoferrate as the cathodic electron acceptor. E0 of methylene blue is 0.01 V at pH 7 and that of ferricyanide is 0.28 V. Thus, the potential difference of these fuel cells that could be expected is approximately 0.27 V (subject to the current generated). These authors, in general, report a potential of about 0.3 V. In some of the above reports, S. cerevisiae cells are described as unsatisfactory as a fuel cell catalyst e.g.‘sluggish’51 and being a ‘sluggish biocatalyst’ and having ‘lethargic’ responses to external substrates.52 Chiao et al.53 suggested that performance of their micromachined yeast fuel cell could be enhanced by changing to use ‘more active bacteria’.
Walker and Walker54 characterised the performance of a standard design batch fuel cell with platinum mesh electrodes. The cell potential was controlled with a potentiostat at either 0.3 V or 0.1 V. They varied yeast cell concentration, temperature, substrate concentration, agitation rate and aerobic and anaerobic conditions. Their results generally confirm what could be predicted from the known characteristics of the yeast. The exception is the performance of the yeast at high temperatures. The maximum temperature for the growth i.e. multiplication of most S. cerevisiae is generally between 35 °C and 37 °C. However, their results at ten minutes show a maximum power density per projected electrode surface area of 32 mW m−2 at 0.3V and power density at 55 °C was still significant (30 mW m−2). Unpublished data from our laboratory shows that current production by S. cerevisiae at 47 °C increased over the period of a 100 min experiment and that power density of the order of 200 mW m−2 was maintained over this time. Temperature tolerant strains are known and N. Kiran Sree et al.55 report the isolation of four strains that can grow and produce ethanol at 44 °C. The yield of biomass and ethanol however decreased significantly as the temperature increased from 35 °C to 44 °C. The observations by Walker and Walker and ourselves suggest that catabolism is maximal at around 45–50 °C whereas Sree et al. found that ethanol production dropped from 35 °C. Findings by Tony D'Amore et al.56 support this observation. This apparent contradiction needs further investigation to fully understand the effect of temperature on the catabolism of S. cerevisiae. A fuel cell operating at high temperature should produce more power and in the case of a pure culture device, reduce the possibility of contamination.
Wilkinson et al.57 propose a mechanism for the interaction of mediators with S. cerevisiae redox centres. They reasoned that there are two sources of electrons within the cell and selected mediators to target NADH and a component of the electron transport chain. Neutral red has an E0 of −0.325 V and is close to the potential of NADH and the potential of methylene blue, E0 = 0.011, is close to the succinate/fumarate (SUC/FUM) complex of the electron transport chain, E0 = 0.03 V. They reasoned that in an anaerobic environment with neutral red only, respiration would shut down because the potential of neutral red is too low to enable it to oxidise any electron transport molecules. The current produced would come from the two NADH per glucose molecule produced in glycolysis and would thus be small. Methylene blue on the other hand interacts with SUC/FUM in the electron transport chain which keeps ETC functioning and makes electrons from the TCA (tricarboxylic acid cycle) degradation of pyruvate available to the mediator resulting in increased current. A combination of both mediators resulted in methylene blue keeping electron transport, and thus TCA, functioning and allowed more efficient collection of electrons directly from TCA NADH by neutral red. The experimental results presented support the hypothesis but unfortunately experiments that could confirm the results were not done e.g. use of agents that block electron flow early in ETC to investigate the role of methylene blue in the proposed system. The idea that methylene blue does not oxidise NADH or NADPH is, however, not supported in the literature and there are many accounts of in vitro studies that describe the reduction of methylene blue by these molecules. There are some accounts of in vivo interactions, for example May et al.58 showed that both NADPH and NADH were oxidised by methylene blue in red blood cells. On the other hand the notion that a mediator can promote respiration in anoxic conditions is supported by Zhao et al.59 who demonstrated that the mediator menadione (E0 = 0.14 V vs. SHE pH 7) was able to promote respiration in yeast in an anaerobic environment whereas two other lipophilic mediators, 2,6 dichlorophenolindophenol (DCPIP, E0 = 0.217 V) and N′N″N′N″-tetramethyl-p-phenylenediamine (TMPD, E0 = 0.25 V) did not. They propose that the proximity of the E0 of menadione to that of coenzyme Q (E0 = 0.045 V) enables menadione to act as a competitor with Coenzyme Q for electrons from NADH and thus provides a terminal electron acceptor in the absence of oxygen, resulting in the continuation of respiration. TMPD and DCPIP on the other hand are not able to act as ETC terminal electron acceptors and the organism loses respiratory activity and becomes fermentative. The signals detected with these two mediators are proposed to be from NADH produced in glycolysis. Some differences exist between the menadione and TMPD results obtained by this group and results reported by Baronian et al.60 It could be that there are strain differences within S. cerevisiae. In any case it seems that further work is required to clarify the way in which various mediators interact and affect catabolism in eukaryotes.
The use of mediators in a flow-through fuel cell of the type proposed for the treatment of dilute wastes such as sewage is impractical and some kind of direct i.e. non-mediated transfer of electrons from the cell to the anode is required. The major problem in the use of yeast as catalysts in flow-through fuel cells is that they are eukaryotes and have the catabolic mechanisms located within the cytoplasm (glycolytic pathways) and in the matrix and the inner membrane of the mitochondria (TCA and catabolic electron transport, respectively), which results in the majority of catabolic electrons not being directly accessible from the exterior of the cell i.e. it would appear that a mediatorless yeast fuel cell is unlikely to be successful. In contrast, prokaryote cells have the catabolic electron transport system located in the cell membrane (the glycolytic pathway and TCA are both located in the cytoplasm), which results in all catabolic electrons being potentially available from the external surface of the cell membrane. In addition bacteria such as P. aeruginosa have been demonstrated to export reduced redox molecules23 and direct transfer of electrons from the cell to an electrode via pili has been demonstrated.29 A third mechanism that enables the transfer of electrons from bacterial cells to an electrode is seen in S. putrefaciens which has redox proteins located in the outer membrane.20 The development of mediatorless bacterial microbial fuel cells has exploited all three of these direct electron transfer mechanisms.
In common with all cells, however, yeast have trans Plasma Membrane Electron Transport systems (tPMET) also referred to as Plasma Membrane Oxido-Reductase systems (PMOR). These systems lie across the membrane and export electrons from reduced cytoplasmic molecules such as NADH and NADPH to an external electron acceptor. The exported electrons are, for example, used to prepare external nutrients for uptake as in the reduction of Fe2+ to Fe3+.61 Some electrons are thus available at the cell membrane surface for either direct or mediated transfer to the electrode. The number of electrons exiting the cell by this route is however very small when compared to total number of electrons available from the catabolism of aerobically grown cells. In some yeasts the rate of glycolysis does increase in anaerobic conditions to compensate for the loss of mitochondrial ATP production resulting in an increase in cytoplasmic NADH/NADPH concentrations and thus the real effect of tPME-only transfer of electrons from yeast in an anaerobic anode may not be as limited as the following results suggest.62Fig. 8 (from Baronian et al.60) shows that the difference in current detected between aerobically grown yeast cells incubated with a hydrophilic mediator and yeast cells incubated with a lipophilic mediator is approximately 400-fold. This difference can be explained as follows: the hydrophilic mediator cannot cross the cell membrane and can only access tPMET proteins on the membrane surface whereas the lipophilic mediator can cross the membranes of the cell and mitochondria to access internal reduced molecules (NADH and NADPH).
 | ||
| Fig. 8 Treatment groups (t g) 1 and 2, current from S. cerevisiae surface redox molecules using a hydrophilic mediator (potassium ferricyanide). Treatment groups 3 and 4, current from S. cerevisiae surface and internal redox processes using a hydrophilic and lipophilic mediator couple (potassium ferricyanide and menadione). Y axis is a logarithmic scale of current in nA. Experimental: Incubation of fresh S. cerevisiae cells for 1 h with: t g 1; 20 mM ferricyanide, t g 2; 20 mM ferricyanide and 7.5 mM glucose, t g 3; 20 mM ferricyanide and 100 μM menadione, t g 4; 20 mM ferricyanide, 7.5 mM glucose and 100 μM menadione. Each treatment was performed in triplicate and each sample was analysed in triplicate. | ||
Potter3 conducted his experiments without the assistance of a redox mediator and it must be assumed that any electron transfer from the cells to the electrode was either by direct electron transfer from the cell surface or by the export of reduced molecules from the cell. This observation has been repeated by Chiao et al.53 who demonstrated a small current in the absence of an anode mediator and more recently by Prasad et al.63 (Fig. 9). Mediatorless electron transport to a gold disk electrode and a gold disk electrode modified with a self-assembled monolayer of cystamine was demonstrated using cyclic votammetry and a maximum power density of 2.9 W m−3 was obtained in a fuel cell. This power density is at the low end of values for a number pure bacterial cultures64 which where as high as 18 W m−3 (Proteus vulgaris).
 | ||
| Fig. 9 Cyclic voltammograms demonstrating the electrochemical activity of Hansenula anomala using electrode assembly II; scan rate 50 mV s−1. (1) Blank CV in phosphate buffer; (2–4) after successive additions of 0.5 ml each of lactate stock solution (0.5 M). From: D. Prasad et al.63 This result shows an essentially non-reversible peak at approximately 0.44 V. We have also seen a similar peak in CVs from Arxula adeninivorans. Preliminary work in our lab indicates that this organism does not export soluble redox molecules but it does produce some power in a microbial fuel cell in the absence of a mediator (K. Baronian, unpublished data). | ||
Although the evidence supports direct electron transfer in yeast, direct electron transfer from tPMET molecules in the yeast cell membrane to an electrode does not seem to be a likely explanation for the findings of Prasad et al.63 and ourselves. The yeast cell wall is very dense65 and the distance from the exterior of the cell membrane to the outside of the cell wall is large (cell wall 100–200 nm, periplasmic space 35–45 Å) and thus direct contact between the cell membrane and an electrode does not seem possible. Wartman et al.66 did however provide evidence for the existence of a ferrireductase on the exterior surface of the cell wall of A. adeninivorans and this may be the explanation for the observed results.
4 Recent advances in MFC research
As mentioned in the introduction, current MFCs deliver a rather low power output that, for the moment, precludes their widespread use outside of niche applications. The above discussions on bacteria and yeasts found in MFCs show that much needs to be learnt about the precise mechanisms of extracellular electron transfer from micro-organisms to an electrode in order to improve MFCs. In this section a selection of the most recent advances in the study of exoelectrogens and in the design of improved MFCs is reviewed.4.1 Alternative cathodes for MFC
In addition to improving the performance of the anaerobic microbial anode and reducing the internal resistance of the fuel cell to a minimum, improving the performance of the cathode is a relevant way to significantly increase power production by MFCs. For convenience and study purposes, or simply to boost power, alternative electron acceptors like ferricyanide or potassium permanganate have been used at the cathode. Again, this approach is costly and is not sustainable as it requires a regular replenishing of electron acceptors in the catholyte and also involves leaching out of pollutants (e.g. metal ions) into the environment. A better suited electron acceptor for MFCs is dioxygen as it is freely and widely available, it is one of the natural electron acceptors in microbial respiration, and in the best-case scenario, it can be thermodynamically reduced at high potential to harmless water (E0 = 0.82 V vs. SHE at pH 7). The kinetic reality is however much less favourable. Dioxygen reduction is notoriously difficult and intense research is being carried out, especially in the fuel cell field, to improve the performance of oxygen cathodes' materials.67 This fact can be simply realized by recording a voltammogram at a carbon working electrode in an O2 saturated electrolyte: even at rather low current densities the overpotential is well over 500 mV. The overpotential is diminished with expensive metal catalysts like platinum but remains significant. Hence, to address this critical problem several approaches have been developed. One approach consists of developing or selecting known molecular O2 reduction catalysts followed by their immobilisation at the surface of the cathode where they retain their catalytic properties. Suitable candidates for that matter are found for example within the extended family of transition metal complexes of porphyrins and phthalocyanines.67,68 These catalysts reduce dioxygen in several steps and, depending on their relative rates and pH conditions, the reduction may stop at the hydrogen peroxide level, which in turn may degrade the electrode and/or the immobilized catalytic centres. An approach that has been successfully followed for some years is the pyrolytic heat treatment of the catalyst in an inert gas, which creates a protective carbon matrix in which the active catalytic centres may be embedded and protected.68 Among other factors, the method is interesting because it relies on relatively low cost transition metal ions such as iron or cobalt. Similar approaches with related coordination complexes include the heat treatment of iron phenanthroline coordination complexes on carbon for example.69 An interesting and original alternative to the use of molecular “bio-mimetic” catalysts is the direct use of dioxygen reducing enzymes (oxygenases) at specifically designed cathodes. These types of cathodes have been developed for miniature enzymatic biofuel cells that may find applications as a low power source for small electronic devices to be implanted in vivo.6 The combination of a refined microbial anode with a robust enzymatic cathode, together with a fuel cell design that minimize internal resistance, might be the perfect mix for high power MFCs. Some oxygenases reduce dioxygen directly to water at very high potentials (very close to the thermodynamic value for O2/H2O). Hence, for example, the blue copper protein70 laccase71–73 or bilirubin oxidase74,75 have been used as the biocatalyst in enzymatic biofuel cells.6 Because of the relative insulation of the enzyme catalytic site within the protein matrix, the use of redox enzyme as catalysts requires redox mediators to achieve significant catalytic current at the electrode. The redox mediator and enzyme may be in solution or co-immobilized at the surface of the electrode, for example through cross-linking of the enzyme with a redox polymer. Such an electrode type, based on laccase, has been shown to be more efficient than platinum for the electroreduction of dioxygen.76 In the case of laccase, ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) is a well known and efficient redox mediator as it is well suited to the active site of the enzyme in terms of size, charge and structure. At the University of Rennes 1, we have compared the performance of a microbial fuel cell with a bare platinum cathode and with the laccase/ABTS catalytic system. We have found that the maximum power output of the MFC increases by one order of magnitude with the enzymatic cathode.77 This very encouraging result may pave the way toward improved hybrid microbial and enzymatic fuel cell devices. The living catalysts at the anode are persistent by nature since they can reproduce. For an enzymatic electrode however, the issue of stability and performance over time is critical. In order to address this problem, recent approaches have recently appeared in the literature.78–81 These include the entrapment of enzyme and mediator into a polymer,78 the covalent attachment79,80 or strong adsorption81 of the enzyme onto a pre-functionalized electrode by electroreduction of appropriate aryldiazonium salts, a popular and versatile method for the covalent functionalization of conducting surfaces, and especially carbon.82–84 The added value of the immobilization routes reported,80-81 is that efficient direct electron transfer was obtained. This means that using these strategies and high surface area electrodes, sufficient current densities may be reached without resorting to any redox mediators.As discussed in the section on bacteria, optimized microbial cathodes are also being developed and may be more interesting for the reduction of polluting inorganic ions, such as nitrate for instance, rather than for reducing O2. As will be discussed in the next section, any electrode surface modification, regardless of its refinement, may be simply clogged or eaten and digested, in a medium where microbial life is thriving. If true, this latter point may be a strong argument for the development of a microbial cathode and/or for seeking much more robust and microbial-resistant surface modifications.
4.2 Redox mediators at MFC electrodes
In this section, the redox mediation concept developed in the preceding section still applies with respect to microbial electrodes. Indeed, it is even more relevant since, in addition to the insulation of the enzyme redox active site within the protein matrix, the protein itself may be buried in a thick lipidic membrane. This certainly seems to preclude even more, any prospect of direct electron transfer between a microbe and an electrode. The efficient (but not sustainable) use of added redox mediators led researchers to attempt the robust and persistent attachment of redox mediators at the surface of electrodes. Recent examples include the covalent functionalisation of gold85 and carbon surface86 with an osmium coordination complex, stable in water in the +II and +III oxidation state. The electrode surface modification protocol takes advantage of thiol chemistry (on gold) or aryl diazonium reduction,82–84 followed by coupling chemistry between the functionalized surface and the functionalized pyridine ligand of the Os complex (Fig. 10). Although this type of redox active surface was shown to be very stable over time (a minimum of six months stability has been demonstrated),86 its behaviour in terms of efficient redox mediator for microbial electrodes and its resistance to microbial degradation remain to be investigated.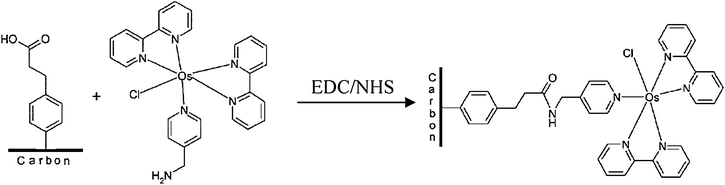
Very recently, Adachi and co-coworkers have reported the incorporation of a sulfonated anthraquinone (or AQDS, a widely used organic redox mediator with E0 = −0.184 V vs. SHE) into a polymer and its use at the anode of a microbial fuel cell.87 The advantage of the redox polymer approach may be the increase of the total amount of surface bound redox active species: the authors calculated a mediator surface density of the order of 10−7 mol cm−2 compared with ca. 10−10 mol cm−2, which is the order of magnitude for a monolayer. Considering the redox mediation between the redox proteins of the microbial membrane and the electrode; the structure and properties of the surface modification (length and flexibility, lipophilicity, etc.) is likely to be more critical than the absolute number of surface redox sites. The positive effect on power output obtained by Adachi and co-workers with their modified anode was also tested over time and a stable current density was claimed for a minimum of four months.87 The engineering and the refining of microbial electrode surfaces modifications will certainly be one the most important challenges in the near future since it can both increase power output and permit the sustainability of the MFC technology.
Despite their significant effect on MFCs power output, it is important to realize that added mediators, be they immobilized or not, are not essential for electron transfer between a microbe and an electrode. The rather recent discovery that some bacteria are able to develop thin filaments (pili) involved in extracellular and intermicrobial electron transfer is relevant.21 These can be seen as the natural refinement of the bacterial membrane that may be complementary to the engineering of the electrode surface suggested above, for an optimized interfacial electron transfer. Indirect proof that pili are indeed involved in electron transfer may come from genetic engineering. Richter and co-workers88 have shown that G. sulfurreducens can exchange electrons with a gold anode, an electrode substrate that was shown to be poorly compatible with S. putrefaciens in MFCs. Interestingly, Richter and co-workers report that no current was obtained if the gene responsible for a structural protein of the pili is deleted.88 Combining genetic studies with surface investigation techniques may yield an even more detailed picture on the structure of these conductive so-called “nano-wires” and of the external architecture of the bacteria cell membrane. Recently, a thin film gold electrode has also been used with G. sulfurreducens in order to combine electrochemical study with ATR-SEIRAS (attenuated total reflection surface enhanced infrared absorbtion spectroscopy). The authors demonstrate electron transfer between the polarized gold electrode (0.2 V vs. Ag/AgCl) and the concomitant detection of infra-red adsorption band assigned to proteins and lipids connecting the electrode surface. A new band at 1600 cm−1 was assigned to the c-type cytochromes known to be involved in extracellular electron transfer. The change in adsorption spectra has been followed as the electrode was polarized from −0.1 to + 0.4 V. A mid point potential was calculated to be 0.17 V from the spectro-electrochemical measurement and was consistent with the electrochemical measurement.89
Direct electron transfer from engineered electrodes and/or evolved microbes, should not hinder the fact that natural secretion of the redox mediator may be quite a simple and widespread mechanism for connecting exoelectrogens to electrodes. In a recent paper by Marsili et al.90 flavins have been identified as naturally secreted redox mediators in Shewanella. The biochemical reason for the secretion of natural redox mediator is open to questions.
4.3 Challenges and new ideas
Approaching the end of this review article, it is important to comment on several important aspects that are not possible to treat comprehensively in a concise format. It is apparent from the discussion at the end of the preceding section and throughout the paper that electrochemical studies are critical and powerful ways to study exoelectrogens and MFCs, especially in combination with other spectroscopic or genetic techniques. Molecular electrochemistry is usually carried out in a pure electrolyte and the pure analyte is often a well behaved electrochemical system. Switching from these comfortable conditions to the study of bioactive sludges can then prove to be quite challenging with ill defined peaks, large capacitive current and so on. However, the different achievements found in the recent literature demonstrate that much is to be learnt provided a reasonable approach is followed and preferably combined with techniques from other fields of chemistry and biology.88–92As discussed at the end of this review surface modification is an important tool to introduce different functionalities at electrodes followed by attachment of redox mediators, for example. Other important properties might be conferred to the electrode surface by this approach like wettability, bio-compatibility or the promotion of bio-adherence, without sacrificing the required conducting properties of the electrode. A problem that might occur at a microbial anode is the building up of protons in the biofilm due to substrate degradation and poor agitation and diffusion.93 Here again, smart electrodes with surface bound acid–base functionalities and channels might provide a solution.
To conclude this account, we would like to mention the development suggested by recent papers of a plant microbial fuel cell.94–96 The principle is shown in Fig. 11 and first involves carbon fixation by plants through photosynthesis. Some of the fixed carbon is used for plant growth but a fraction of it, which can be quite high in some species, is exudated through the roots as rhizodeposits. These organic molecules would then in turn be used as fuel and oxidized back to CO2 at the microbial anode of an MFC. Using a plant–MFC prototype, D. Strik et al. were able to establish this principle and reported a maximum power output of 67 mW per square meter of anode. Similarly, a sediment type MFC was recently shown to increase its power output by one order of magnitude in the presence of living rice plants.95

5 Conclusions
The field of microbial fuel cells is experiencing rapid growth with the merging of approaches from different fields: microbiology, biochemistry, electrochemistry, material science and so on. This translates to much more detailed and fundamental insights into the intimate mechanism of extracellular microbial electron transfer and a consistent improvement of microbial fuel cell design and performance. Microbial fuel cell research is attractive because it is multidisciplinary, and because it holds much promise in terms of the current energy and environmental crises. MFC researchers will do their best to contribute to the meeting of societal and political demands on these issues. What they will not do, however, is break the laws of thermodynamics. The present time is therefore not only a blessing for this field, but it also holds a risk of overselling and disappointment. It is the opinion of the authors that this fascinating field of research should continue to focus on fundamental and high standard science and develop constructive dialogue between different specialists who, at times, seem to have very different scientific cultures. We hope that the present review modestly contributes to this dialogue. Following this approach, we are confident that MFCs will indeed contribute to future energy production and to wastewater treatment plants, and that this research will also be fruitful in related fields such as bioprocesses, biosensing and surface science, for example.Acknowledgements
O. S. thanks ADEME and Région Bretagne for a studentship. The France–New Zealand Dumont d'Urville exchange program is gratefully acknowledged.References
- B. E. Logan, Microbial Fuel Cells, Wiley, New York, 2008 Search PubMed.
- Z. He and L. T. Angenent, Electroanalysis, 2006, 18, 2009–2015 CrossRef CAS.
- M. C. Potter, Proc. R. Soc. London, Ser. B, 1912, 84, 260–276 CAS.
- K. Rabaey and J. Keller, Water Sci. Technol., 2008, 57, 655–659 Search PubMed.
- S. K. Chaudhuri and D. R. Lovley, Nat. Biotechnol., 2003, 21, 1229–1232 CrossRef.
- A. Heller, Phys. Chem. Chem. Phys., 2004, 6, 209–2162 RSC.
- L. M. Tender, S. A. Gray, E. Groveman, D. A. Lowy, P. Kauffman, J. Melhado, R. C. Tyce, D. Flynn, R. Petrecca and J. Dobarro, J. Power Sources, 2008, 179, 571–575 CrossRef CAS.
- At the First International Symposium on Microbial Fuel Cells held at Penn State University, USA, May 27–29, 2008, all papers dealt with bacteria and none was given that involved yeasts. A sample of presentations given at this meeting is available at http://www.microbialfuelcell.org/ together with much useful other information on MFCs.
- A. Porat, S.-H. Cho and J. Beckwith, Resour. Microbiol., 2004, 155, 617–622 Search PubMed.
- M. Rosenbaum, F. Zhao, U. Schröder and F. Scholz, Angew. Chem., Int. Ed., 2006, 45, 6658–6661 CrossRef CAS.
- U. Schröder, Phys. Chem. Chem. Phys., 2007, 9, 2619–2629 RSC.
- B. E. Logan and J. M. Regan, Environ. Sci. Technol., 2006, 40, 5172–5180 CAS.
- B. E. Logan, B. Hamelers, R. Rozendal, U. Schröder, J. Keller, S. Freguia, P. Aelterman, W. Verstraete and K. Rabaey, Environ. Sci. Technol., 2006, 40, 5181–5182 CrossRef CAS.
- Environ. Sci. Technol., 2006, 40(17). Special Section on Microbial Fuel Cells in this issue Search PubMed.
- Z. Du, H. Li and T. Gu, Biotechnol. Adv., 2007, 25, 464–482 CrossRef CAS.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W.H. Freeman and Company, 5th edn, 2001 Search PubMed.
- J. Lee, N. T. Phung, I. S. Chang, B. H. Kim and H. C. Sung, FEMS Microbiol. Lett., 2003, 223, 185–191 CrossRef CAS.
- N. Kim, Y. Choi, S. Jung and S. Kim, Biotechnol. Bioeng., 2000, 70, 109–114 CrossRef CAS.
- C. R. Myers and K. H. Nealson, J. Bacteriol., 1990, 172, 6232–6238 CAS.
- B. H. Kim, H.-J. Kim, M.-S. Hyun and D. H. Park, J. Microbiol. Biotechnol., 1999, 9, 127–131 CAS.
- Y. A. Gorby, S. Yanina, J. S. McLean, K. M. Rosso, D. Moyles, A. Dohnalkova, T. J. Beveridge, I. S. Chang, B. H. Kim, K. S. Kim, D. E. Culley, S. B. Reed, M. F. Romine, D. A. Saffarini, E. A. Hill, L. Shi, D. A. Elias, D. W. Kennedy, G. Pinchuk, K. Watanabe, S. Ishii, B. Logan, K. H. Nealson and J. K. Fredrickson, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11358–11363 CrossRef CAS.
- Y. Qiao, C. M. Li, S.-J. Bao, Z. Lu and Y. Hong, Chem. Commun., 2008, 11, 1290–1292 RSC.
- K. Rabaey, N. Boon, S. D. Siciliano, M. Verhaege and W. Verstraete, Appl. Environ. Microbiol., 2004, 70, 5373–5382 CrossRef CAS.
- C. A. Pham, S. J. Jung, N. T. Phung, J. Lee, I. S. Chang, B. H. Kim, H. Yi and J. Chun, FEMS Microbiol. Lett., 2003, 223, 129–134 CrossRef CAS.
- D. E. Holmes, D. R. Bond and D. R. Lovley, Appl. Environ. Microbiol., 2004, 70, 1234–1237 CrossRef CAS.
- D. R. Bond, D. E. Holmes, L. M. Tender and D. R. Lovley, Science, 2002, 295, 483–485 CrossRef CAS.
- B. Min, S. Cheng and B. E. Logan, Water Res., 2005, 39, 1675–1686 CrossRef CAS.
- D. R. Bond and D. R. Lovley, Appl. Environ. Microbiol., 2003, 69, 1548–1555 CrossRef CAS.
- G. Reguera, K. D. McCarthy, T. Mehta, J. S. Nicoll, M. T. Tuomimen and D. R. Lovley, Nature, 2005, 435, 1098–1101 CrossRef CAS.
- D. E. Holmes, J. S. Nicoll, D. R. Bond and D. R. Lovley, Appl. Environ. Microbiol., 2004, 70, 6023–6030 CrossRef CAS.
- K. Rabaey, N. Boon, M. Höfte and W. Verstraete, Environ. Sci. Technol., 2005, 39, 3401–3408 CrossRef CAS.
- K. Venkateswaran, D. P. Moser, M. E. Dollhopf, D. P. Lies, D. A. Saffarini, B. J. MacGregor, D. B. Ringelberg, D. C. White, M. Nishijima, H. Sano, J. Burghardt, E. Stackebrandt and K. H. Nealson, Int. J. Syst. Bacteriol., 1999, 49, 705–724 Search PubMed.
- C. R. Myers and J. M. Myers, J. Bacteriol., 1997, 179, 1143–1152 CAS.
- C. R. Myers and J. M. Myers, Biochim. Biophys. Acta, 1997, 1326, 307–318 CAS.
- A. S. Beliaev and D. A. Saffarini, J. Bacteriol., 1998, 180, 6292–6297 CAS.
- D. K. Newman and R. Kolter, Nature, 2000, 405, 94–97 CrossRef CAS.
- B. R. Ringeisen, E. Henderson, P. K. Wu, J. Pietron, R. Ray, B. Little, J. C. Biffinger and J. M. Jones-Meehan, Environ. Sci. Technol., 2006, 40, 2629–2634 CrossRef CAS.
- H. S. Park, B. H. Kim, H. S. Kim, H. J. Kim, G. T. Kim, M. Kim, I. S. Chang, Y. K. Park and H. I. Chang, Anaerobe, 2001, 7, 297–306 CrossRef CAS.
- D. R. Bond and D. R. Lovley, Appl. Environ. Microbiol., 2005, 71, 2186–2189 CrossRef CAS.
- Y.-H. Jia, H.-T. Tran, D.-H. Kim, S.-J. Oh, D.-H. Park, R.-H. Zhang and D.-H. Ahn, Bioprocess. Biosyst. Eng., 2008, 31, 315–321 Search PubMed.
- F. C. Boogerd and J. P. M. de Vrind, J. Bacteriol., 1987, 169, 489–494 CAS.
- A. Rhoads, H. Beyenal and Z. Lewandowski, Environ. Sci. Technol., 2005, 39, 4666–4671 CrossRef CAS.
- A. Lopez-Lopez, E. Exposito, J. Anton, F. Rodriguez-Valera and A. Aldaz, Biotechnol. Bioeng., 1999, 63, 79–86 CrossRef CAS.
- C. Dumas, R. Basseguy and A. Bergel, Electrochim. Acta, 2008, 53, 2494–2500 CrossRef CAS.
- K. B. Gregory, D. R. Bond and D. R. Lovley, Environ. Microbiol., 2004, 6, 596–604 CrossRef CAS.
- P. Clauwaert, D. van der Ha, N. Boon, K. Verbeken, M. Verhaege and W. Verstraete, Environ. Sci. Technol., 2007, 41, 7564–7569 CrossRef CAS.
- P. Clauwaert, K. Rabaey, P. Aelterman, L. de Schamphelaire, T. H. Pham, P. Boeckx, N. Boon and W. Verstraete, Environ. Sci. Technol., 2007, 41, 3354–3360 CrossRef CAS.
- G.-W. Chen, S.-J. Choi, T.-H. Lee, G.-Y. Lee, J.-H. Cha and C.-W. Kim, Appl. Microbiol. Biotechnol., 2008, 79, 379–388 CrossRef CAS.
- D. H. Park, M. Laivenieks, M. V. Guettler, M. K. Jain and J. G. Zeikus, Appl. Environ. Microbiol., 1999, 65, 2912–2917 CAS.
- D. H. Park and J. G. Zeikus, J. Bacteriol., 1999, 181, 2403–2410 CAS.
- H. P. Bennetto, Biotechnol. Educ., 1990, 1, 163–168 Search PubMed.
- S. Wilkinson, Auton. Robot., 2000, 9, 99 CrossRef.
- M. Chiao, K. B Lam and L. Lin, Proceedings of the IEEE Conference on Micro Electro Mechanical System, MEMS 2003, Kyoto, Japan, 2003, 383–386 Search PubMed.
- A. L. Walker and C. W. Walker, J. Power Sources, 2006, 160, 123–129 CrossRef CAS.
- N. K. Sreea, M. Sridhara, K. Suresha, I. M. Banatb and L. V. Rao, Bioresour. Technol., 2000, 72, 43–46 CrossRef.
- T. D'Amore, G. I. CelottoRussell and G. G. Stewart, Enzyme Microb. Technol., 1989, 11, 411–416 CrossRef CAS.
- S. Wilkinson, J. Klar and S. Applegarth, Electroanalysis, 2006, 18, 2001–2007 CrossRef CAS.
- J. M. May, Z. Qu and C. E. Cobb, Am. J. Physiol., 2004, 286, C1390–C1398 CrossRef CAS.
- J. Zhao, M. Wang, Z. Yang, Z. Wang, H. Wang and Z. Yang, Anal. Chim. Acta, 2007, 597, 67–74 CrossRef CAS.
- K. H. R. Baronian, A. J. Downard, R. K. Lowen and N. Pasco, Appl. Microbiol. Biotechnol., 2002, 60, 108–113 CrossRef CAS.
- E. Lesuisse and P. Labbe, Plant Physiol., 1992, 100, 769–777 CrossRef CAS.
- G. M. Walker, Yeast Physiology and Biotechnology, John Wiley and Sons,Chichester, 1998 Search PubMed.
- D. Prasad, S. Arun, M. Murugesan, S. Padmanaban, R. S. Satyanarayanan, S. Berchmans and V. Yegnaraman, Biosens. Bioelectron., 2007, 22, 2604–2610 CrossRef CAS.
- K. Rabaey and W. Verstraete, Trends Biotechnol., 2005, 23, 291–298 CrossRef CAS.
- F. M. Klis, A. Boorsma and P. W. J. De Groot, Yeast, 2006, 23, 185–202 CrossRef CAS.
- T. Wartmann, U. W. Stephan, I. Bube, E. Böer, M. Melzer, R. Manteuffel, R. Stoltenburg, L. Guengerich, G. Gellissen and G. Kunze, Yeast, 2002, 19, 849–862 CrossRef CAS.
- M. L'Her, Redox properties, electrochemistry of oxygen, in Encyclopedia of Electrochemistry: Inorganic electrochemistry, ed. A. J. Bard, M. Stratman, F. Scholz, C. J. Pickett, Wiley–VCH, Weinheim, 2006, vol. 7a, 119–142 Search PubMed.
- F. Zhao, F. Harnisch, U. Schroder, F. Scholz, I. Bogdanoff and I. Herrmann, Environ. Sci. Technol., 2006, 40, 5193–5199 CrossRef CAS.
- M. Bron, J. Radnik, M. Fieber-Erdmann, P. Bogdanoff and S. Fiechter, J. Electroanal. Chem., 2002, 535, 113–119 CrossRef CAS.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2577 CrossRef.
- V. Soukharev, N. Mano and A. Heller, J. Am. Chem. Soc., 2004, 126, 8368–8369 CrossRef CAS.
- F. Barrière, Y. Ferry, D. Rochefort and D. Leech, Electrochem. Commun., 2004, 6, 237–241 CrossRef.
- F. Barrière, P. Kavanagh and D. Leech, Electrochim. Acta, 2006, 51, 5187–5192 CrossRef CAS.
- S. Tsujimura, K. Kano and T. Ikeda, Electrochemistry, 2002, 70, 940 CAS.
- N. Mano, F. Mao and A. Heller, ChemBioChem, 2004, 5, 1703 CrossRef CAS.
- N. Mano, J. L. Fernandez, Y. Kim, W. Shin, A. J. Bard and A. Heller, J. Am. Chem. Soc., 2003, 125, 15290–15291 CrossRef CAS.
- O. Schaetzle, University of Rennes 1, unpublished work.
- A. Habrioux, G. Merle, K. Servat, K. B. Kokoh, C. Innocent, M. Cretin and S. Tingry, J. Electroanal. Chem., 2008 DOI:10.1016/j.jelechem.2008.05.011.
- M. Pellissier, F. Barrière, A. J. Downard and D. Leech, Electrochem. Commun., 2008, 10, 835–838 CrossRef CAS.
- C. F. Blandford, R. S. Heath and F. A. Armstrong, Chem. Commun., 2007, 1710–1712 RSC.
- C. Vaz-Dominguez, S. Campuzano, O. Rüdiger, M. Pita, M. Gorbacheva, S. Shleev, V. M. Fernandez and A. L. De Lacey, Biosens. Bioelectron., 2008 DOI:10.1016/j.bios.2008.05.002.
- A. J. Downard, Electroanalysis, 2000, 12, 1085 CrossRef CAS.
- J. Pinson and F. Podvorica, Chem. Soc. Rev., 2005, 34, 429 RSC.
- F. Barrière and A. J. Downard, J. Solid State Electrochem., 2008, 12, 1231–1244 CrossRef CAS.
- A. Ricci, C. Rolli, S. Rothacher, L. Baraldo, C. Bonazzola, E. Calvo, N. Tongalli and A. Fainstein, J. Solid State Electrochem., 2007, 11, 1511–1520 CrossRef CAS.
- S. Boland, F. Barrière and D. Leech, Langmuir, 2008, 24, 6351–6358 CrossRef CAS.
- M. Adachi, T. Shimomura, M. Komatsu, H. Yakuwa and A. Miya, Chem. Commun., 2008, 2055–2057 RSC.
- H. Richter, K. McCarthy, K. P. Nevin, J. P. Johnson, V. M. Rotello and D. Lovley, Langmuir, 2008, 24, 4376–4379 CrossRef CAS.
- J. P. Busalmen, A. Esteve-Núñez, A. Berná and J. M. Feliu, Angew. Chem., Int. Ed., 2008, 47, 4874–4877 CrossRef CAS.
- E. Marsili, D. B. Baron, I. D. Shikhare, D. Coursolle, J. A. Gralnick and D. R. Bond, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3968–3973 CrossRef CAS.
- K. Fricke, F. Harnisch and U. Schröder, Energy Environ. Sci., 2008, 1, 144–147 RSC.
- T. Zhang, C. Cui, S. Chen, H. Yang and P. Shen, Electrochem. Commun., 2008, 10, 293–297 CrossRef CAS.
- C. I. Torres, A. K. Marcus and B. E. Rittmann, Biotechnol. Bioeng., 2008, 100, 872–881 CrossRef.
- D. P. B. T. B. Strik, H. V. M. Hamelers, J. F. H. Snel and C. J. N. Buisman, Int. J. Energy Res., 2008, 32, 870–876 CrossRef CAS.
- L. De Schamphelaire, L. Van Den Bossche, H. S. Dang, M. Höfte, N. Boon, K. Rabaey and W. Verstraete, Environ. Sci. Technol., 2008, 42, 3053–3058 CrossRef CAS.
- N. Kaku, N. Yonezawa, Y. Kodama and K. Watanabe, Appl. Microbiol. Biotechnol., 2008, 79, 43–49 CrossRef CAS.
- D. H. Park and J. G. Zeikus, Appl. Microbiol. Biotechnol., 2002, 59, 58–61 CrossRef CAS.
- D. Xing, Y. Zuo, S. Cheng, J. M. Regan and B. E. Logan, Environ. Sci. Technol., 2008, 42, 4146–4151 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |